

Background: PAS is an antimycobacterial whose mechanism(s) of action remains elusive.
Results: PAS is incorporated into the folate pathway by DHPS-DHFS, generating an anti-metabolite. DHFS and RibD are associated with PAS resistance.
Conclusion: Hydroxyl dihydrofolate inhibits DHFR. folC and ribD are drug target genes for identification of clinical PAS resistance.
Significance: Metabolite analog incorporation into essential biosynthetic pathways is promising for developing antibacterials.
Keywords: Antibiotic Resistance, Antibiotic Action, Drug Development, Folate Metabolism, Microbiology, Mycobacterium tuberculosis
Abstract
para-Aminosalicylic acid (PAS) is one of the antimycobacterial drugs currently used for multidrug-resistant tuberculosis. Although it has been in clinical use for over 60 years, its mechanism(s) of action remains elusive. Here we report that PAS is a prodrug targeting dihydrofolate reductase (DHFR) through an unusual and novel mechanism of action. We provide evidences that PAS is incorporated into the folate pathway by dihydropteroate synthase (DHPS) and dihydrofolate synthase (DHFS) to generate a hydroxyl dihydrofolate antimetabolite, which in turn inhibits DHFR enzymatic activity. Interestingly, PAS is recognized by DHPS as efficiently as its natural substrate para-amino benzoic acid. Chemical inhibition of DHPS or mutation in DHFS prevents the formation of the antimetabolite, thereby conferring resistance to PAS. In addition, we identified a bifunctional enzyme (riboflavin biosynthesis protein (RibD)), a putative functional analog of DHFR in a knock-out strain. This finding is further supported by the identification of PAS-resistant clinical isolates encoding a RibD overexpression mutation displaying cross-resistance to genuine DHFR inhibitors. Our findings reveal that a metabolite of PAS inhibits DHFR in the folate pathway. RibD was shown to act as a functional analog of DHFR, and as for DHFS, both were shown to be associated in PAS resistance in laboratory strains and clinical isolates.
Introduction
Mycobacterium tuberculosis, a human pathogen causing tuberculosis, claims 1.2–1.5 million lives each year, and it is estimated that one third of the global population is latently infected by the bacilli (1). With the alarming rise of multidrug-resistant tuberculosis, there is an urgent need for new anti-tuberculosis agents to combat the disease (2). Understanding the molecular mechanism(s) of action and resistance of current drugs will provide new perspectives and approaches for drug discovery. para-Aminosalicylic acid (PAS)5 is one of the first antituberculosis agents found to be effective in the 1940s (3). Although PAS has been widely used clinically, its precise mode of action remains largely speculative (4–6). The structural similarity between PAS and para-aminobenzoic acid (pABA) has led to the speculation that it might inhibit dihydropteroate synthase (DHPS) in the folate biosynthetic pathway. This speculation is founded on the well studied mechanism of action of sulfonamides, another group of pABA structural analogs. However, unlike the sulfonamides, PAS has not been shown to be an inhibitor of DHPS (4). Nevertheless, mutations within the thymidylate synthase gene thyA have been associated with resistance to PAS in M. tuberculosis (7), suggesting that PAS may interfere with the folate pathway through an unknown mechanism. Recently, Chakraborty et al. (8) demonstrated that PAS acts as an alternative substrate for folate metabolism in M. tuberculosis. However, the specific site of action in the folate pathway that results in growth inhibition remains to be elucidated (8). The folate pathway generates tetrahydrofolate in prokaryotes and eukaryotes, which is a ubiquitous one-carbon carrier involved in the biosynthesis of purine, thymidine, glycine, methionine, and pantothenate. In bacteria, tetrahydrofolate is also required for the synthesis of formylmethionyl tRNAfMet, which is essential for the initiation of protein synthesis. In the folate pathway, DHPS catalyzes the condensation of pABA and 6-hydroxymethyl-7,8-dihydropterin pyrophosphate into 7,8-dihydropteroate, which is then converted by the dihydrofolate synthase (DHFS) into dihydrofolate and reduced to generate the cofactor tetrahydrofolate by the enzyme dihydrofolate reductase (DHFR). DHFR is the target of several important anticancer and antibacterial drugs. For example, methotrexate targets human DHFR and is used as an anticancer drug, whereas both trimethoprim and pyrimethamine are inhibitors of bacterial and protozoal DHFR (9). WR99210 is another potent DHFR inhibitor and antimalarial whose clinical development was terminated due to unacceptable side effects (10). Interestingly, WR99210 is also a potent inhibitor of DHFR and a growth inhibitor in M. tuberculosis (11). Despite the pharmacological validation of DHFR as a drug target for malaria and cancer, it has not yet been fully exploited in the battle against M. tuberculosis. Here we provide evidence that PAS is a prodrug and targets DHFR in M. tuberculosis. DHFR in M. tuberculosis is encoded by dfrA. We show that the folate pathway is essential for the conversion of PAS into an active antimetabolite. Overexpression of dfrA was sufficient to confer resistance to PAS in M. tuberculosis. We demonstrated that PAS must be metabolized by an intact folate pathway before it can inhibit DHFR activity. We found that spontaneous mutants overexpressing a previously uncharacterized protein RibD are resistant to PAS or other DHFR inhibitors. When expressed at high level, RibD can act as an alternative dihydrofolate reductase and compensate for a genetic deletion of dfrA in M. tuberculosis, reinforcing the notion that DHFR is the target of PAS. Finally, we identified a similar polymorphism in ribD in M. tuberculosis clinical isolates that are associated with PAS resistance.
EXPERIMENTAL PROCEDURES
Strains and Growth Conditions
M. tuberculosis H37Rv and Mycobacterium bovis BCG Pasteur strains and their derivatives were maintained in Middlebrook 7H9 broth medium supplemented with 0.2% glycerol, 0.05% Tween 80, and 10% ADS (albumin-dextrose-saline) supplement or Middlebrook 7H11 agar. Culture medium was supplemented with hygromycin (50 μg/ml) or kanamycin (25 μg/ml) when necessary.
Drug Susceptibility Testing
Bacterial drug susceptibility was determined in 7H9 medium as described previously (12). Briefly, compounds dissolved in 90% DMSO were 2-fold serial-diluted in duplicates and spotted in 96-well clear plates, resulting in 10–12 dilutions of each compound. A volume of 200 μl of M. tuberculosis or BCG culture (final A600 = 0.02) was added to each well, and the assay plates were incubated at 37 °C for 5 days. A600 values were recorded using a SpectraMax M2 spectrophotometer, and minimum inhibition concentration curves were plotted using GraphPad Prism 5 software.
Protein Purification and Analysis
M. tuberculosis H37Rv dfrA, folP (DHPS), and ribD were expressed as His6-tagged proteins in Escherichia coli BL21 as described previously and purified by standard procedures (13). Proteins were analyzed with Western blot. Antibody against RibD was raised in rabbits against keyhole limpet hemocyanin-conjugated peptides (RibD, qrqhrqargqsevp) by GenScript. The antibodies were affinity-purified by using specific peptides as ligands. Antibody against ClpC1 (14) was used as an internal control.
DHFR Enzymatic Assay
The DHFR enzymatic assay utilizes a tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS). Briefly, 2 μl of test compound in 90% (v/v) DMSO was added into 96-well plates to incubate with 50 μl of 60 nm DHFR for 30 min at room temperature. Then 50 μl of 200 μm NADPH was added and incubated for another 10 min. The enzymatic reaction was started by adding 100 μl of a mixture containing 10 μm dihydrofolate (5 μm final concentration) and 300 μm MTS (150 μm final concentration) diluted in DHFR assay buffer (100 mm HEPES, pH 7.5, + 50 mm potassium chloride + 0.05% CHAPS). The reaction was monitored at an absorbance of 490 nm with kinetic reading for 10 min. For the DHFR enzymatic assay with cell lysate flow-through (CLFT), bacterial cultures were grown to an A600 of ∼0.6 in 7H9 medium, and antibiotics were added at concentrations of 10-fold higher than the minimum inhibition concentration (MIC50) achieved with drug susceptibility testing. Cultures were shaken at 60 rpm for varying periods as indicated. Bacterial cells were pelleted and washed once with PBS + 0.05% Tween 80. The pellet was resuspended with 1.5 ml of DHFR enzymatic assay buffer. The cells were then broken using 0.1-mm glass beads with Precellys 24 (Bertin Technologies), and cell lysates were collected after centrifugation. Samples were normalized by measuring protein concentration and then passed through a 10-kDa cut-off membrane to remove the native DHFR protein. The flow-through (CLFT) was then used in the DHFR enzymatic assay. To perform the assay, 10 μl of 300 nm DHFR was mixed with 90 μl of bacterial lysates and incubated at room temperature for 30 min, and 50 μl of 20 μm NADPH was then added and kept at room temperature for another 10 min. Fifty microliters of 20 μm DHF/600 μm MTS mixture in DHFR assay buffer was added to start the reaction.
DHPS Enzymatic Assay and Identification of Hydroxyl-7,8-dihydropteroate by LC/MS/MS
The incorporation of PAS into the folate pathway was examined by DHPS assay as described previously (15). Pyrophosphate released by the DHPS reaction is cleaved into monophosphate by the addition of inorganic pyrophosphatase to allow the formation of a green complex in the presence of malachite green and molybdate. Briefly, 6-hydroxymethyl-7,8-dihydropterin-pyrophosphate (H2PtPP) (30 μm), purified recombinant M. tuberculosis DHPS (0.6 μg), and inorganic pyrophosphatase (0.1 units/ml) were incubated with various amounts of pABA or PAS (0, 2, 4, 8, 16, 32, 64, and 128 μm) in 20 mm Tris-HCl (pH 8.8), 200 mm NaCl, 5 mm MgCl2, 1 mm dithiothreitol in 500 μl at 37 °C for 30 min. After reaction, 100 μl of sample was transferred to a 96-well plate, and 20 μl of malachite green assay solution was added. All further steps were carried out according to the manufacturer's protocol for the malachite green assay. Samples from the DHPS reaction were used for analysis by a triple quadruple Quattro PremierTM mass spectrometer (Waters, Milford, MA) with electrospray ionization. Instruments were controlled by MassLynx software (Version 4.0, Waters, Milford, MA). In brief, sample aliquots from the DHPS reaction were extracted with acetonitrile. After the removal of proteins by centrifugation at 13,200 rpm for 10 min at 4 m, the supernatant was subjected to HPLC separation with tandem mass spectrometric detection. The mobile phases were (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid at a flow rate of 0.4 ml/min. The LC conditions were 5% B at 0 min, a linear gradient from 5 to 50% B over 0.5 min, held at 50% for 0.25 min and then ramped from 50 to 95% over 0.25 min followed by 95% B for 0.75 min and back to 5% B over 0.25 min and then held at 5% B for the remaining 0.75 min.
Whole Genome Sequencing
Whole genome sequencing was performed as described previously (16).
Mutant Construction and Gene Overexpression
Disruption of mycobacterial dfrA gene was obtained by allelic exchange as described previously (17). The mycobacterial mutant was constructed by using a modified plasmid pYUB854. Briefly, a sacB-lacZ cassette was excised from the pGOAL17 plasmid (18) and ligated into the PacI site of pYUB854. About 1-kb flanking sequences of the gene of interest were cloned into the derivative plasmid on each side of the hygromycin cassette. The resulting plasmid was introduced into mycobacterial cells by electroporation. Deletion mutants were confirmed by PCR or Southern blotting. For gene overexpression in mycobacterium, the genes of interest were amplified and cloned into plasmid pMV262 (19). The constructs were then electroporated into mycobacterial strains.
Southern Blot Analysis
Bacterial genomic DNA was digested with PvuII with restriction endonuclease. Southern blotting was performed with the digoxigenin DNA labeling kit, digoxigenin wash, and block buffer set and nitroblue tetrazolium salt (NBT)/5-bromo-4-chloro-3-indolyl phosphate, toluidinium salt (BCIP) stock solution (Roche Diagnostics). Transfers of DNA to nylon membranes, hybridization, and visualization were as recommended by the manufacturer's protocol.
RESULTS
Overexpression of dfrA Leads to PAS Resistance in M. tuberculosis
Because the folate pathway is known to be associated with the mechanism of PAS resistance, we tested whether the overexpression of the key enzymes DHFS (encoded by folC) and DHFR (dfrA) would result in PAS resistance. M. tuberculosis H37Rv growth was inhibited by PAS with an MIC50 of 0.4 μm. H37Rv overexpressing dfrA was fully resistant to PAS, with no inhibition observed at PAS concentrations up to 100 μm. Conversely, overexpression of folC had no impact on the susceptibility of H37Rv to PAS (Fig. 1A). These results suggested that DHFR is a potential target of PAS. However, in an in vitro enzymatic assay, PAS did not show any inhibition of recombinant M. tuberculosis DHFR (rDHFR), whereas the known DHFR inhibitor WR99210 inhibited rDHFR as expected (Fig. 1B).
FIGURE 1.

dfrA contributes to PAS resistance in M. tuberculosis. A, H37Rv overexpressing dfrA but not folC is resistant to PAS. OD, optical density. B, PAS does not inhibit rDHFR enzymatic activity as compared with the specific DHFR inhibitor WR99210 with the DHFR assay. The values represent the means ± S.D. from one representative experiment performed with triplicate samples.
DHFR Is Inhibited by PAS-treated Mycobacterial Cell Lysates
The observation that PAS did not inhibit DHFR in vitro led us to speculate that PAS might be a prodrug that requires metabolic activation to manifest its inhibitory effect. To test this hypothesis, we made an attempt to bioactivate PAS in mycobacteria. M. bovis BCG was treated with PAS for 3 h, and the cell lysate was isolated, normalized to the protein concentration, and passed through a 10-kDa cut-off membrane to remove any DHFR protein produced by mycobacterial cells. The CLFT was then used as a potential source of bioactivated PAS to examine the inhibition of rDHFR enzymatic activity. CLFT from PAS-treated cells significantly inhibited rDHFR activity. In contrast, CLFT from streptomycin-treated cells did not show any inhibitory effect (Fig. 2A). Similar results were obtained in M. tuberculosis H37Rv (Fig. 2B). Intracellular activation of PAS was proven to be time-dependent, with maximum rDHFR inhibition (∼60%) observed following 6 h of PAS treatment prior to CLFT isolation and testing in the enzymatic assay (Fig. 2C). These results suggest that PAS is activated within mycobacterial cells and that its bioconversion product inhibits DHFR.
FIGURE 2.

PAS is activated intracellularly to target DHFR. A and B, CLFTs from PAS-treated M. bovis BCG (A) and M. tuberculosis (B) inhibit rDHFR activity. WR99210 and streptomycin (Strep) were used as positive and negative controls, respectively. OD, optical density. C, PAS intracellular activation is time-dependent. D, PAS inhibition was decreased upon the addition of pABA. E, providing pABA in PAS-treated mycobacterial cells neutralized the inhibition of CLFT on rDHFR. The values represent the means ± S.D. from one representative experiment performed with triplicate samples. The experiments were carried out in three independent biological replicates resulting in the same conclusion.
Because PAS is structurally similar to pABA, the natural substrate of DHPS, we hypothesized that PAS might be incorporated into the folate pathway by competing with pABA in the reaction catalyzed by DHPS, the product of which is further processed by DHFS to generate hydroxyl dihydrofolate (Fig. 3). This derivative could be the active form of PAS, which would subsequently interact with DHFR to inhibit its enzymatic activity and eventually bacterial growth (Fig. 3). A prediction based on this hypothesis is that the exogenous addition of pABA should reduce PAS-mediated growth inhibition of Mycobacterium and the formation of the PAS-derived antimetabolite. Indeed, the PAS-mediated mycobacterial growth inhibition in M. tuberculosis (20) and M. bovis BCG was alleviated in the presence of pABA (Fig. 2D). In addition, the formation of the PAS-derived antimetabolite was significantly reduced in the presence of pABA (Fig. 2E). Thus these results indicate that PAS is activated within mycobacterial cells and competes with pABA for its bioactivation, the product of which targets DHFR.
FIGURE 3.

A model of PAS mechanism of action in Mycobacterium. The normal folate pathway is depicted on the left side. As a pABA analog, PAS is incorporated into the folate pathway by competing with pABA in the reaction catalyzed by DHPS, the product of which is further processed by DHFS to generate hydroxyl dihydrofolate (right side). This antimetabolite in turn inhibits DHFR activity (denoted by the T bar) and thus blocks the folate pathway (denoted by the cross).
PAS Acts as a Substrate of DHPS and Is Incorporated into the Folate Pathway
We next sought to test whether PAS is recognized as a direct substrate by DHPS. A nonradioactive enzymatic assay for the recombinant mycobacterial DHPS revealed that PAS is indeed recognized as a substrate by DHPS, and the affinity of PAS to DHPS was comparable with that of pABA in the presence of the cofactor H2PtPP. Under our experimental conditions, pABA had a Michaelis-Menten constant (Km) of 11.4 ± 0.1 for DHPS, whereas PAS had a Km of 17.7 ± 0.1 (Fig. 4, A and B). The condensation of PAS with the DHPS cofactor H2PtPP should theoretically lead to the synthesis of hydroxyl dihydropteroate. The mixture reactions were then analyzed by LC/MS/MS. A peak at 329 (m/z) corresponding to hydroxyl dihydropteroate was identified from the reaction mixture by the Q1 full scan (Fig. 4C), which is not observed with the reaction lacking DHPS. The ion was further fragmented into two peaks at 176 (m/z) and 151 (m/z) corresponding to H2PtPP and PAS, respectively, in the Q3 scan (Fig. 4D). The chromatograms monitoring this ion demonstrated a single peak (Fig. 4E), suggesting that hydroxyl dihydropteroate was indeed produced by DHPS with PAS and H2PtPP as substrate. These data showed that PAS can be incorporated into the folate pathway and bioactivated into an antimetabolite at the level of DHPS.
FIGURE 4.

PAS is incorporated into the folate pathway and bioactivated into an antimetabolite at the level of DHPS. A and B, DHPS catalyzes the reaction of H2PtPP and its natural substrate pABA (A) and PAS (B) with a similar affinity. The Michaelis-Menten values (Km values) of pABA and PAS to DHPS are labeled. O.D., optical density. C, LC/MS/MS chromatography revealed a peak at 329 (m/z) corresponding to hydroxyl dihydropteroate identified from the reaction mixture by the Q1 full scan. D, the ion was further fragmented into two peaks at 176 (m/z) and 151 (m/z) corresponding to H2PtPP and PAS, respectively, in the Q3 scan. E, the chromatograms monitoring this ion demonstrated a single peak, suggesting that hydroxyl dihydropteroate was produced by DHPS with PAS and H2PtPP as substrate.
Enzymes from the Folate Pathway Are Required for PAS Bioactivation
According to our proposed antimetabolite model for the PAS mechanism of action (Fig. 3), the activation of PAS requires functional DHPS and DHFS. Any interruption of their function should therefore impair hydroxyl dihydrofolate synthesis and thus give rise to PAS resistance. To test this hypothesis, we first examined whether inhibition of DHPS would result in PAS resistance. Depletion of DHPS fails to phenocopy treatment with sulfonamides, a presumed inhibitor of the enzyme, probably because DHPS is present in considerable excess or the activity of the enzyme has to be reduced by ∼99% to prevent growth (21–24). We thus treated mycobacterium with PAS in the presence of different concentrations of sulfonamide, and bacterial growth was measured after 5 days of treatment. In agreement with our hypothesis, the addition of sulfathiazole to bacterial cultures indeed decreased M. tuberculosis susceptibility to PAS, with complete neutralization of PAS-mediated growth inhibition at sulfathiazole concentrations above 40 μm (Fig. 5A). Similar results were observed with other sulfonamides (data not shown). Furthermore, the addition of sulfathiazole to PAS-treated mycobacterial cultures suppressed the inhibition of rDHFR by CLFT (Fig. 5B).
FIGURE 5.

Enzymes in the folate pathway are required for PAS activation. A, inhibition of DHPS antagonizes PAS-mediated inhibition, as shown by the growth inhibition curves of PAS in the presence of increasing sulfathiazole (Sul) concentrations. OD, optical density. B, the addition of sulfathiazole to PAS-treated mycobacterial cultures suppresses the inhibition of rDHFR activity by CLFT. C, mutation in folC (folCE40A) reduces M. tuberculosis susceptibility to PAS, and complementation with wild-type copy of folC (in pMV262) restores its susceptibility to wild-type level. The values in B and C represent the means ± S.D. from one representative experiment performed with triplicate samples.
The other enzyme predicted to be required for generating the proposed hydroxyl dihydrofolate is DHFS (folC). Because there are no known DHFS inhibitors, we resorted to a genetic approach to demonstrate the requirement of DHFS for PAS activation. By screening for spontaneous PAS-resistant mutants, we isolated one M. tuberculosis mutant (folCE40A) displaying high level resistance to PAS (Fig. 5C). This mutant harbored a mutation in folC resulting in a substitution of DHFS residue Lys-40 by Ala. Lys-40 is located in a mobile loop of both M. tuberculosis and E. coli DHFS and has been shown to be important for pteroate binding in E. coli (25, 26). Complementation of folCE40A with wild-type folC restored PAS susceptibility to wild-type levels (Fig. 5C), confirming that a mutation in the folC gene is associated with resistance to PAS. Collectively, these results indicate that PAS-mediated growth inhibition is DHPS- and DHFS-dependent.
RibD Is a Putative Functional Analog of DHFR
To date, the isolation of spontaneous PAS-resistant mutants has not revealed mutations in DHFR (7). This may suggest that the fitness cost of a mutation in DHFR is higher than for any other genes of the folate pathway, at least in vitro. We thus set out to select spontaneous M. tuberculosis mutants resistant to a specific DHFR inhibitor. In the context of a target-based drug discovery project, we synthesized a series of WR99210 analogs, one of which (NITD344) inhibited both DHFR enzymatic activity and M. tuberculosis growth (Fig. 6, A and B). Analysis of 20 spontaneous M. tuberculosis mutants resistant to the DHFR inhibitor NITD344 demonstrated no mutations in dfrA. To get an insight into the genetic determinant of the resistance to NITD344, the whole genome of the NITD344-resistant mutant M. tuberculosis R7 was sequenced. The results revealed a Gly to Ala substitution located 11 bp upstream of the translational start codon of RibD (Rv2671), causing increased RibD expression (Fig. 6C). Although RibD is annotated as a riboflavin biosynthesis protein, its C-terminal reductase domain shares strong sequence similarities with dihydrofolate reductases (27, 28) (data not shown). Because overexpression of RibD caused resistance to a specific DHFR inhibitor, we tested whether it also conferred resistance to PAS. Interestingly, M. tuberculosis R7 was cross-resistant to PAS (Fig. 6, D and E). Moreover, overexpression of RibD into M. tuberculosis also conferred resistance to PAS (Fig. 6, B and C). In contrast, overexpression of another classical riboflavin biosynthesis protein RibG did not affect mycobacterial growth inhibition by NITD344 or PAS (data not shown). Importantly, 3 out of 30 distinct M. tuberculosis clinical isolates (7) resistant to PAS harbored the same regulatory mutation found in the strain M. tuberculosis R7, showing that this mutation is associated with PAS resistance in humans.
FIGURE 6.

RibD is responsible for M. tuberculosis PAS resistance. A, chemical structure of NITD344. B, NITD344 inhibits M. tuberculosis rDHFR enzymatic activity demonstrated in the DHFR enzyme inhibition assay. OD, optical density. C, Western blot showing increased RibD expression in spontaneous NITD344-resistant mutant M. tuberculosis R7. D and E, both M. tuberculosis R7 (red squares) and H37Rv overexpressing ribD in pM262 (blue triangles) are resistant to NITD344 (D) and PAS (E). The values in B, D, and E represent the means ± S.D. from one representative experiment performed with triplicate samples.
The observation that RibD overexpression confers resistance to a specific DHFR inhibitor led us to speculate that RibD might function as a dihydrofolate reductase when expressed at high levels. Through genetic studies, we showed that dfrA is essential (29). However, the gene could successfully be knocked out in M. tuberculosis when RibD was overexpressed from a multicopy plasmid (Fig. 7, A and B). These results indicate that RibD, when expressed at high levels, can provide sufficient one-carbon carriers to sustain bacterial survival in the absence of dfrA.
FIGURE 7.
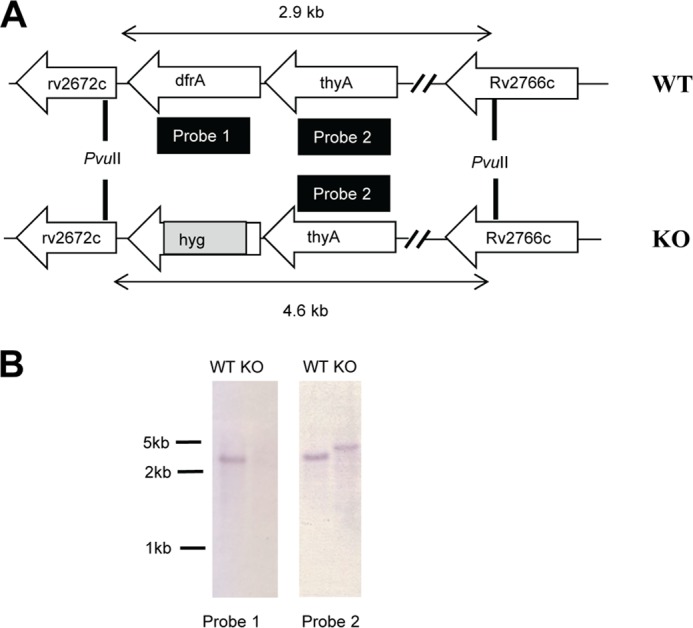
Overexpression of RibD compensates for the knock-out of DHFR in Mycobacterium. A, schematic representation of the genome region of the H37Rv wild type (WT) and knock-out mutant (KO) showing the relative gene organization of dfrA and its flanking genes, the locations of the probes for Southern blot analyses and PvuII restriction sites, and the corresponding fragment lengths. B, dfrA could be successfully knocked out only while overexpressing RibD in M. tuberculosis. The knock-out mutant of dfrA was confirmed by Southern blot using probes 1 and 2.
DISCUSSION
The folate biosynthetic pathway is essential in the production of tetrahydrofolate and derivatives required as cofactors in the biosynthesis of purines, thymidylate, serine, and methionine (30). Unlike vertebrates, which utilize exogenous sources of folates, many prokaryotes and protozoa must synthesize these essential cofactors de novo. The folate pathway thus has been targeted for the development of new antibiotics. Nevertheless, none of the enzymes in the folate pathway has conclusively been shown to be the target of current antimycobacterials. Recently, PAS, an antimycobacterial drug that has been in clinical use for more than 60 years, has been linked to the folate pathway (5). However, its exact molecular target and mechanism(s) of action are still not clear. Here our results indicate that PAS acts as a metabolic precursor that, when incorporated in the folate pathway by DHPS and DHFS, generates a toxic dihydrofolate analog that subsequently poisons the folate pathway.
During the synthesis of tetrahydrofolate, DHPS in the folate pathway catalyzes the condensation of pABA and 6-hydroxymethyl-7,8-dihydropterin pyrophosphate to form 7,8-dihydropteroate (31), which is then used as a substrate of DHFS in the synthesis of DHF. Reduction of dihydrofolate to tetrahydrofolate is carried out in most bacteria and eukaryotes by the enzyme DHFR. Our results support that PAS is misincorporated into the folate pathway as a pABA surrogate. Rather than directly inhibiting DHPS as speculated previously (4), PAS is recognized by DHPS as a substrate and converted into hydroxyl dihydropteroate. However, the hydroxyl dihydropteroate does not block the folate pathway itself. Instead, it functions as a substrate of DHFS in the synthesis of hydroxyl dihydrofolate through the incorporation of glutamate. The close structural analogy between hydroxyl dihydrofolate and dihydrofolate suggests that these compounds possibly compete for the active site of DHFR (Fig. 3). The binding of hydroxyl dihydrofolate to DHFR likely blocks its enzymatic activity. This leads to a depletion of tetrahydrofolates essential for protein synthesis, resulting in inhibition of bacterial growth and death. However, the specific effects of this interaction remain elusive. A proof of principle for this mechanism is to assess the activity of hydroxyl dihydrofolate in the rDHFR enzyme assay. However, our attempt to synthesize hydroxyl dihydrofolate was impaired by the production of unstable intermediaries. Note that consistent with our hypothesis, we demonstrated that PAS was bioactivated into an antimetabolite at the level of DHPS to generate its precursor hydroxyl dihydropteroate. Moreover, the fact that CLFT from PAS-treated cells inhibits rDHFR as well as the fact that inhibition of DHPS or mutation in DHFS (folC) decreases M. tuberculosis susceptibility to PAS further supports our antimetabolite model for the mechanism of action of PAS.
Although we have been unable to identify spontaneous mutants resistant to PAS or NITD344 encoding mutations in DHFR (dfrA), polymorphisms in other enzymes of the folate pathway, notably ThyA (thymidylate synthase), are well known to contribute to PAS resistance (7) in addition to the contributions of DHFS and RibD identified in this study. These observations are not at odds with our model of DHFR being a target of a PAS-derived antimetabolite. Treatment of bacteria with PAS inhibits DHFR, resulting in reduction of tetrahydrofolate production. As thymidylate synthase is a major consumer of reduced folates, mutations in thyA are likely to cause a decrease in the utilization of tetrahydrofolate derivatives. Thus more reduced folates become available for other essential one-carbon addition reactions, leading to increased bacterial survival (32). Mutations in the thyA are permissible likely due to the fact that M. tuberculosis encodes ThyX, a highly active functional analog of thyA that utilizes tetrahydrofolate as a carrier of single-carbon groups but not as a cofactor. Mutations in the thyA are found in clinical PAS-resistant isolates but not in the dfrA (7). Interestingly, RibD provides an additional gene target for PAS resistance of clinical relevance. We found that RibD overexpression replaced the functional activity of dfrA in a knock-out dfrA recombinant strain, clearly indicating that RibD either possesses DHFR-like activity or is able to catalyze an alternative reaction that complements the classical DHFR. However, RibD is ordinarily present in low concentrations or has poor DHFR activity, and thus it cannot completely replace the essential function of dfrA unless overexpressed. Our findings suggest that in addition to thyA, folC and ribD are candidates for the rapid identification of PAS resistance in clinical studies.
PAS or its metabolite acts as substrate for an essential enzyme. Although it does seem possible to have mutations in the activating enzyme DHFS, which no longer catalyzes the bioconversion of PAS, most resistance in clinical isolates seems to result from metabolic remodeling of the cell. In other bacteria, drug synergy can occur by inhibition of multiple steps in the folate synthetic pathway, e.g. through the combined use of trimethoprim and sulfonamide drugs (33). Our results suggest that there are at least two mechanisms to evade killing even by such a combination, either through decreased utilization of folates (thyA mutations) or through up-regulation of an alternate pathway (RibD overexpression mutations). On the other hand, such adaptations could well incur significant fitness costs yet to be explored, particularly when growing in the hostile environment of the host.
In conclusion, our results indicate that PAS is a prodrug and that it acts as a metabolic precursor that, when incorporated in the folate pathway by DHPS and DHFS, generates a toxic dihydrofolate analog that subsequently inhibits DHFR activity. In this model, the antibiotic is converted to an active form intracellularly by a metabolic pathway, but unlike classical prodrugs, it mimics the substrate of an essential enzyme, leading to poisoning of the metabolic pathway itself by its bioactivation product.
Supplementary Material
Acknowledgments
We acknowledge several scientists at Novartis: Bryan Yeung, Ravinder Kondreddi, and Ranga Rao for providing NITD344; Boon Heng Lee and Koh Sin for technical help with MIC assays and LC/MS analysis; Whitney Barnes for whole genome sequencing; Veronique Dartois for critical review of the manuscript; and Paul Smith for support and stimulating discussions. The Institute Pasteur-Korea receives support from the Korean Ministry of Education, Science and Technology.
This article was selected as a Paper of the Week.

This article contains supplemental Methods.
- PAS
- para-aminosalicylic acid
- pABA
- para-aminobenzoic acid
- DHFR
- dihydrofolate reductase
- DHF
- dihydrofolate
- rDHFR
- recombinant M. tuberculosis DHFR
- DHPS
- dihydropteroate synthase
- DHFS
- dihydrofolate synthase
- RibD
- riboflavin biosynthesis protein
- ThyA
- thymidylate synthase
- DMSO
- dimethyl sulfoxide
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- MS/MS
- tandem mass spectrometry
- CLFT
- cell lysate flow-through
- H2PtPP
- 6-hydroxymethyl-7,8-dihydropterin-pyrophosphate
- MIC
- minimum inhibition concentration.
REFERENCES
- 1. World Health Organization (2011) WHO Report: Global Tuberculosis Control, pp. 9–26, World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Dye C., Williams B. G., Espinal M. A., Raviglione M. C. (2002) Erasing the world's slow stain: strategies to beat multidrug-resistant tuberculosis. Science 295, 2042–2046 [DOI] [PubMed] [Google Scholar]
- 3. Lehmann J. (1946) para-Aminosalicylic acid in the treatment of tuberculosis. Lancet 1, 15. [DOI] [PubMed] [Google Scholar]
- 4. Nopponpunth V., Sirawaraporn W., Greene P. J., Santi D. V. (1999) Cloning and expression of Mycobacterium tuberculosis and Mycobacterium leprae dihydropteroate synthase in Escherichia coli. J. Bacteriol. 181, 6814–6821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rengarajan J., Sassetti C. M., Naroditskaya V., Sloutsky A., Bloom B. R., Rubin E. J. (2004) The folate pathway is a target for resistance to the drug para-aminosalicylic acid (PAS) in mycobacteria. Mol. Microbiol. 53, 275–282 [DOI] [PubMed] [Google Scholar]
- 6. Ratledge C., Brown K. A. (1972) Inhibition of mycobactin formation in Mycobacterium smegmatis by p-aminosalicylate. A new proposal for the mode of action of p-aminosalicylate. Am. Rev. Respir. Dis. 106, 774–776 [DOI] [PubMed] [Google Scholar]
- 7. Mathys V., Wintjens R., Lefevre P., Bertout J., Singhal A., Kiass M., Kurepina N., Wang X. M., Mathema B., Baulard A., Kreiswirth B. N., Bifani P. (2009) Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 53, 2100–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chakraborty S., Gruber T., Barry C. E., 3rd, Boshoff H. I., Rhee K. Y. (2013) para-Aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schweitzer B. I., Dicker A. P., Bertino J. R. (1990) Dihydrofolate reductase as a therapeutic target. FASEB J. 4, 2441–2452 [DOI] [PubMed] [Google Scholar]
- 10. Hekmat-Nejad M., Rathod P. K. (1997) Plasmodium falciparum: kinetic interactions of WR99210 with pyrimethamine-sensitive and pyrimethamine-resistant dihydrofolate reductase. Exp. Parasitol. 87, 222–228 [DOI] [PubMed] [Google Scholar]
- 11. Gerum A. B., Ulmer J. E., Jacobus D. P., Jensen N. P., Sherman D. R., Sibley C. H. (2002) Novel Saccharomyces cerevisiae screen identifies WR99210 analogues that inhibit Mycobacterium tuberculosis dihydrofolate reductase. Antimicrob. Agents Chemother. 46, 3362–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurabachew M., Lu S. H., Krastel P., Schmitt E. K., Suresh B. L., Goh A., Knox J. E., Ma N. L., Jiricek J., Beer D., Cynamon M., Petersen F., Dartois V., Keller T., Dick T., Sambandamurthy V. K. (2008) Lipiarmycin targets RNA polymerase and has good activity against multidrug-resistant strains of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 62, 713–719 [DOI] [PubMed] [Google Scholar]
- 13. Freuler F., Stettler T., Meyerhofer M., Leder L., Mayr L. M. (2008) Development of a novel Gateway-based vector system for efficient, multiparallel protein expression in Escherichia coli. Protein Expr. Purif. 59, 232–241 [DOI] [PubMed] [Google Scholar]
- 14. Schmitt E. K., Riwanto M., Sambandamurthy V., Roggo S., Miault C., Zwingelstein C., Krastel P., Noble C., Beer D., Rao S. P., Au M., Niyomrattanakit P., Lim V., Zheng J., Jeffery D., Pethe K., Camacho L. R. (2011) The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew Chem. Int. Ed. Engl. 50, 5889–5891 [DOI] [PubMed] [Google Scholar]
- 15. Gengenbacher M., Xu T., Niyomrattanakit P., Spraggon G., Dick T. (2008) Biochemical and structural characterization of the putative dihydropteroate synthase ortholog Rv1207 of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 287, 128–135 [DOI] [PubMed] [Google Scholar]
- 16. Pethe K., Sequeira P. C., Agarwalla S., Rhee K., Kuhen K., Phong W. Y., Patel V., Beer D., Walker J. R., Duraiswamy J., Jiricek J., Keller T. H., Chatterjee A., Tan M. P., Ujjini M., Rao S. P. S., Camacho L., Bifani P., Mak P. A., Ma I., Barnes S. W., Chen Z., Plouffe D., Thayalan P., Ng S. H., Au M., Lee B. H., Tan B. H., Ravindran S., Nanjundappa M., Lin X. H., Goh A., Lakshminarayana S. B., Shoen C., Cynamon M., Kreiswirth B., Dartois V., Peters E. C., Glynne R., Brenner S., Dick T. (2010) A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun. 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bardarov S., Bardarov Jr. S., Jr., Pavelka Jr. M. S., Jr., Sambandamurthy V., Larsen M., Tufariello J., Chan J., Hatfull G., Jacobs Jr. W. R., Jr. (2002) Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG, and M. smegmatis. Microbiology 148, 3007–3017 [DOI] [PubMed] [Google Scholar]
- 18. Parish T., Stoker N. G. (2000) Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146, 1969–1975 [DOI] [PubMed] [Google Scholar]
- 19. Stover C. K., de la Cruz V. F., Fuerst T. R., Burlein J. E., Benson L. A., Bennett L. T., Bansal G. P., Young J. F., Lee M. H., Hatfull G. F., et al. (1991) New use of BCG for recombinant vaccines. Nature 351, 456–460 [DOI] [PubMed] [Google Scholar]
- 20. Youmans G. P., Raleigh G. W., Youmans A. S. (1947) The tuberculostatic action of para-aminosalicylic acid. J. Bacteriol. 54, 409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Achari A., Somers D. O., Champness J. N., Bryant P. K., Rosemond J., Stammers D. K. (1997) Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat. Struct. Biol. 4, 490–497 [DOI] [PubMed] [Google Scholar]
- 22. Ong W., Sievers A., Leslie D. E. (2010) Mycobacterium tuberculosis and sulfamethoxazole susceptibility. Antimicrob. Agents Chemother. 54, 2748; author reply 2748–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei J.-R., Krishnamoorthy V., Murphy K., Kim J.-H., Schnappinger D., Alber T., Sassetti C. M., Rhee K. Y., Rubin E. J. (2011) Depletion of antibiotic targets has widely varying effects on growth. Proc. Natl. Acad. Sci. U.S.A. 108, 4176–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woong Park S., Klotzsche M., Wilson D. J., Boshoff H. I., Eoh H., Manjunatha U., Blumenthal A., Rhee K., Barry C. E., 3rd, Aldrich C. C., Ehrt S., Schnappinger D. (2011) Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 7, e1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young P. G., Smith C. A., Metcalf P., Baker E. N. (2008) Structures of Mycobacterium tuberculosis folylpolyglutamate synthase complexed with ADP and AMPPCP. Acta Crystallogr. D Biol. Crystallogr. D64, 745–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mathieu M., Debousker G., Vincent S., Viviani F., Bamas-Jacques N., Mikol V. (2005) Escherichia coli FolC structure reveals an unexpected dihydrofolate binding site providing an attractive target for anti-microbial therapy. J. Biol. Chem. 280, 18916–18922 [DOI] [PubMed] [Google Scholar]
- 27. Chen S. C., Chang Y. C., Lin C. H., Lin C. H., Liaw S. H. (2006) Crystal structure of a bifunctional deaminase and reductase from Bacillus subtilis involved in riboflavin biosynthesis. J. Biol. Chem. 281, 7605–7613 [DOI] [PubMed] [Google Scholar]
- 28. Li R., Sirawaraporn R., Chitnumsub P., Sirawaraporn W., Wooden J., Athappilly F., Turley S., Hol W. G. (2000) Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J. Mol. Biol. 295, 307–323 [DOI] [PubMed] [Google Scholar]
- 29. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 30. Brown G. M., Williamson J. M. (1982) Biosynthesis of riboflavin, folic acid, thiamine, and pantothenic acid. Adv. Enzymol. Relat. Areas Mol. Biol. 53, 345–381 [DOI] [PubMed] [Google Scholar]
- 31. Shiota T., Disraely M. N., McCann M. P. (1964) The enzymatic synthesis of folate-like compounds from hydroxymethyldihydropteridine pyrophosphate. J. Biol. Chem. 239, 2259–2266 [PubMed] [Google Scholar]
- 32. Green J. M., Nichols B.P., Matthews R.G. (1996) Folate biosynthesis, reduction, and polyglutamation. in Escherichia coli and Salmonella, Vol. 1 (Neidhardt F. C., ed), pp. 665–673, American Society for Microbiology Press, Washington, D. C. [Google Scholar]
- 33. Forgacs P., Wengenack N. L., Hall L., Zimmerman S. K., Silverman M. L., Roberts G. D. (2009) Tuberculosis and trimethoprim-sulfamethoxazole. Antimicrob. Agents Chemother. 53, 4789–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.