

Abstract
Significance: The biological mechanisms at the heart of the aging process are a long-standing mystery. An influential theory has it that aging is the result of an accumulation of molecular damage, caused in particular by reactive oxygen species produced by mitochondria. This theory also predicts that processes that protect against oxidative damage (involving detoxification, repair, and turnover) protect against aging and increase lifespan. Recent Advances: However, recent tests of the oxidative damage theory, many using the short-lived nematode worm Caenorhabditis elegans, have often failed to support the theory. This motivates consideration of alternative models. One new theory, conceived by M.V. Blagosklonny, proposes that aging is caused by hyperfunction, that is, overactivity during adulthood of processes (particularly biosynthetic) that contribute to development and reproduction. Such hyperfunction can lead to hypertrophy-associated pathologies, which cause the age increase in death. Critical Issues: Here we assess whether the hyperfunction theory is at all consistent with what is known about C. elegans aging, and conclude that it is. In particular, during adulthood, C. elegans shows a number of changes that may reflect pathology and/or hyperfunction. Such changes seem to contribute to death, at least in some cases (e.g., yolk accumulation). Future Directions: Our assessment suggests that the hyperfunction theory is a plausible alternative to the molecular damage theory to explain aging in C. elegans. Antioxid. Redox Signal. 19, 321–329.
Introduction
What is aging? According to one theory, it is caused by molecular damage, particularly oxidation by reactive oxygen species (ROS). Here the superoxide free radical (O2−), hydrogen peroxide (H2O2), and their derivatives, generated as by-products of mitochondrial respiration, have been considered major culprits (22, 47).
This theory predicts that interventions that reduce levels of oxidative damage should retard aging, while those that increase levels of oxidative damage should accelerate aging. During the last decade, stringent tests of these predictions have been performed using several model organisms. Although the results have been mixed, a growing number of findings do not support the theory (39, 42).
This has created an exciting challenge to researchers: to conceive and test fundamentally different ideas about how aging occurs. In this essay, we explore one such new idea about the biology of aging that it is the result of continued activity (hyperfunction) during adulthood of pathways and processes that evolved to optimize development to adulthood. This idea argues that the resulting overgrowth causes pathologies that limit lifespan (4, 5). We will first briefly outline the crisis in the oxidative damage theory, then describe the hyperfunction theory, and finally ask whether it could explain aging in one key model organism, the nematode Caenorhabditis elegans.
The Oxidative Damage Theory in Crisis
Many studies that have used C. elegans to test the oxidative damage theory of aging have been previously surveyed (17, 52). In many cases, their results have been difficult to reconcile with the oxidative damage theory. For example, treatment with the inhibitor of glycolysis 2-deoxyglucose increases C. elegans lifespan, apparently by increasing mitochondrial ROS production (44). Similarly, lifespan is increased by treatment with low concentrations of the O2− generators juglone (23) and paraquat (59). Other irreconcilable studies involve the gene sod-2, which encodes the major mitochondrial Mn superoxide dismutase (SOD) in C. elegans. SOD is an antioxidant enzyme that detoxifies O2−, converting it into H2O2. sod-2 mutants are hypersensitive to oxidative stress and show elevated levels of protein oxidation, yet their lifespan is unchanged (13, 60) or even increased (51). Even worms in which all five sod genes are deleted do not show any reduction in lifespan (53).
One interpretation of these studies is that oxidative damage is a part of a broader spectrum of molecular damage that causes aging (16). However, a more radical interpretation that we consider here is that molecular damage accumulation is not the primary cause of aging at all.
Aging As Quasi-Programmed Hyperfunction
The hyperfunction theory, proposed by Mikhail Blagosklonny, encompasses central elements of the biology of aging, including interventions that slow aging (dietary, pharmacological, and genetic), the oxidative damage theory, cellular senescence, the relationship between aging and disease, and the evolutionary theory of aging. In broad terms, what Blagosklonny has done is to remove certain elements from the existing conceptual framework of biogerontology, add some new ideas, and rearrange the rest. The result is a radically new, coherent integrated theory that presents a very different big picture of aging (4–6).
This conceptual reorganization hinges on two ideas in particular. First, that aging is not the result of molecular damage. Second, that the real cause of aging is hyperfunction, especially excess biosynthesis (hypertrophy). This hypertrophy is driven by the nutrient-sensitive signaling network that controls growth (and thereby, reproduction), and includes the insulin, insulin-like growth factor 1 (IGF-1), and in particular, the target of rapamycin (TOR) kinase pathways (8). Hypertrophy leads to a broad spectrum of pathologies in late life that cause the age increase in mortality (Fig. 1A, B).
FIG. 1.

The hyperfunction theory of aging. (A) Simplified representation of molecular damage theory (including oxidative damage theory). (B) Simplified representation of hyperfunction theory. (C) Mode of action of target of rapamycin (TOR) pathway on aging, based on the molecular damage theory. (D) Mode of action of TOR pathway on aging, based on the hyperfunction theory. Derived from (5). IIS, insulin/IGF-1 signaling.
Of the signaling network controlling growth, TOR is viewed as the critical element controlling aging. This is reasonable, particularly since inhibition of the TOR pathway increases lifespan in budding yeast, C. elegans, Drosophila, and mice (28). Moreover, TOR appears to mediate effects on lifespan of dietary restriction (DR) in yeast, worms, and flies. Pharmacological inhibition of TOR with rapamycin increases lifespan in animal models (including mice), raising the possibility that it may do so in humans.
Crucially, the hyperfunction theory proposes mechanistic explanations for how TOR controls aging, for example, by regulating biomass within cells. TOR inhibits autophagy and promotes translation of mRNA into protein. Model organism studies have shown that autophagy is required for life extension in a number of contexts, and that inhibition of protein synthesis can increase lifespan (28). The molecular damage theory was able to explain why autophagy promotes longevity: it removes damaged molecules. However, why protein synthesis should promote aging has been a puzzle.
Autophagy disposes of damaged cellular constituents. It also reduces biomass and recycles cellular components during times of nutrient deprivation to assure survival. The hyperfunction theory suggests that it is reduction of biomass that is critical to longevity assurance, rather than removal of damaged molecules (Fig. 1C, D). The theory also provides a simple explanation for why protein synthesis promotes aging, because it contributes to hypertrophy. Inhibition of protein translation reduces hypertrophy, thereby retarding the development of pathology and delaying age-related mortality.
According to the hyperfunction theory, the life-threatening diseases of aging should typically be driven by hypertrophy. Moreover, this hypertrophy should be associated with overactivity of TOR. In fact, this is often the case (5, 49). For example, in mammals, insulin resistance tends to increase with age, which to some extent is driven by overactivation of TOR in fat and skeletal muscle. TOR-dependent hyperactivity in osteoclasts contributes to osteoporosis, leading to bone fractures. Various forms of hypertrophy contribute to the loss of blood supply that leads to myocardial infarction, atherosclerosis, cardiomyocyte hypertrophy, increased coagulation and platelet hyperactivity, hypertension, and inflammation. Cancer, of course, is a hyperplastic disease. Plausibly, the reduced cancer levels in long-lived rodents (mutant or under-DR) reflect suppression of hyperplasia rather than reduced levels of founder mutations. In short, there is an association between TOR signaling, hyperfunction, and age-related disease in mammals.
However, does molecular damage really not contribute to aging? Blagosklonny suggests that there are two relationships between damage and aging. First, he observes that molecular damage surely does accumulate with time, but it makes little contribution to mortality. This is because hyperfunction causes death long before damage has had time to accumulate to a level sufficient to cause life-threatening pathology. Second, he suggests that major contributors to the observed age increase in levels of damage are the pathologies caused by hyperfunction.
Hyperfunction and the Evolution of Aging
The evolutionary theory provides a convincing account of how aging evolves. It is a consequence of the reduced force of natural selection on the effects of genotype on phenotype at late ages at which, in the wild, little or no reproduction occurs (38). Here, an outcome is that mutant alleles that increase early life fitness, but have deleterious effects late in life, may increase overall fitness. Such alleles that exhibit antagonistic pleiotropy (with good effects in the young, but bad in the old) will therefore increase in frequency in the population (57).
Discovering the biochemical and cellular causes of aging will hopefully allow an understanding of the mechanisms underlying the tradeoffs between early and late life traits. An important early attempt to integrate evolutionary and mechanistic theories of aging yielded the disposable soma theory (31). This incorporated the view that aging is caused by accumulation of molecular damage, whereas longevity is enhanced by somatic maintenance processes (detoxification, repair, and turnover). The disposable soma theory hinges ingeniously upon the assumption that somatic maintenance is costly in resource terms (e.g., energy). Consequently, to optimize fitness, a given species must optimally partition resources between reproduction and somatic maintenance. Such optimization will maximize reproductive output, while ensuring organismal (somatic) survival only to the end of the reproductive period. The result is a short-lived, disposable soma.
The development of the disposable soma theory was an important landmark in biogerontology. Critically, it provides a template for integrated evolutionary mechanistic theories of aging. However, according to the hyperfunction model, aging is not driven by molecular damage, nor do somatic maintenance processes protect against aging (except in the sense that they are needed acutely to keep an organism alive) (6). In this case, the disposable soma theory cannot be true (6, 7).
The disposable soma theory has also been applied to try to explain the effects of DR, which extends lifespan and, typically, reduces fertility. It has been suggested that organisms under DR increase resource investment into somatic maintenance (36) (Fig. 2A). As Blagosklonny points out, this is paradoxical (6). Why, under conditions of unrestricted food, should organisms limit investment of resources into somatic maintenance? Arguably, the hyperfunction theory provides a simpler explanation that low food reduces signaling via the growth-promoting pathways (particularly TOR). This suppresses hypertrophy, thereby retarding aging (Fig. 2B).
FIG. 2.

Explanation for effects of dietary restriction (DR) in terms of the bloated soma theory. (A) Prior interpretation in terms of disposable soma theory. (B) New interpretation in terms of bloated soma theory (incorporating hyperfunction theory). Based on (6). IGF-1, insulin-like growth factor 1.
One might say that Blagosklonny has substituted the idea of a disposable soma with one of a bloated soma. In evolutionary terms, bloated soma makes a lot of sense. A major challenge to organismal survival is fluctuation in the food supply. To optimize fitness in the face of such fluctuation, organisms have evolved a dynamic and extensive phenotypic plasticity that allows rapid switching between states of growth and reproduction on the one hand (ample food), and starvation resistance and diapause on the other (scarce food). Bloated soma presents a plausible biological mechanism for the evolution of aging by antagonistic pleiotropy. Mutations that boost growth in early life given ample food (e.g., by increasing TOR signaling) will increase rates of biosynthesis, and thereby of growth and reproduction. They will also, in later life, exacerbate hypertrophic pathologies, thereby accelerating aging.
The hyperfunction theory can also resolve the old question of whether or not aging is programmed. According to the evolutionary theory, aging does not evolve for a purpose and in that sense cannot be genetically programmed. Yet, genetic studies have identified scores of genes where mutation leads to increased lifespan (29). This shows that aging is under genetic control, that is, programmed. According to the hyperfunction theory, insofar as aging rate can be altered by growth control pathways, it is quasi-programmed. Here, quasi-program means an unintended continuation of the developmental program (4, 6). The importance of the prefix quasi- is that the evolved function of the program is not to cause aging. Blagosklonny draws an analogy between development and running a bath. If you leave the water running, the program for filling your bath becomes a quasi-program to flood your bathroom. In fact, open faucet models have been suggested previously in relation to aging in C. elegans (24, 35).
Could Hyperfunction Cause Aging in C. elegans?
Despite intensive research on the biology of aging in C. elegans in many laboratories over the last 20 years, the mechanisms of aging in this creature remain obscure. Could the hyperfunction theory provide the necessary illumination? The theory predicts that when C. elegans die of old age, it is as the result of pathologies associated with hyperfunction, particularly hypertrophy. However, is there any evidence that this is the case?
In broad anatomical terms, C. elegans consists of an outer tube that includes a cuticle with underlying hypodermis and body wall muscle, within which run two smaller tubes: the alimentary canal (pharynx and intestine) and the gonad. Within the worm anatomy, a number of changes occur during aging that are suggestive of both pathology and hypertrophy (summarized in Fig. 3).
FIG. 3.

Evidence for hypertrophy during aging in Caenorhabditis elegans. This figure shows six examples of age change that involves hypertrophy, as follows: (i) accumulation and redistribution of yolk (shaded area, high levels of yolk [VIT-2::GFP fluorescence]); (ii) stacking of oocytes in the gonad; (iii) appearance of tumor-like, intrauterine masses, a likely consequence of runaway endoreduplication in unfertilized oocytes (O, oocyte; T, tumor); (iv) cuticular hypertrophy (C, cuticle; H, hypodermis); (v) neurite outgrowth (NO, neurite outgrowth); and (vi) lipid droplet accumulation (here in aging muscle, L, lipid droplet; M, muscle). In each part, young adult worm is shown above and an older worm below. Redrawn after published observations (12, 15, 18, 24, 27, 37, 48).
Aging in the gonad arms
In the adult hermaphrodite gonad, mitotic germ cells proliferate and then enter meiosis, yielding mature oocytes. The latter are fertilized in the spermatheca, and then pushed into the uterus where embryogenesis begins (20). During aging, the gonad undergoes two kinds of gross anatomical change: atrophy of the germline and hypertrophy within the uterus.
In the absence of mating, the hermaphrodites' own sperm deplete by day 3–5 of adulthood. After this, oocyte production carries on for a period before grinding to a halt. Initially, oocytes continue to pass through the spermatheca and uterus, and are laid unfertilized. Such oocytes are polyploid as the result of multiple rounds of endoreduplication, and have large nuclei (56). Excess oocytes also accumulate within the gonad, and become stacked (squashed together) (Fig. 3), such that cell density more than doubles (27). Thus, cessation of reproduction leads to hypertrophy in the germline, at least initially.
From around day 5 of adulthood, the gonad distal to the spermatheca starts to deteriorate. First, the density of nuclei decreases; next, the gonad begins to shrivel (15), and then disintegrate (Fig. 4), leaving a small number of large mature oocytes that remain adrift in the body cavity. Such germline disintegration could be consistent with the hyperfunction theory, as follows. In younger animals, during starvation-induced adult reproductive diapause, reversible germline atrophy occurs, which can reduce the number of germline nuclei from ∼125 per gonad arm to as few as 35 (3). This atrophy is a programmed process, blocked by mutation of the cell death gene ced-3, which abrogates apoptosis, and which contributes to reproductive fitness after diapause. Moreover, in fully fed worms, some 50% of oocytes are destroyed by apoptosis, and prevention of this by ced mutations reduces oocyte quality (2). These observations suggest that continued (quasi-programmed) apoptosis might lead to age-related germline disintegration.
FIG. 4.

Age-associated gonadal atrophy. (A–D) progressive stages of atrophy of hermaphrodite gonad. (A) 1-day-old adult, healthy, full-sized gonad; (B) 5-day-old adult, gonad slightly atrophied; (C) 7-day-old adult, shrunken, but largely intact gonad; (D) 9-day-old adult, fragmented gonad.
Aging in the uterus
From around day 9 of adulthood, the hermaphrodite uterus shows a marked increase in DNA content (18). This is associated with the appearance of intrauterine masses (Fig. 3), which the authors of the study liken to tumors, and may be the result of runaway endoreduplication in unfertilized oocytes (18). This is another salient example of hypertrophy during aging in C. elegans. Development of uterine masses can be suppressed by feeding worms with fluorodeoxyuridine (FUdR), which blocks DNA replication. However, FUdR administration does not usually increase C. elegans lifespan (14), implying that these uterine growths do not contribute to mortality. More broadly, age changes in the gonad seem not to have major effects on organismal aging, since removal of the entire gonad does not increase lifespan, at least not in wild-type fully fed animals (25, 58).
Age changes in yolk and lipid droplets
A number of studies have reported age changes in levels and distribution of yolk and lipid in C. elegans. The protein component of yolk (vitellogenin) is encoded by the genes vit-2 to vit-6. Yolk is synthesized in the intestine, and then transported across the body cavity to the gonad, where it provisions developing oocytes (30).
Depletion of sperm and cessation of oogenesis trigger a striking redistribution of yolk within the worm, as visualized using a transgene expressing VIT-2 tagged with green fluorescent protein (GFP). In worms with sperm, VIT-2::GFP is seen only in the intestine and oocytes. When sperm depletes, yolk rapidly appears throughout the worm, particularly in the body cavity, where it accumulates to high levels (12, 15, 24, 37) (Fig. 3). This suggests that intestinal yolk production continues after oocyte production stops, resulting in excess yolk that has effectively nowhere to go. Distribution of lipid also seems to change in aged worms. Electron micrographs reveal an age increase in lipid inclusions in the intestine, muscle (Fig. 3), and hypodermis in C. elegans (24).
Age changes in the level and/or distribution of yolk and lipid may well contribute to mortality. For example, knockdown of expression of vit-2 and vit-5 increased lifespan (40), and yolk levels are greatly reduced in long-lived daf-2 insulin/IGF-1 receptor mutants (12). Similarities have been noted between worm yolk (lipoprotein) particles and mammalian apoB-dependent low-density lipoprotein (LDL) (9, 45). In mammals, LDL particles contribute to atherosclerosis, suggesting distant parallels between the pathology of aging in worms and mammals. Moreover, the appearance of lipid inclusions in multiple tissues during aging (24) (Fig. 3) is something that happens during mammalian aging (54). One mechanism by which obesity is thought to cause metabolic syndrome is ectopic deposition of lipid in nonadipose tissue (steatosis), for example, in the muscle and liver, causing lipid-induced pathology (or lipotoxicity) (50, 54).
Interestingly, a number of recent studies have reported that genes promoting lipolysis or fatty acid desaturation also promote longevity (11, 19, 33, 55). Potentially, this could reflect altered energy metabolism, or changes to lipid composition, membrane fluidity, and signaling pathways (10, 11). A further possibility is that they retard the accumulation of pathogenic forms of lipid (e.g., lipotoxic inclusions) (1).
Aging in the intestine
In the laboratory, C. elegans are typically cultured on agar plates with a lawn of Escherichia coli as a food source. In elderly worms, the lumen of the entire alimentary canal (pharynx and intestine) can become clogged up (constipated) with packed E. coli (15). Aging intestinal cells also show marked deterioration, including atrophy, abnormalities in nuclear morphology (e.g., affecting the nuclear lamina), and loss of nuclei and microvilli (15, 18, 21, 24, 37). Although constipation is a problem of E. coli hyperproliferation, there is no obvious evidence that hyperfunction within intestinal cells causes their degeneration. However, the process of organismal death involves a systemic cascade of cellular necrosis, which passes in a wave along the intestine, propagated by calcium signaling (C. Coburn and D. Gems, unpublished observations). This could constitute a quasi-program.
Aging in the nervous system and musculature
It was recently discovered that aging neurons often sprout additional neurite branches (41, 48) (Fig. 3). By day 15, ∼70% of animals showed extraneuronal processes in mechanosensory (touch) neurons, and ∼25% in GABAergic motor neurons (48). This is a striking example of hypertrophy during aging in C. elegans. Such neurite branching was found to affect neuronal function, but not organismal mortality.
Although age changes in muscle are not as marked as those in the gonad and intestine, some deterioration has been observed during aging in both pharyngeal and body wall muscles. Aging pharynges show various signs of decline (15). Body wall muscles show some atrophy and loss of organization of sarcomeres, and lipid inclusions appear within the muscles of older worms (24). Whether these or other factors contribute to the rapid age decline in the rate of pharyngeal pumping and locomotion remains unclear.
Aging in the cuticle and hypodermis
Ultrastructural studies have shown a marked thickening with age of the cuticle. In some places, the cuticle of elderly worms is up to 10 times thicker than those of young worms (24) (Fig. 3). From this, the authors deduce that cells of the underlying hypodermis continue to synthesize cuticle collagens well into adulthood, another example of hypertrophy.
Conclusions
In this review, we have explored the possibility that aging in C. elegans is not caused by molecular damage, but by hyperfunction. In particular, we have asked whether aging in C. elegans is accompanied by anatomical changes consistent with the occurrence of hyperfunction-driven pathologies. A number of previous reports provide evidence that supports this idea. In particular, the age increases in levels of yolk and lipid inclusions, neurite outgrowths, cuticle thickness, and uterine tumors are consistent with the occurrence of hypertrophy (or bloating of the soma) during aging. In some cases (e.g., yolk accumulation), there is evidence that these changes contribute to age-related mortality. The hyperfunction theory also furnishes alternative explanations for the effects on aging of autophagy and protein synthesis. We conclude that this new theory provides an account of aging in C. elegans that is at least as plausible as the accumulation of molecular damage. Further studies of the role of hypertrophy (and atrophy) in aging in C. elegans (and other model organisms such as Drosophila) are warranted.
In fact, observation of age changes led the authors of one of the anatomical studies cited here to draw conclusions that anticipate the hyperfunction theory, as follows. “Overall, the extensive accumulation of macromolecules, such as yolk proteins, cuticle proteins and lipid, suggest that in postreproductive animals, a life-stage that has been subject to little (if any) natural selection pressure over time, biosynthesis/protein turnover is not tightly regulated like it is in developmental and reproductive phases. […] We speculate that nonregulated biosynthesis might contribute to the senescent decline of C. elegans by causing the continued production of irrelevant macromolecules” (24).
The very short natural lifespan of C. elegans suggests that whatever mechanisms drive aging are active to a high degree in this organism. As described here, observations of pathology in aging worms suggest the presence of intense hyperfunction. One possibility is that the relative contribution to aging of damage and hyperfunction varies between species. For example, in mammals, DNA damage contributes to cancer, a major disease of aging, though hyperfunction also plays an important role (4, 5). By contrast, there have been no reports of somatic mutation-induced hyperplasia (i.e., true cancer) in C. elegans. Adult C. elegans are relatively insensitive to the life-shortening effects of UV radiation, which likely reflects the lack of somatic cell division during adulthood (32). Only doses of ionizing radiation above 100 krad begin to shorten C. elegans lifespan (26). These findings suggest that lifespan in this organism is not limited by DNA damage. We postulate that while DNA damage contributes to aging in mammals, molecular damage makes little contribution to aging in C. elegans (Fig. 5).
FIG. 5.
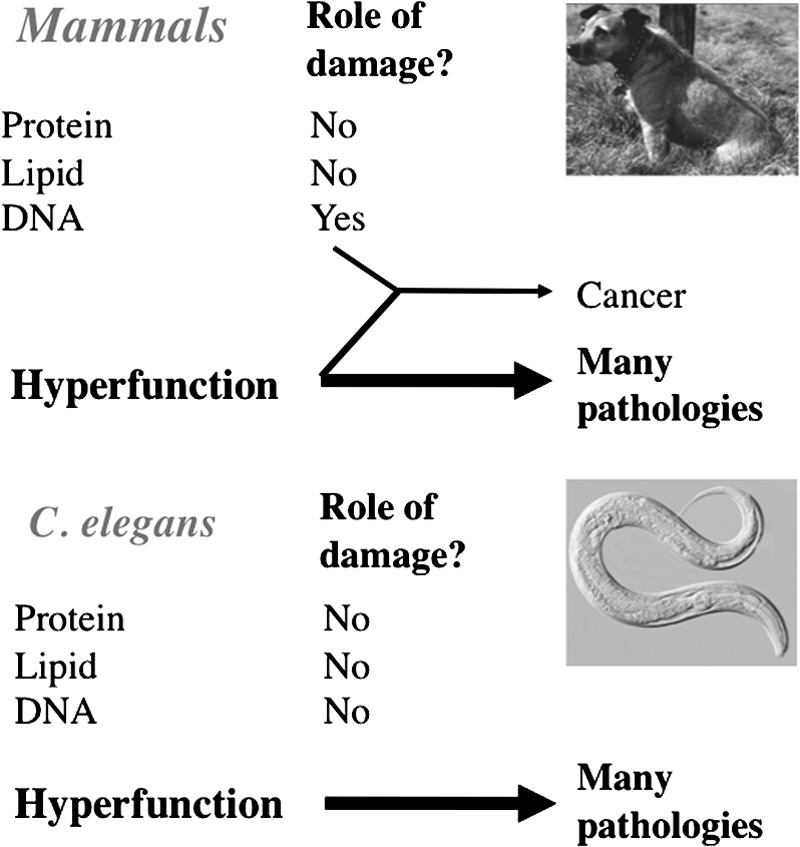
Relative contributions of damage and hyperfunction in mammals and nematodes. This scheme describes the hypothesis that DNA damage and hyperfunction are predominant primary determinants of mammalian aging, while in C. elegans hyperfunction is particularly severe, such that molecular damage plays no primary role at all.
This by no means implies that mitochondria play no role in worm aging. Many interventions that disrupt mitochondrial function in C. elegans lead to increased lifespan, often accompanied by delayed development—the so-called Mit phenotype (43). One possibility is that hyperfunction is suppressed in Mit mutants, thereby increasing lifespan. Under nutrient-replete conditions, high fuel influx will cause mitochondria to be fully polarized leading to increased production of not only ROS but also intermediates for anabolism. Thus, perhaps it is the latter rather than the former that accelerates aging rate, by promoting hyperfunction.
One unresolved issue from tests of the ROS theory in C. elegans is how conditions that increase oxidative stress lead to increased lifespan (23, 44, 59). If aging in C. elegans is the result of hyperfunction, then one possibility is that oxidative stress ameliorates hyperfunction. This could occur either indirectly, via effects on signaling (e.g., HIF-1 activation) (34), or directly on proximal hyperfunction mechanisms.
Finally, if the molecular damage theory really is incorrect, how could we have all got it so wrong for so long? Here, a historical perspective may be helpful. In the absence of fundamental knowledge about a topic, the human intellect is bound to generate plausible theories to fill the void. Widespread acceptance of such ideas may owe more to the attractiveness of the theory to intuition (or common sense) than to any careful empirical investigations. For example, before the 17th Century, it was believed that the Sun revolved around the Earth.
Like the geocentric theory, the molecular damage theory is plausible and intuitively appealing. After all, living systems are intricate nanomachines, and all forms of man-made machinery wear out eventually, even Volkswagen Beetles. Such primordial theories derived from guesswork and common sense have been referred to as folk science (46). Historically, scientific advance often really kicks off when the fruits of empirical research lead to replacement of the founding folk theories. In contrast to folk theories, such empirically based concepts are often counterintuitive. For example, the heliocentric theory required the acceptance of the initially incredible idea of a world that both spins around on its axis, and hurtles through space at a terrific speed. Perhaps, the molecular damage theory will prove to be the great folk theory of biogerontology.
Abbreviations Used
- DR
dietary restriction
- FUdR
fluorodeoxyuridine
- GABA
gamma-aminobutyric acid
- GFP
green fluorescent protein
- IGF-1
insulin-like growth factor 1
- LDL
low-density lipoprotein
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TOR
target of rapamycin
Acknowledgments
We thank Linda Partridge and members of the Gems laboratory for useful discussion, and Ivana Bjedov, Mikhail Blagosklonny, Filipe Cabreiro, Lazaros Foukas, Nick Lane, and Jennifer Tullet for comments on the manuscript. We acknowledge funding from the European Union (FP6-036894 and FP6-518230), el Instituto para la Formación y Aprovechamiento de Recursos Humanos, y la Secretaria Nacional de Ciencia, Tecnología e Innovación (Panamá)(to Y. de la G.), and the Wellcome Trust (Strategic Award).
References
- 1.Ackerman D. Gems D. The mystery of C. elegans aging: an emerging role for fat. Bioessays. 2012;34:466–471. doi: 10.1002/bies.201100189. [DOI] [PubMed] [Google Scholar]
- 2.Andux S. Ellis RE. Apoptosis maintains oocyte quality in aging Caenorhabditis elegans females. PLoS Genet. 2008;4:e1000295. doi: 10.1371/journal.pgen.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angelo G. Van Gilst MR. Starvation protects germline stem cells and extends reproductive longevity in C. elegans. Science. 2009;326:954–958. doi: 10.1126/science.1178343. [DOI] [PubMed] [Google Scholar]
- 4.Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006;5:2087–2102. doi: 10.4161/cc.5.18.3288. [DOI] [PubMed] [Google Scholar]
- 5.Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008;7:3344–3354. doi: 10.4161/cc.7.21.6965. [DOI] [PubMed] [Google Scholar]
- 6.Blagosklonny MV. Paradoxes of aging. Cell Cycle. 2007;6:2997–3003. doi: 10.4161/cc.6.24.5124. [DOI] [PubMed] [Google Scholar]
- 7.Blagosklonny MV. Why the disposable soma theory cannot explain why women live longer and why we age. Aging. 2010;2:884–887. doi: 10.18632/aging.100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blagosklonny MV. Hall MN. Growth and aging: a common molecular mechanism. Aging. 2009;1:357–362. doi: 10.18632/aging.100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandt BW. Zwaan BJ. Beekman M. Westendorp RG. Slagboom PE. Shuttling between species for pathways of lifespan regulation: a central role for the vitellogenin gene family? Bioessays. 2005;27:339–346. doi: 10.1002/bies.20161. [DOI] [PubMed] [Google Scholar]
- 10.Branicky R. Desjardins D. Liu J. Hekimi S. Lipid transport and signaling in Caenorhabditis elegans. Dev Dyn. 2010;239:1365–1377. doi: 10.1002/dvdy.22234. [DOI] [PubMed] [Google Scholar]
- 11.Chen S. Whetstine J. Ghosh S. Hanover J. Gali R. Grosu P. Shi Y. The conserved NAD(H)-dependent corepressor CTBP-1 regulates Caenorhabditis elegans life span. Proc Natl Acad Sci U S A. 2009;106:1496–1501. doi: 10.1073/pnas.0802674106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Depina A. Iser W. Park S. Maudsley S. Wilson M. Wolkow C. Regulation of Caenorhabditis elegans vitellogenesis by DAF-2/IIS through separable transcriptional and posttranscriptional mechanisms. BMC Physiol. 2011;11:11. doi: 10.1186/1472-6793-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doonan R. McElwee JJ. Matthijssens F. Walker GA. Houthoofd K. Back P. Matcheski A. Vanfleteren JR. Gems D. Against the oxidative damage theory: Superoxide dismutases protect against oxidative stress but have little or no effect on lifespan in C. elegans. Genes Dev. 2008;22:3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandhi S. Santelli J. Mitchell DG. Stiles JW. Raosanadi D. A simple method for maintaining large, aging populations of Caenorhabditis elegans. Mech Ageing Dev. 1980;12:137–150. doi: 10.1016/0047-6374(80)90090-1. [DOI] [PubMed] [Google Scholar]
- 15.Garigan D. Hsu A. Fraser A. Kamath R. Ahringer J. Kenyon C. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics. 2002;161:1101–1112. doi: 10.1093/genetics/161.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gems D. Doonan R. Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle. 2009;8:1681–1687. doi: 10.4161/cc.8.11.8595. [DOI] [PubMed] [Google Scholar]
- 17.Gems D. Doonan R. Oxidative stress and aging in the nematode Caenorhabditis elegans. In: Miwa S, editor; Beckman K, editor; Muller F., editor. Oxidative Stress in Aging. Totowa, NJ: Humana Press; 2008. pp. 81–110. [Google Scholar]
- 18.Golden T. Beckman K. Lee A. Dudek N. Hubbard A. Samper E. Melov S. Dramatic age-related changes in nuclear and genome copy number in the nematode Caenorhabditis elegans. Aging Cell. 2007;6:179–188. doi: 10.1111/j.1474-9726.2007.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goudeau J. Bellemin S. Toselli-Mollereau E. Shamalnasab M. Chen Y. Aguilaniu H. Fatty acid desaturation links germ cell loss to longevity through NHR-80/HNF4 in C. elegans. PLoS Biol. 2011;9:e1000599. doi: 10.1371/journal.pbio.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenstein D. WormBook. The C. elegans Research Community; 2005. Control of oocyte meiotic maturation and fertilization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haithcock E. Dayani Y. Neufeld E. Zahand AJ. Feinstein N. Mattout A. Gruenbaum Y. Liu J. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2005;102:16690–16695. doi: 10.1073/pnas.0506955102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 23.Heidler T. Hartwig K. Daniel H. Wenzel U. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology. 2010;11:183–195. doi: 10.1007/s10522-009-9239-x. [DOI] [PubMed] [Google Scholar]
- 24.Herndon L. Schmeissner P. Dudaronek J. Brown P. Listner K. Sakano Y. Paupard M. Hall D. Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- 25.Hsin H. Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 1999;399:362–366. doi: 10.1038/20694. [DOI] [PubMed] [Google Scholar]
- 26.Johnson TE. Hartman PS. Radiation effects on life span in Caenorhabditis elegans. J Gerontol. 1988;43:B137–B141. doi: 10.1093/geronj/43.5.b137. [DOI] [PubMed] [Google Scholar]
- 27.Jud M. Razelun J. Bickel J. Czerwinski M. Schisa JA. Conservation of large foci formation in arrested oocytes of Caenorhabditis nematodes. Dev Genes Evol. 2007;217:221–226. doi: 10.1007/s00427-006-0130-3. [DOI] [PubMed] [Google Scholar]
- 28.Kapahi P. Chen D. Rogers AN. Katewa SD. Li PW. Thomas EL. Kockel L. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11:453–465. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenyon C. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 30.Kimble J. Sharrock WJ. Tissue-specific synthesis of yolk proteins in Caenorhabditis elegans. Dev Biol. 1983;96:189–196. doi: 10.1016/0012-1606(83)90322-6. [DOI] [PubMed] [Google Scholar]
- 31.Kirkwood TBL. Evolution of ageing. Nature. 1977;270:301–304. doi: 10.1038/270301a0. [DOI] [PubMed] [Google Scholar]
- 32.Klass MR. Aging in the nematode Caenorhabditis elegans: major biological and environmental factors influencing life span. Mech Ageing Dev. 1977;6:413–429. doi: 10.1016/0047-6374(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 33.Lapierre L. Gelino S. Meléndez A. Hansen M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol. 2011;21:1507–1514. doi: 10.1016/j.cub.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SJ. Hwang AB. Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lund J. Tedesco P. Duke K. Wang J. Kim S. Johnson T. Transcriptional profile of aging in C. elegans. Curr Biol. 2002;12:1566–1573. doi: 10.1016/s0960-9822(02)01146-6. [DOI] [PubMed] [Google Scholar]
- 36.Masoro E. Austad S. The evolution of the antiaging action of dietary restriction: a hypothesis. J Gerontol A Biol Sci Med Sci. 1996;51:B387–B391. doi: 10.1093/gerona/51a.6.b387. [DOI] [PubMed] [Google Scholar]
- 37.McGee MD. Weber D. Day N. Vitelli C. Crippen D. Herndon LA. Hall DH. Melov S. Loss of intestinal nuclei and intestinal integrity in aging C. elegans. Aging Cell. 2011;10:699–710. doi: 10.1111/j.1474-9726.2011.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medawar PB. An Unsolved Problem Of Biology. London: H.K. Lewis; 1952. [Google Scholar]
- 39.Muller FL. Lustgarten MS. Jang Y. Richardson A. Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 40.Murphy CT. McCarroll SA. Bargmann CI. Fraser A. Kamath RS. Ahringer J. Li H. Kenyon CJ. Genes that act downstream of DAF-16 to influence the lifespan of C. elegans. Nature. 2003;424:277–284. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 41.Pan C. Peng C. Chen C. McIntyre S. Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc Natl Acad Sci U S A. 2011;108:9274–9279. doi: 10.1073/pnas.1011711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perez VI. Bokov A. Van Remmen H. Mele J. Ran Q. Ikeno Y. Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rea SL. Metabolism in the Caenorhabditis elegans Mit mutants. Exp Gerontol. 2005;40:841–849. doi: 10.1016/j.exger.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 44.Schulz TJ. Zarse K. Voigt A. Urban N. Birringer M. Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 45.Shibata Y. Branicky R. Landaverde IO. Hekimi S. Redox regulation of germline and vulval development in Caenorhabditis elegans. Science. 2003;302:1779–1782. doi: 10.1126/science.1087167. [DOI] [PubMed] [Google Scholar]
- 46.Smith Churchland P. Neurophilosophy: Towards a Unified Science of the Mind-Brain. Cambridge: MIT Press; 1986. [Google Scholar]
- 47.Sohal RS. Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tank EM. Rodgers KE. Kenyon C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J Neurosci. 2011;31:9279–9288. doi: 10.1523/JNEUROSCI.6606-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsang CK. Qi H. Liu LF. Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 50.Unger RH. Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab. 2010;21:345–352. doi: 10.1016/j.tem.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Raamsdonk JM. Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Raamsdonk JM. Hekimi S. Reactive oxygen species and aging in Caenorhabditis elegans: causal or casual relationship? Antioxid Redox Signal. 2010;13:1911–1953. doi: 10.1089/ars.2010.3215. [DOI] [PubMed] [Google Scholar]
- 53.Van Raamsdonk JM. Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A. 2012;109:5785–5790. doi: 10.1073/pnas.1116158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Virtue S. Vidal-Puig A. It's not how fat you are, it's what you do with it that counts. PLoS Biol. 2008;6:e237. doi: 10.1371/journal.pbio.0060237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang MC. O'Rourke EJ. Ruvkun G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 2008;322:957–960. doi: 10.1126/science.1162011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ward S. Carrel JS. Fertilization and sperm competition in the nematode Caenorhabditis elegans. Dev Biol. 1979;73:304–321. doi: 10.1016/0012-1606(79)90069-1. [DOI] [PubMed] [Google Scholar]
- 57.Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 58.Yamawaki TM. Arantes-Oliveira N. Berman JR. Zhang P. Kenyon C. Distinct activities of the germline and somatic reproductive tissues in the regulation of Caenorhabditis elegans' longevity. Genetics. 2008;178:513–526. doi: 10.1534/genetics.107.083253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang W. Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang W. Li J. Hekimi S. A measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics. 2007;177:2063–2074. doi: 10.1534/genetics.107.080788. [DOI] [PMC free article] [PubMed] [Google Scholar]