

Diamond-Blackfan anemia (DBA) is a congenital bone marrow failure syndrome, characterized by red blood cell aplasia, macrocytic anemia, variable malformations, and increased risk of malignancy.1–3 DBA has been associated with heterozygous mutations or large deletions in ribosomal protein (RP) genes in more than 50% of patients, except for rare germline GATA1 mutations reported in three X-linked DBA families.4 We recently identified two novel causative RP genes, RPS27 and RPL27, by whole-exome sequencing (WES) analysis of 48 patients without known causative mutations.5 To date, 14 RP genes (RPS7, RPS10, RPS17, RPS19, RPS24, RPS26, RPS27, RPS28, RPS29, RPL5, RPL11, RPL26, RPL27, and RPL35A) have been reported to be responsible for DBA.5–12 These mutations have been reported in up to 60% of DBA patients. However, the molecular etiology of many DBA patients remains to be clarified.
In this report, we performed WES to survey novel DBA disease-causing genes in a family with multiple individuals affected with DBA. We identified a novel DBA causative gene, RPS15A, and we used a human erythroid cell line and zebrafish as model systems to determine the consequences of the DBA-associated mutation. The proband was a girl born at a gestational age of 37 weeks to unrelated parents. Her birth weight was 2,155 g and her height was 47 cm. She had total anomalous pulmonary venous connection and bilateral acetabular dysplasia. She had a family history of anemia; her mother and older sister presented anemia during childhood. They do not have physical abnormalities. She was diagnosed with DBA at the age of three months. Peripheral blood analysis at diagnosis showed hemoglobin 88 g/L, mean corpuscular volume (MCV) 88.5 fL, white blood cell (WBC) 5.1 × 109/L with normal differential counts, platelet count 306 × 109/L, and 0.1% reticulocytes. Bone marrow aspiration showed severe selective erythroid hypoplasia (0%), but otherwise normal cellularity. She developed macrocytosis (MCV 113.0) at 14 years old. The proband, as well as her sister and mother, responded to the initial corticosteroid therapy, and eventually became steroid-independent. The clinical characteristics of the affected family members are presented in Online Supplementary Table S1. All of them were negative for mutations or deletion in 11 known DBA causative genes (RPL5, RPL11, RPL27, RPL35A, RPS7, RPS10, RPS17, RPS19, RPS24, RPS27 and GATA1) and RPS14, which is implicated in the 5q- myelodysplastic syndrome characterized by a defect in erythroid differentiation.5,13
We then performed whole-exome sequencing (WES) of all family members. All clinical samples were obtained with informed consent from the patients and family. The Ethics Committee of Hirosaki University Graduate School of Medicine and the University of Tokyo approved this study. Genomic DNA (gDNA) was extracted from peripheral blood leucocytes with the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany), enriched for protein-coding sequences with a SureSelect Human all Exon V5 kit (Agilent Technologies, Santa Clara, CA, USA) and followed by massive parallel sequencing with the HiSeq 2000 platform with 100 bp paired-end reads (Illumina, San Diego, CA, USA) as described previously.5,14 To validate the detected mutations, we performed direct sequencing analysis using gDNA and primers described in Online Supplementary Methods.
WES revealed a putative splicing mutation at the third exon of RPS15A (c.213G>A, p.K71K) in the affected members, but not the unaffected father.
The mutation had not been registered in the SNP database either of the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/) or the Tohoku Medical Megabank Organization (https://ijgvd.megabank.tohoku.ac.jp/). The mutations were validated by direct sequencing analysis (Figure 1A). The estimate of the prevalence of RPS15A mutation is 1 of 141 families (0.7%) in our cohort.
Figure 1.
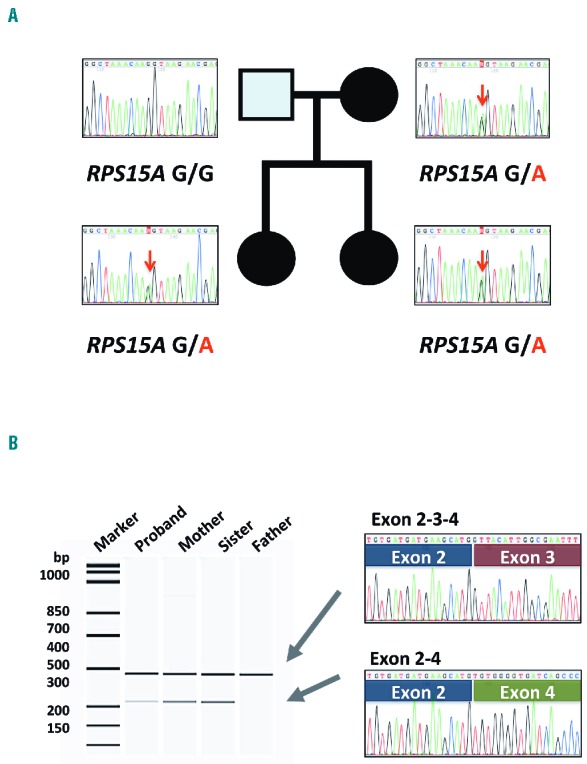
Pedigree of a Diamond-Blackfan anemia family with RPS15A mutation. (A) Each electropherogram indicates the gDNA sequence including the boundary between the third exon and IVS-3 of the RPS15A gene. Red arrows indicate the position of nucleotide substitution c.213G>A. (B) Reverse-transcriptase polymerase reaction (RT-PCR) analysis using the primer set located on the second and fourth exons of the RPS15A gene. Arrows indicate PCR products for the full-length variant and alternative splicing form lacking the third exon of RPS15A. Both transcripts were expressed only in the affected family members.
To confirm the effect of the mutation, we performed reverse transcriptase polymerase chain reaction (RT-PCR) analysis of all family members. We found a shorter transcript lacking the third exon by alternative splicing in addition to the full-length transcript only in the affected family members (Figure 1B).
We then performed CRISPR/Cas9 genome editing in the human erythroid cell line K562 to validate the effect of the RPS15A-deficiency on erythroid lineage cells. The CRISPR/Cas9 genome editing technique was performed using GeneArt CRISPR Nuclease Vector kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. Using two kinds of ssODNs (see Online Supplementary Methods), we introduced the mutation observed in the patients (c.213G>A) into K562 cells. We also used a silent mutation (c.207A>C) as a negative control. These transfected cells were cultured for 12 days continuously. We analyzed allele frequency in bulk DNA of each culture by targeted next generation sequencing with Miseq (Illumina) once every 3 days, up to 12 days. Paired t-test showed that the allele frequency of the DBA-associated RPS15A mutation (c.213G>A) was significantly decreased (see Online Supplementary Methods, Table S2 and Figure S1). These results suggested that the RPS15A mutation observed in the patients suppressed cell proliferation.
Several studies showed that the heterozygous mutations in RP genes observed in DBA disturb the synthesis of either the small or large subunit through defects in pre-ribosomal RNA (pre-rRNA) processing.6,8,9,15 To determine if the haploinsufficiency for RPS15A would affect pre-rRNA processing in erythroid lineage cells, we applied CRISPR/Cas9 technology to introduce mutations in one copy of RPS15A in K562 cells. We established a stable cell line (#75) with a heterozygous RPS15A mutation by limiting dilution after genome editing. This cell line expressed normal and alternative splicing forms lacking the second exon of RPS15A (Online Supplementary Figure S2). Northern blotting analysis showed the decrease of 18S-E pre-rRNA expression and the accumulation of 30S pre-rRNA in #75 cell line as well as RPS15A, RPS19 or RPS27 knocked-down cells by siRNAs (Figure 2) (see Online Supplementary Methods).5 No differences were discernable other than the known effect of RPL5 knockdown on increased 32S. These results suggested that haploinsufficiency for RPS15A would disturb processing of 18S rRNA.
Figure 2.
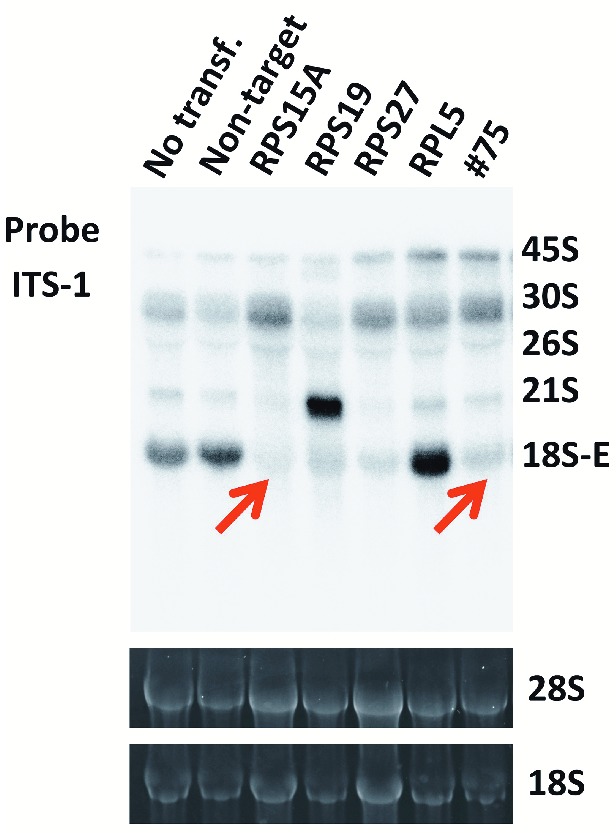
Perturbation of pre-rRNA processing by knockdown or heterozygous knockout of RPS15A. Northern blot analysis of pre-rRNA processing in K562 cells in which we knocked down RPS15A, RPS19, RPS27 or RPL5 by siRNAs or heterozygously knocked out RPS15A by CRISPR/Cas9 gene editing (#75). The 5′-extremities of the internal transcribed spacer 1 (ITS-1) were used as probes to detect the precursors to the 18S rRNA associated with the small subunit. The UV images of the gels were used to judge loading consistency. The decrease of 18S-E pre-rRNA was detected by the ITS-1 probe in RPS15A knockdown cells and #75 cell line (red arrows) as well as RPS19 and RPS27 knockdown cells.
Because a limited number of RP genes have been shown to cause DBA, it is likely that haploinsufficiency for some RP genes is either harmless or lethal. We showed that knockdown of RPS15A expression with siRNA disturbed pre-ribosomal RNA processing in the human erythroid cell line K562. However, most knockdowns with siRNA will decrease newly synthesized RP by well over 50%, and it is very difficult to control the knockdown levels. To overcome this problem, we successfully introduced a heterozygous RPS15A mutation into K562 cells by CRISPR/Cas9 genome editing and established a stable cell line showing disturbed pre-ribosomal RNA processing (Figure 2). This cell model system might be very useful for functional analysis of these variants.
Finally, to investigate the effects of the RPS15A mutations in vivo, we knocked down the zebrafish ortholog (rps15a) using a morpholino antisense oligonucleotide (MO), after which we analyzed the morphology and erythropoietic states during embryonic development. The coding region of rps15a shares 81% of the nucleotides and 99% of the amino acid identities with its human ortholog. MO was designed on the 5′-splice site of the second intron of rps15a that corresponds to the position at which the mutation was identified in the patients (Figure 3A). Injection of 0.5–2.5 μg/μL rps15a MO into the one-cell stage embryos perturbed the splicing as observed in the patients and resulted in exclusion of exon 3 (Figure 3B). The amount of the shorter transcript was increased with increases in MO concentration.
Figure 3.
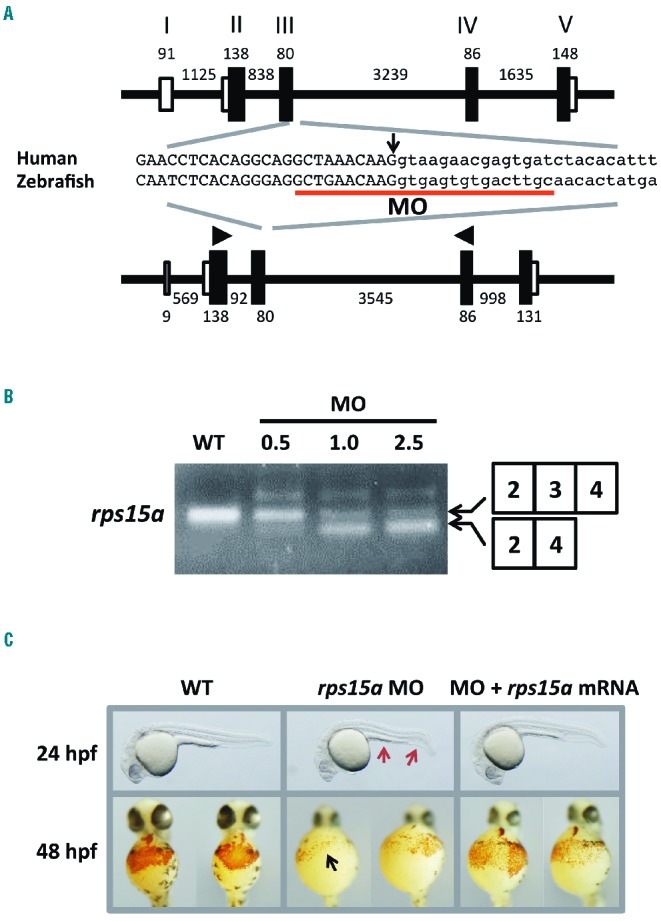
Morphological features and hemoglobin staining of Rps15a-deficient zebrafish. (A) The gene structures of human RPS15A (NM_001019.4) and zebrafish rps15a (NM_212762.1). The MO target site is underlined. The arrow indicates the position of the mutated nucleotide in the patients. Arrowheads show the primer positions for the RT-PCR. Black and white boxes indicate coding and non-coding exons, respectively. Black lines indicate introns. (B) The results of RT-PCR of rps15a in wild-type and MO-injected embryos. A smaller transcript without exon 3 was observed in the morphants as seen in the patients at a comparable level. The amount of smaller transcripts correlated with the concentration of injected MOs. (C) Morphological features of wild-type and MO-injected embryos. A thin yolk sac extension and a bent tail (red arrows) were prominent in the morphants injected with 1.0 μg/μL MO targeted against rps15a, whereas these features were rescued in the embryos injected with rps15a mRNA at 400 ng/μL. Compared to the wild-type embryos, rps15a morphants injected with MO showed a drastic reduction in the number of hemoglobin-stained blood cells (black arrow). Morphants co-injected with rps15a mRNA showed recovery of the stained cells.
We compared the MO-injected embryos (morphants) with wild-type embryos in terms of their morphological features and erythropoietic development. The morphants showed abnormal phenotypes, such as a thin yolk sac extension and a bent tail at 24 hours post fertilization (hpf) (Figure 3C). These phenotypes were similar to other known DBA morphants. We performed hemoglobin staining at 54 hpf and found a marked reduction of erythrocyte production in the cardiac vein of the morphants (Figure 3C). These abnormalities were more prominent in the morphants that were injected with MO at higher concentrations (Online Supplementary Figure S3). All these abnormalities were rescued by the simultaneous injection of rps15a mRNA into the embryos, indicating that the abnormal phenotypes were caused by the aberrant splicing of rps15a in zebrafish. These results suggested that the splice site mutation identified in human RPS15A could be responsible for the pathogenesis of DBA.
In conclusion, WES analysis identified RPS15A loss-of-function mutations in a DBA family. We demonstrated that the mutation caused splicing aberrations. The RPS15A mutation observed in the patients led to suppressed cell proliferation in K562 cells. Heterozygous knock-in of the mutation by CRISPR/Cas9 gene editing disturbed pre-rRNA processing for the small subunit. The zebrafish model of RPS15A mutation showed impairment of erythrocyte production. These results suggested that RPS15A plays an important role in erythropoiesis and that haploinsufficiency of RPS15A, like haploinsufficiency of other ribosomal protein genes, caused Diamond-Blackfan anemia. This discovery adds another RP gene to the growing list of mutated genes in DBA patients and supports DBA as a disorder of the ribosome.
Supplementary Material
Acknowledgments
The authors would like to thank H. Kudo Y. Kudo and A. Mikami for their technical assistance.
Footnotes
Funding: this work was supported by Practical Research Project for Rare/Intractable Diseases (15ek0109133) and Grant-in-Aids (15ek0109099h001) from the Japan Agency for Medical Research and Development (AMED) and the Research on Measures for Intractable Diseases Project and Health and Labor Sciences Research grants (Research on Intractable Diseases) from the Ministry of Health, Labour and Welfare and JSPS KAKENHI Grant Numbers JP2591003, JP15K09656.
The online version of this paper contains a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ito E, Konno Y, Toki T, et al. Molecular pathogenesis in Diamond-Blackfan anemia. Int J Hematol. 2010;92(3):413–418. [DOI] [PubMed] [Google Scholar]
- 3.Vlachos A, Rosenberg PS, Atsidaftos E, et al. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012;119(16):3815–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klar J, Khalfallah A, Arzoo PS, et al. Recurrent GATA1 mutations in Diamond-Blackfan anaemia. Br J Haematol. 2014;166(6):949–951. [DOI] [PubMed] [Google Scholar]
- 5.Wang R, Yoshida Y, Toki T, et al. Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond-Blackfan Anemia. Br J Haematol. 2015;168(6):854–64. [DOI] [PubMed] [Google Scholar]
- 6.Doherty L, Sheen MR, Vlachos A, et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2010;86(2):222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Draptchinskaia N, Gustavsson P, Andersson B, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nature Genetics. 1999;21(2):169–175. [DOI] [PubMed] [Google Scholar]
- 8.Gazda HT, Grabowska A, Merida-Long LB, et al. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79(6):1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gazda HT, Preti M, Sheen MR, et al. Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in diamond-blackfan anemia. Hum Mutat. 2012;33(7):1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerrard G, Valgañón M, Foong HE, et al. Target enrichment and high-throughput sequencing of 80 ribosomal protein genes to identify mutations associated with Diamond-Blackfan anaemia. Br J Haematol. 2013;162(4):530–536. [DOI] [PubMed] [Google Scholar]
- 11.Konno Y, Toki T, Tandai S, et al. Mutations in the ribosomal protein genes in Japanese patients with Diamond-Blackfan anemia. Haematologica. 2010;95(8):1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirabello L, Macari ER, Jessop L, et al. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multi-case Diamond-Blackfan anemia families. Blood. 2014;124(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008; 451(7176):335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunishima S, Okuno Y, Yoshida K, et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet. 2013;92(3):431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choesmel V, Bacqueville D, Rouquette J, et al. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood. 2007;109(3):1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.