

Background: The contribution of chlorite dismutases to heme metabolism is not well characterized.
Results: Inactivation of cld/hemQ makes S. aureus a heme auxotroph and SCV. HemQ is inactivated via its redox-sensitive heme.
Conclusion: HemQ may have a regulatory role controlling heme biosynthesis in response to redox or other stresses.
Significance: HemQ proteins may regulate heme biosynthesis and/or cell growth in many Gram-positive organisms.
Keywords: Bacteria, Heme, Peroxidase, Redox Regulation, Staphylococcus aureus, Superoxide Ion, Chlorite Dismutase
Abstract
The chlorite dismutases (C-family proteins) are a widespread family of heme-binding proteins for which chemical and biological roles remain unclear. An association of the gene with heme biosynthesis in Gram-positive bacteria was previously demonstrated by experiments involving introduction of genes from two Gram-positive species into heme biosynthesis mutant strains of Escherichia coli, leading to the gene being renamed hemQ. To assess the gene product's biological role more directly, a Staphylococcus aureus strain with an inactivated hemQ gene was generated and shown to be a slow growing small colony variant under aerobic but not anaerobic conditions. The small colony variant phenotype is rescued by the addition of exogenous heme despite an otherwise wild type heme biosynthetic pathway. The ΔhemQ mutant accumulates coproporphyrin specifically under aerobic conditions. Although its sequence is highly similar to functional chlorite dismutases, the HemQ protein has no steady state reactivity with chlorite, very modest reactivity with H2O2 or peracetic acid, and no observable transient intermediates. HemQ's equilibrium affinity for heme is in the low micromolar range. Holo-HemQ reconstituted with heme exhibits heme lysis after <50 turnovers with peroxide and <10 turnovers with chlorite. The heme-free apoprotein aggregates or unfolds over time. IsdG-like proteins and antibiotic biosynthesis monooxygenases are close sequence and structural relatives of HemQ that use heme or porphyrin-like organic molecules as substrates. The genetic and biochemical data suggest a similar substrate role for heme or porphyrin, with possible sensor-regulator functions for the protein. HemQ heme could serve as the means by which S. aureus reversibly adopts an SCV phenotype in response to redox stress.
Introduction
Heme proteins are essential for many cellular processes, including respiration, small molecule sensing, biosynthesis, and biodegradations. The chlorite dismutases (Clds2 or C-family proteins) are a widespread but poorly understood group of microbial, histidine-ligated heme proteins found in at least 13 bacterial and 3 archaeal phyla (1). These proteins catalyze the detoxification of chlorite (ClO2−) in perchlorate-respiring Proteobacteria via its conversion to O2 and Cl− (2). This unusual reaction, only the second known O–O bond-forming process in biology, has been studied via a variety of mechanistic and structural approaches (3–12).
A cld gene, however, is found in many microbes, including important pathogenic species such as Staphylococcus aureus that have no known ability or environmental demand to detoxify chlorite. The absence of a known need for chlorite metabolism has led to questions about the gene's biological role. Genetic and bioinformatic studies have suggested possible functions in heme metabolism and/or respiratory biochemistry. In Actinobacteria, cld is co-operonic with an apparently complete set of hem genes, which encode the eight-step biosynthesis of heme. In at least two cases, cld genes form fusions with the gene encoding ferrochelatase, which catalyzes the pathway's final Fe(II)/protoporphyrin IX coupling step. In the Halobacteriaceae, the cld genes form fusions with genes annotated as “isdG-like” (13). The latter encode heme oxygenases used by Gram-positive bacteria for the oxidative scission and subsequent reclamation of nutritional iron from ingested heme (14).
An association between cld and heme metabolism was experimentally confirmed by Dailey et al. (15) by introducing cld genes from two Gram-positive bacteria (Bacillus subtilis and Mycobacterium tuberculosis) into Escherichia coli inactivated for the ultimate and penultimate steps of the heme biosynthetic pathway. It was found that a Gram-positive cld gene was required for the final two steps of the pathway to function, leading to the renaming of the gene as hemQ. However, in the absence of an identifiable enzymatic function for HemQ and because E. coli lacks an endogenous cld, it was not possible to determine by what means HemQ supports heme biosynthesis. A potential catalase role was proposed. In addition to heme biosynthesis, functional links to anaerobic metabolism or the switch between aerobic/anaerobic lifestyles have also been experimentally demonstrated for the B. subtilis hemQ (previously referred to as ywfL) using proteomic and transcriptomic methods (16, 17).
SaHemQ is representative of a large subgroup of Cld-family proteins, including all Clds from phylum Firmicutes, which have a conserved glutamine residue at the position of a distal Arg that we have observed is critical for efficient reactivity with anionic oxidants (1, 3, 8). Unlike typical peroxidases, Clds likewise lack a distal base and other residues important for stabilizing polar reaction intermediates (Fig. 1) (20). We therefore hypothesized that SaHemQ's spectroscopic and catalytic properties would differ profoundly from those of its O2-evolving homolog from Dechloromonas aromatica (DaCld) or structurally related peroxidases, which have been the subject of ongoing detailed investigation by our group.
FIGURE 1.
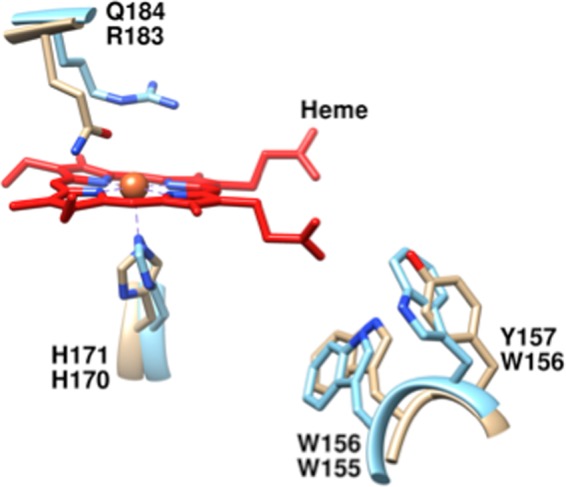
Heme-binding site of the heme-free Cld from G. stearothermophilus (carbon tan) a close sequence relative of HemQ, overlaid with the heme-bound Cld from D. aromatica (carbon blue). His-171 (G. stearothermophilus numbering) is a strictly conserved residue among all Cld/HemQ proteins that aligns with the heme-ligating His-170 from DaCld. Trp-156 is likewise strictly conserved among the entire family and is important for protein stability. Gln-184 aligns with DaCld-Arg-184. A glutamine at this position is strictly conserved among Clds/HemQ proteins from phylum Firmicutes. Figure was generated using PyMOL from Protein Data Bank codes 1T0T and 3Q08.
A role for the hemQ gene in heme metabolism is directly confirmed here using a ΔhemQ strain of S. aureus. Despite having an apparently complete heme biosynthetic pathway, this strain behaves like a heme auxotroph, growing as a small colony variant (SCV). A comprehensive study of the catalytic and biochemical properties of the SaHemQ gene product from S. aureus reveals a weakly heme binding, conditionally stable protein with little catalytic activity toward redox substrates. Bioinformatics analysis shows close evolutionary and structural relationships between SaHemQ and the S. aureus IsdG and related cofactor-independent monooxygenases. Based on the available evidence, we propose a model in which SaHemQ uses heme or porphyrin as a substrate rather than a cofactor during heme biosynthesis, where heme binding and lysis may serve a regulatory role over heme biosynthesis, the SCV phenotype, or both.
EXPERIMENTAL PROCEDURES
Materials
Diluted stocks of reagent grade hydrogen peroxide (H2O2) (35%, Acros) and peracetic acid (PAA) (Sigma) were freshly made, and their concentrations routinely determined by iodometric titration. The one-electron reductant used for the peroxidase assay was 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid (ABTS) (TCI-Ace Co.). All porphyrin standards and substrates were purchased from Frontier Scientific (Logan, UT). All solvents used were HPLC grade from Fluka Analytical.
Bacterial Strains, Growth Conditions, and Plasmid Construction
The strains used in this study are derivatives of the human isolate S. aureus Newman (21). Generation of the hemB mutant, which is not an in-frame mutant, has been described previously.3 The hemQ in-frame deletion mutant was constructed using previously described methods (23) and the following primers: R1-L, 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTGCTTCTAAACTTGGGAATACAA-3′; R1-R, 5′-GTCGACTTGACTCATGTTTATCGCTCC-3′; R2-L, 5′-GTCGACATTTCTTAATACATTGGTACGTTT-3′; and R2-R, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGTTAAAACATCAATCAATCTACC-3′.
The hemQ complementation plasmid was constructed by amplifying hemQ (locus NWMN_0550) via standard PCR techniques and the following primers that anneal to it: 5′-GCGCTCGAGAGTCAAGCAGCCGAAACATTA-3′ and 5′-GCGGGATCCTTAAGAAATCGCAAAGAATTGATC-3′. The PCR product was cloned into the plasmid pOS1 containing the constitutive promoter Plgt using the BamHI and XmaI restriction sites that were engineered into the primers, respectively. Strains were routinely grown in tryptic soy broth (TSB) or TSB-agar, and plasmid selection for the complement was maintained by using chloramphenicol at 10 μg ml−1. Hemin (Sigma) was prepared in a stock concentration of 10 mm by dissolution in 0.1 n NaOH.
Bacterial Culture Growth
TSB-agar plates were streaked from glycerol frozen cell stocks and incubated for 2 days at 37 °C (hemB and ΔhemQ) or overnight (WT). Single colonies were used to inoculate starter cultures (5 ml of TSB, 15-ml Falcon tubes) grown on an Innova shaker incubator (37 °C, 180 rpm). To control for the density of cells added to initiate subsequent cultures monitored for their growth kinetics, seed cultures were allowed to freshly saturate (∼42 h for the mutants; 12 h for WT). For the anaerobic cultures, seed cultures were grown to saturation in sealed culture tubes (∼14 h for the mutants; 8 h WT). This allowed the cultures to consume the O2 in the medium and to shift to anaerobic growth prior to inoculation into septum-sealed anaerobic (N2-purged) culture tubes.
All growth curves were measured in 50 ml of TSB media inoculated 1:100 from starter cultures. Aerobic cultures were grown in 125-ml glass Erlenmeyer flasks and anaerobic cultures in sealed 50-ml plastic Falcon tubes. Growth was monitored via optical density at 600 nm (A600 nm) measured on 0.5-ml aliquots withdrawn every 2 h over a 12-h period using a biophotometer (Eppendorf). Septum-sealed anaerobic cultures were sampled via sterile air-tight syringes. Cultures were supplemented with 5 μm hemin (from 1 mm stock in 0.1 m NaOH) as noted.
Porphyrin Metabolite Extraction and Analysis
Cells were grown as described above, pelleted via gentle centrifugation (Beckman J-10, 7,000 × g, 15 min, 4 °C), and frozen at −80 °C prior to analysis. Pellets were subsequently thawed and resuspended in ∼5 ml of 1:1 1 m HCl/DMSO. The suspensions were lysed by sonication on ice using a micro-tip fitted Branson ultrasonifier at 65% for 5 min (3-s pulses, 2-s intervals). Cellular debris was removed by centrifugation (Beckman JA-20, 17,000 × g, 30 min, 4 °C). The soluble fraction was diluted to 50 ml with sterile deionized water and loaded onto a washed and pre-equilibrated C18 Sep-Pak column (Waters). A colored band due to porphyrin metabolites was readily observed in the column resin and eluted in 2 ml of acetonitrile (Fluka). The solution was filtered using a 0.45-μm syringe filter (VWR Scientific) and concentrated to 200 μl by vacuum centrifugation. To each sample, 0.8 μm of internal porphyrin standard (2-vinyl-4-hydroxymethyl-deuteroporphyrin IX) was added prior to analysis to control for run-to-run variability in instrument performance.
Samples were injected onto a Dionex Ultimate 3000 ultra-HPLC instrument. Separation was achieved by linear gradient elution at 4 ml/min on a BDS Hypersil C18 column (particle size = 2.4 μm) (ThermoScientific) at 50 °C transitioning from 100% solvent A (0.1% formic acid in water) to 100% solvent B (0.1% formic acid in methanol) over 20 min. Visible spectra were measured continuously over 390–420 nm. The ultra-HPLC was coupled to a Bruker MicroTOF-Q11 electrospray mass analyzer operating in positive ion mode. The mass spectrometer used a capillary voltage of 4500 V at 180 °C, and the nebulizing gas was set at 6.0 ml/min. The instrument was operated, and data were analyzed using the Hystar software package. Metabolites were identified via their exact molecular weights determined with accuracies of ± 0.005 atomic mass units.
A mix of porphyrin metabolites and heme intermediates (Frontier Scientific) was used to determine elution times and to construct a standard curve for each metabolite via its measured extracted ion chromatogram peak area. Extracted ion chromatogram peak areas for the metabolites in the samples were referenced to these standard curves to determine their concentrations. (Data are not shown here but are contained in Ref. 24.) Standard curves for uroporphyrin III (m/z = 830.23; 0.14–2.8 μm), coproporphyrin III (m/z = 645.27; 0.14–2.8 μm), protoporphyrin IX (m/z = 562.26; 0.18–3.5 μm), hemin (m/z = 616.18; 0.6–7.5 μm), and the internal standard (2-vinyl-4-hydroxymethyl-deuteroporphyrin IX) (m/z = 567.26; 0.13–2.6 μm) were generated from triplicate measurements. All stock solutions were made and diluted in analytical grade acetonitrile (Fluka Analytical).
Catalase Activity in Lysates
ΔhemQ, hemB, and WT S. aureus strains were grown aerobically for 12 h, pelleted, resuspended in sodium phosphate buffer, pH 6.8, sonicated, and centrifuged to isolate the clarified lysate. The clarified lysates were centrifuge-concentrated (10,000 kDa) at 4 °C to eliminate small molecules and to increase the protein concentration. Catalase activity was detected by continuously monitoring oxygen production polarographically on a Clark-type O2 electrode. Reactions were carried out in a 1.5-ml chamber in which buffer alone was equilibrated for 5 min under N2 gas to lower the starting O2 concentration to between 0 and 50 μm. The probe was inserted into the solution, and measurements were initiated. Varying concentrations of H2O2 (10 and 50 mm) were added via gas-tight Hamilton syringe, and background reactivity was measured. Finally, 5–10 μl of clarified lysate was added via a gas-tight syringe. Initial rates were determined from the first 5–10% of the linear portion of the O2 production curve, relative to background O2 production measured in the absence of added proteins. Initial velocities were referenced per total protein as measured via the Bradford colorimetric assay (Bio-Rad).
Cloning, Expression, and Purification of HemQ Protein
The hemQ gene from S. aureus strain COL was obtained from TIGR. A QuikChange® PCR mutagenesis kit (Stratagene) was used to insert an NdeI cut site upstream of the gene using the following primer, 5′-TGTACAAAAAAGCAGGCTTCATATGAGTCAAGCAGCCGAAAC-3′, and its reverse complement. Additionally, an NdeI cut site in the gene itself was silently mutated using a second primer, 5′-CCTAATCCCTACATATTCATACGTATCAGTCATTGAATTGAGC-3′, and its reverse complement. The amplification product was cloned following digestion with NdeI/BsrGI restriction enzymes (Promega) into a pET41(a) (PT7, KanR, Novagen) expression vector. The expression construct was sequenced and used to transform competent E. coli Tuner (DE3) cells (Novagen) via heat shock for protein overexpression. Cell stocks were maintained and stored as frozen glycerol (30%) stocks at −80 °C.
Overexpression of HemQ was achieved in a manner similar to that used for DaCld (7). Briefly, a freshly saturated culture of E. coli Tuner (DE3) cells containing the pET-41(a)-hemQ construct was prepared from a single colony from an LB agar (50 mg of kanamycin/ml) plate inoculated (1:100) with 50 ml of Terrific Broth. The 50-ml culture was grown overnight at 37 °C on a shaker incubator (250 rpm, Innova) and used to inoculate 1 liter of Terrific Broth in a 2-liter Fernbach flask. 1-Liter cultures were grown at 30 °C, 250 rpm, until the A600 nm reached ∼0.4–0.6 and induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside. The temperature was lowered to 20 °C and protein expressed for 24 h. Cells were harvested by centrifugation (Beckman J-2 centrifuge: JA-10 rotor, 7000 × g, 30 min, 4 °C) and pellets stored at −80 °C until used.
For protein purification, ∼10 g of wet cell pellet was resuspended in 5 volumes of 0.1 m potassium phosphate buffer, pH 7.4, supplemented with 1 mm of the protease inhibitor phenylmethylsulfonyl fluoride (PMSF). Cells were lysed on ice by sonication using a micro-tip fitted Branson ultrasonifier at 65% power (3-s pulses; 2-s intervals). Cellular debris was sedimented by centrifugation (JA-20 rotor, 17,000 × g, 50 min, 4 °C), and the soluble fraction was dialyzed against 20 mm Tris-Cl buffer (pH 8.6, 4 °C, 10,000 MWCO tubing (Fisher)) with three buffer exchanges at ≥3 h per cycle.
The dialysate was loaded at 4 ml/min onto a pre-equilibrated (20 mm Tris-Cl, pH 8.6) Sepharose-Q Fast Flow anion exchange column (GE Healthcare) using an AKTA Prime protein purification system (GE Healthcare). Weakly bound proteins were washed from the column with 250 mm NaCl in 20 mm Tris-Cl, pH 8.6. HemQ was eluted with 300 mm NaCl in 20 mm Tris-Cl, pH 8.6. Fractions enriched in the 29-kDa monomer (viewed by SDS-PAGE) were concentrated under N2 gas pressure in a stirred cell (Amicon) with a 10,000 MWCO YM-20 membrane. Concentrated protein (∼10 ml) was loaded onto a 2.6 × 240-cm gel filtration column (S-200 Sephacryl, GE Healthcare) by gravity and run at 0.4 ml/min in 0.1 m potassium phosphate buffer, pH 6.8. Fractions were collected and screened for purity using SDS-PAGE. Pure protein was pooled and concentrated in 10,000 MWCO centrifuge concentrators (Millipore) (JA-10, 3000 × g, 20 min, 4 °C). Aliquots were flash-frozen in liquid N2 and stored at −80 °C.
Two methods for enhancing the fraction of as-purified holo-HemQ were tried. First, expression was carried out in the presence of added δ-aminolevulinic acid, a heme precursor commonly used to enhance the abundance of heme inside expressed heme proteins. Second, an E. coli protein expression strain capable of taking up exogenously added heme was used to express the protein in the presence of heme-supplemented growth media (25). Although both methods increased the fraction of as-isolated holo-HemQ, the magnitude of the increase was marginal.
Protein and Heme Concentration
To determine protein concentrations, the Bradford method was used with a standard curve generated using bovine serum albumin (Bio-Rad). Final heme concentrations were determined by the pyridine-hemochrome method of Berry and Trumpower (26) using horse heart myoglobin as a standard (Sigma).
Reconstitution of Apo-HemQ with Heme
As-purified holoprotein represented a minority (≤25%) of the pure protein described above, eluting from the gel filtration column independently from the apo-HemQ. This fraction was collected and characterized. In addition, purified apoprotein was incubated at 4 °C for several hours with ∼2 eq of hemin (from 1 mm stock dissolved in 0.1 m NaOH) (Frontier Scientific) per protein monomer to increase its heme content. Excess hemin was added to the cell pellets immediately prior to homogenization (27), as with DaCld, but the former method gave a higher yield of heme incorporation. Excess weakly bound or unbound heme was observed as a shoulder to the Soret band corresponding to the absorbance due to hemin dissolved in buffer. To avoid interference from weakly bound heme with the measurements of activity, the protein was dialyzed against buffer containing 1 mm EDTA, which resulted in the disappearance of the shoulder. The protein with heme reincorporated is referred to as heme-reconstituted holo-HemQ. This protein has ASoret/A280 ratios less than 0.5, reflecting substoichiometric heme binding (also quantified by pyridine hemochrome assay).
Molecular Weight Determination
The molecular weights of the reconstituted holo-HemQ and apo-HemQ proteins were estimated by size exclusion chromatography using a 1.6 × 18-cm analytical gel filtration column (S-200 Sephacryl, GE Healthcare) run at 0.5 ml/min in 0.05 m potassium phosphate, 150 mm NaCl, pH 7, at 4 °C. Molecular weight standards (Bio-Rad) were used to make a standard curve based on elution volumes as follows: protein aggregate (void), thyroglobulin (670 kDa), α-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (13.5 kDa). The method assumes globular protein structures and is consequently approximate. The HemQ monomer molecular weight was further confirmed by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry using a Bruker Autoflex III spectrometer.
UV-visible pH Titrations
All UV-visible spectra were measured on a Varian Cary 50 spectrometer with temperature control from a Peltier cooler. The holo-enzyme was titrated in 0.1 m potassium phosphate buffer starting at pH 6.8, 25 °C, and finishing at pH 5.1 or 10.2. Each titration was conducted in a 2-ml cuvette with the protein at ∼8 μm heme-containing monomer, where the heme concentration was determined via the pyridine hemochrome assay. Solutions were continuously stirred, and the pH was monitored using a pH meter with a glass electrode (Corning, pH 430). Small volumes of HCl (1 m) or NaOH (1 m) were added to the solution; the pH was measured, and UV-visible spectra were recorded. All spectra were adjusted for dilution.
Heme and Ligand Equilibrium Binding
A 1 mm stock of heme was made in 0.1 m NaOH and diluted to working concentrations in 0.1 m potassium phosphate buffer, pH 6.8. All other ligands were prepared in the same buffer and corrected for any pH changes. Equilibrium dissociation constants were determined via UV-visible titration of 5–10 μm holo- or apoprotein with increasing concentrations of ligand (0–500 μm for HCN; 0–10 mm for imidazole; 0–50 μm for heme). Separate samples were prepared for each ligand concentration and were allowed to incubate several hours in tubes to achieve full equilibration. UV-visible spectra were recorded for each sample, and equilibrium constants were determined from plots of the change in absorbance at wavelengths of maximal change as a function of ligand concentration. For heme, separate titrations were performed in the absence and presence of protein, and the difference between the two spectra was plotted as a function of heme concentration, to correct for absorbance contributions due to free heme. The data were fit to Equation 1, the equilibrium isotherm appropriate for weakly binding ligands,
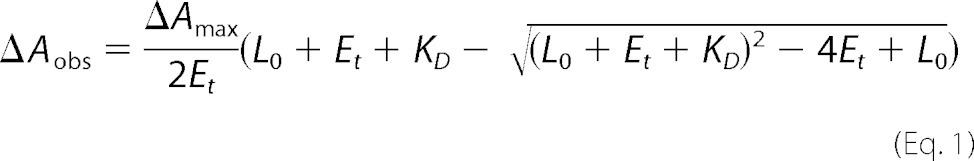 |
L0, Et, KD, and ΔAmax are the initial ligand concentration, total enzyme or protein concentration, the equilibrium dissociation constants, and the maximum change in absorbance, respectively.
Titrations using fluorescence spectroscopy were carried out on a Jobin Yvon Horiba FluoroMax-3 fluorimeter in scanning mode. Apo-HemQ (5 μm) was diluted in 0.1 m potassium phosphate buffer, pH 6.8, and titrated with stocks of heme (1 mm in 0.1 m NaOH) and protoporphyrin IX (1 mm in DMSO) each diluted to 500 μm at 25 °C. The percent of fluorescence quenched was plotted as a function of ligand concentration and fit using Equation 1 with ΔA substituted by % quenched.
Circular Dichroism (CD)
Electronic CD absorption spectra of heme-reconstituted holo- and apo-HemQ were measured using a Jasco circular dichroism spectrometer. 1 μm solutions of apo- and holo-HemQ were scanned from 190 to 250 nm (0.1 m potassium phosphate buffer, pH 6.8, 25 °C). The data were analyzed using the web-based software, to estimate the content of α-helix, β-sheet, and turns/loop segments (28).
Transient Kinetics
For transient kinetic measurements of the reaction of reconstituted holo-HemQ with peracetic acid (PAA) or hydrogen peroxide, a Hi-Tech SF-61DX2 stopped-flow system in single-mixing mode was used. Briefly, ∼8 μm of -ontaining monomer was rapidly mixed with varying concentrations of oxidants in 0.1 m potassium phosphate buffer, pH 6.8, 25 °C. Reactions were monitored over various time scales from 320 to 700 nm using diode array detection.
Steady State Kinetics
Peroxidase activity was studied by measuring initial rates of 2,2′-azino-bis[3-ethylbenzthiazoline-6-sulfonic acid (ABTS) oxidation using H2O2 (0–30 mm) or PAA (0–10 mm). Reactions were monitored spectroscopically at 25 °C in a 1-cm pathlength quartz cuvette in 0.1 m potassium phosphate buffer, pH 6.8. ABTS oxidation (0.005–10 mm) was observed at 414 nm (ϵ414 = 36 mm−1cm−1). Reactions were initiated by the addition of 1–2 μm of heme-containing monomer. Initial rates were determined from the first 5–10% of the linear portion of the absorbance curve relative to background oxidation measured in the absence of enzyme (Kaleidagraph).
Oxygen production due to chlorite decomposition was assayed using a Clark-type O2 electrode (YSI Inc.) with a custom-built water bath jacket for temperature control. The electrode was equilibrated to 25 °C and calibrated against air-saturated Milli-Q water (Millipore) using the O2 concentration of air-saturated water as a reference. Sodium chlorite (0–20 mm) was added from freshly prepared stocks and continuously stirred. Reactions were initiated via syringe injection of 5 μl of protein (final concentration = 1–2 μm in heme-containing monomer).
Catalase reactivity was likewise monitored via Clark-type O2 electrode. Equilibration and calibration were performed as described above. Reactions were carried out in a 1.5-ml chamber in which buffer was equilibrated for 5 min under N2 gas to lower the starting O2 concentration to 0–50 μm. Varying concentrations of H2O2 (0–50 mm) were added via a gas-tight Hamilton syringe, and background reactivity was measured. 1–2 μm heme-containing monomer was added via a gas-tight syringe to initiate each reaction. Initial rates were determined from the first 5–10% of the linear portion of the O2 production curve, relative to background O2 production measured in the absence of enzyme. All steady state kinetic parameters were obtained from plots of initial rate/[heme-containing monomer] versus [substrate] fit to the Michaelis-Menten equation (Kaleidagraph).
Heme Oxidation and Oxygenation Assays
The redox stability of the HemQ-bound heme was assessed in the ferric state in the presence of chlorite, peracetic acid, H2O2, and dithionite/O2. For the dithionite/O2 reactions, conditions were carefully controlled to distinguish whether heme lysis can occur via heme monooxygenation originating from the coordinated ferrous-O2 state or via initially noncoordinated Fe(III) and H2O2 (29). For the latter reactions, ∼30–40 μm solutions of holo-HemQ (both as-isolated and reconstituted with heme) in 20 mm potassium phosphate buffer (pH 6.8, 25 °C) were titrated with stock solutions of each oxidant prepared in the same buffer. Solutions were added in 0.5–2-μl aliquots containing 2–8 eq of oxidant and allowed to equilibrate until the UV-visible spectrum of the protein ceased changing. Excess oxidant was added at the end of the titration to be certain that the end point had been reached.
For the heme oxygenation reactions, HemQ was diluted to a final concentration of 30–40 μm in 20 mm potassium phosphate buffer, pH 6.8, in a sealed and N2-purged cuvette. >5 mg each of superoxide dismutase from bovine erythrocytes (>3000 units/mg Sigma) and catalase from Aspergillus niger (2000 units/mg, MP Biomedical) were added to the solution to rapidly detoxify any superoxide or peroxide produced in the presence of HemQ, reductant, and O2. In the first experiment, an excess (∼2500 eq) of N2-purged 1 mm dithionite was added to the protein solution, which was subsequently purged with air. UV-visible spectral changes were monitored over time (Cary 50). Spectra were adjusted for volume and holo-HemQ concentration. A no-HemQ control experiment was carried out under identical conditions to assess whether the spectral changes may have been due to the added catalase, which has a strongly absorbing Soret band (Soret λmax = 406 nm) that overlaps with the Soret band from HemQ. As an additional control, the experiment was carried out in the absence of superoxide dismutase/catalase to visualize the reaction in the absence of the competing absorbance from the catalase. In this case, dithionite was added titrimetrically from an anaerobic stock to an aerobic sample of HemQ, and the spectral changes were monitored.
Coproporphyrinogen Oxidase Assays
Oxidized coproporphyrin III (Frontier) was reduced under continuous argon purge using excess sodium amalgam (Sigma) and 0.05 m potassium hydroxide. The resulting reduced porphyrinogen was resuspended at an approximate concentration of 6.7 mm in argon-purged 250 mm Tris-HCl buffer supplemented with 1 mm EDTA and 100 mm sodium thioglycolate for stabilization of the porphyrin in its reduced state (30). Three 500-μl reactions were prepared aerobically in 15-ml culture tubes with varying concentrations of coproporphyrinogen (100, 500, and 1000 μm) and 0.4 mg/ml apo-HemQ per reaction mixture. Reactions were shaken in the dark at 37 °C and 180 rpm for 90 min.
For product analysis, 10 μl of trifluoroacetic acid (TFA) was added to 400 μl of each reaction mixture to precipitate the protein. 30 μl of each reaction was injected onto a C-18 high performance liquid chromatography (HPLC) column (Vydac TP Protein and Peptide C18 column, 10-mm inner diameter, 250-cm length, 5-μm particle diameter, Grace Davison Discovery Sciences) with run parameters as follows: 5 min at 100% Buffer A (water with 0.01% TFA); 35-min linear gradient from 0 to 100% Buffer B (methanol with 0.01% TFA); 5 min at 100% Buffer B. The HPLC traces for the reaction products were compared with traces measured on known concentrations of standards, as well as a no-HemQ control.
Network Analysis of Clds and Their Closest Sequence Relatives
25,802 Cld/HemQ sequences and their closest homologs belonging to clan CL0032 were downloaded from the Pfam database and filtered to 60% pairwise sequence identities using CD-Hit. The resulting 5923 sequences were used to generate sequence similarity networks using previously published methods (31) and visualized using the Cytoscape program. Nodes in the network represent sequences, and edges represent BLAST E-values. An edge is drawn between two sequences only if the statistical significance of the similarity score between them is less than (i.e. better than) a defined E-value cutoff. Multiple E-values were sampled to assess the effects of increasing the level of stringency on the resulting sequence groupings. The organic layout method, which treats two sequences as two masses attached by a spring and E-values as force constants, was used to generate the final groupings.
RESULTS
Genetic Knock-out of hemQ Is a Small Colony Variant
S. aureus ΔhemQ colonies grown on TSA are significantly smaller in size (<1 mm) in comparison with WT colonies (2–3 mm) (Fig. 2). Complementation of the ΔhemQ strain with a plasmid-borne copy of the hemQ gene reversed the small colony phenotype on plates and the slow growing phenotype in liquid cultures (data not shown; liquid media cultures described below). An SCV phenotype is observed for respiration-defective S. aureus strains, including the well studied heme and menaquinone biosynthesis mutants, hemB and menB (32). The hemB gene encodes the tetrapyrrole biosynthesis enzyme porphobilinogen synthase, which catalyzes an early, required step in the common pathways leading to all tetrapyrroles, including the various hemes and chlorophyll (Scheme 1) (33). SCVs typically have defects in their respiratory electron transport pathways, often due to menadione or heme auxotrophy (34). Hence, the role of O2 in mediating the slow growing ΔhemQ phenotype and the effect of heme supplementation in modulating the observed knock-out phenotypes were probed as described below. The hemB strain has previously been characterized as an SCV and heme auxotroph. The hemB gene has a clear and singular role in tetrapyrrole biosynthesis. The absence of its gene product should result in the absence of all endogenously produced heme. This strain was therefore used for comparison with the ΔhemQ knock-out in addition to the WT strain (32).
FIGURE 2.
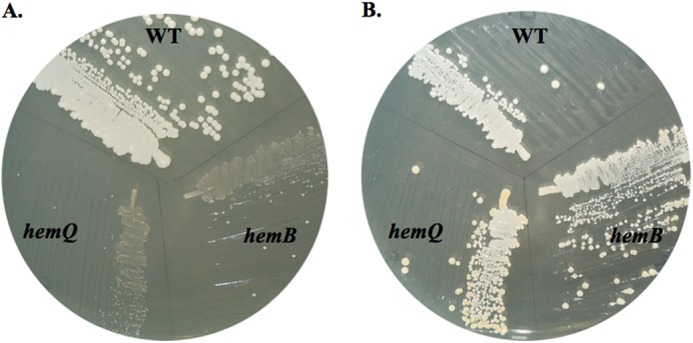
ΔhemQ mutant is a small colony variant that is rescued by addition of heme. WT S. aureus Newman, the hemB, mutant, and the ΔhemQ mutant were grown on TSA overnight at 37 °C in the absence (A) or the presence (B) of 5 μm hemin.
SCHEME 1.

Heme biosynthesis pathway in bacteria.
Exogenous Heme Restores ΔhemQ Culture Growth under Aerobic Conditions
Liquid cultures of both hemB and ΔhemQ grow slowly relative to WT (TSB, 37 °C, and 180 rpm). Five-ml cultures of both mutant strains required ∼48 h to reach full saturation while WT cultures saturated overnight. When inoculated from a freshly saturated culture, a 50-ml culture of WT reached saturation (A600 nm = 3.0) after 6 h. The hemB and ΔhemQ cultures, by contrast, grew slowly, experiencing an initial lag and saturating at a lower A600 nm = 1.0 after 12 h (Fig. 3A). Addition of heme (5 μm) to both the hemB and ΔhemQ cultures completely restored WT-like growth kinetics (Fig. 3B). Addition of the same concentration of a weakly ligated iron complex (iron (III) citrate, FeC3H5O(COO)3) provided some stimulation of the growth of both strains but did not completely restore growth. Addition of 5 μm of the heme precursor protoporphyrin IX did not restore growth to either strain. (Data not shown (24).) These results suggest that HemQ plays a role in the late stages of heme biosynthesis.
FIGURE 3.
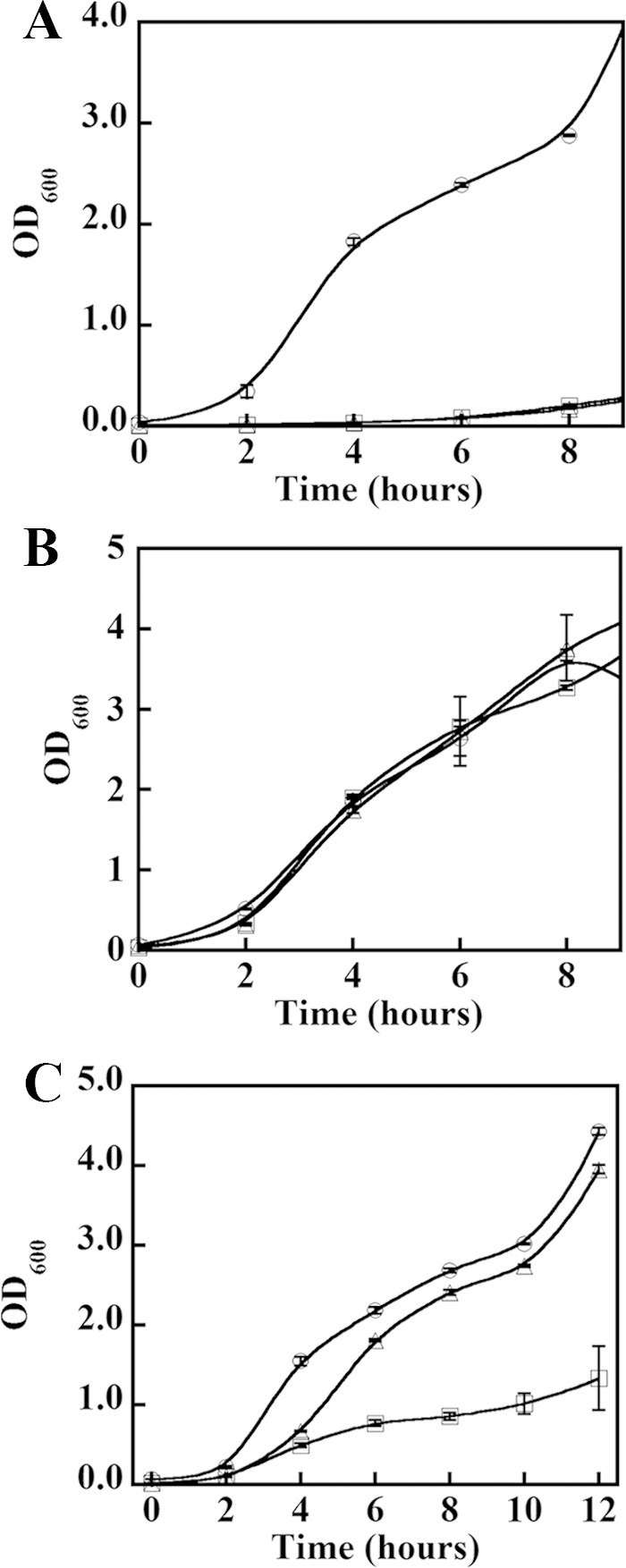
Heme rescues slow growth of ΔhemQ under aerobic conditions, but iron does not. WT, hemB, and ΔhemQ strains of S. aureus Newman were grown in the absence of supplements (A), the presence of exogenous heme (B), and the presence of an equivalent concentration of exogenous Fe (C). Wild type (○), hemB (▵), and ΔhemQ (□) cultures were grown in TSB and monitored for 12 h in 50 ml of TSB in 150-ml Erlenmeyer flasks. Each point is an average of duplicate or triplicate measurements. Cultures in B were supplemented with 5 μm hemin (from 1 mm stock in 0.1 m NaOH). Cultures in C were supplemented with 5 μm Fe(III) citrate and 10 μm sodium citrate.
Growth Deficit Relative to WT Is Not Observed for hemB and ΔhemQ Cultures under Anaerobic Fermentative Conditions
The SCV phenotype has been attributed to defects in respiratory electron transport chains (35). Additionally, the hemQ gene showed functional links to the anaerobic metabolism or the switch between aerobic/anaerobic lifestyles in B. subtilis (16, 17). We therefore examined the hemB and ΔhemQ growth phenotypes under anaerobic fermentative conditions with glucose as the carbon source and no added respiratory electron acceptors. Aerobic culture tubes were seeded with freshly saturated overnight cultures and the culture tubes were sealed, and growth was subsequently monitored. WT cultures of S. aureus grew more slowly than under aerobic conditions, reaching saturation at about 8 h and attaining somewhat lower final culture densities (A600 nm = 1.0–1.5). Under the same conditions, the hemB and ΔhemQ cultures began to grow only after a lag, subsequently doubling at a rate similar to WT and reaching comparable final culture densities (data not shown) (24). The same cultures were allowed to grow to saturation and then were used to inoculate rigorously anaerobic, N2-purged culture flasks. When the growth of these cultures was monitored optically (or via colony counts on solid media), the WT, hemB, and ΔhemQ cultures appeared to perform similarly to one another (24). Hence, the observed growth lag in the initial inoculates was likely due to the transition from aerobic to anaerobic growth.
Cellular Catalase Activity Is Significantly Diminished and Peroxide Sensitivity Increased for Both ΔhemQ and hemB Mutants
A global defect in heme metabolism for both mutants is further indicated by parallel deficiencies in catalase activity and peroxide resistance. Catalase is a major recipient of cellular heme in B. subtilis and presumably in related Gram-positive bacteria (36). Total cellular catalase activity decreases 71- and 77-fold in ΔhemQ and hemB cultures relative to WT, respectively, in the presence of 10 mm added H2O2. By the same token, WT cultures grown with 50 μm added H2O2 exhibit no discernible growth defect. However, growth in 50 μm H2O2 is almost completely abrogated for either mutant strain. See Ref. 24 for more details.
Porphyrin Metabolite Profiles Indicate Coproporphyrin Accumulation by ΔhemQ Cultures
WT, ΔhemQ, and hemB cultures were quantitatively analyzed for heme and its precursors via exact mass LC-MS. The results (ion chromatograms not shown but available in Ref. 24) indicate substantially different profiles for each strain grown under aerobic and anaerobic conditions (Fig. 4). Aerobically, the most abundant porphyrin species in WT cells was iron protoporphyrin IX (heme b), with ∼1.9 ± 0.4 nmol/g of cell pellet. The hemB cells contained significantly less heme b (0.73 ± 0.1 nmol/g cell pellet), although they were not entirely heme-free despite the fact that the gene for an early and presumably nonredundant step in the heme biosynthetic pathway had been inactivated. The most likely sources of heme in this strain are the heme and heme precursors from the rich TSB growth medium; heme is an essential cofactor that S. aureus avidly scavenges. The ΔhemQ cells contained less heme b on average than the WT (1.2 ± 0.4 nmol/g) but a statistically similar amount to the hemB cells. This suggests that this strain also avidly scavenges heme from the growth medium and that the ability to acquire heme limits growth.
FIGURE 4.

ΔhemQ cultures accumulate coproporphyrin under aerobic conditions. The bar graphs show measured amounts of heme (A) and coproporphyrin (B) in nanomoles of metabolite/g of wet cell pellet for wild type (white), ΔhemQ (dark gray), and hemB (black) strains. Error bars are the result of standard deviations taken from analysis of three separate cell cultures. Analyzed cell cultures (50 ml) were grown on a shaker incubator in TSB at 37 °C, 180 rpm, for ∼16 h before harvesting. Aerobic samples were grown in 150-ml Erlenmeyer flasks and anaerobic samples in filled, air-equilibrated 50-ml Falcon tubes that were subsequently sealed. Harvested cells were lysed and porphyrins extracted and quantitatively analyzed by exact mass LCMS as described in the text.
Cells grown under anaerobic/fermentative conditions had significantly less heme b. Only WT S. aureus had detectable levels (0.27 ± 0.07 nmol/g).
Among the heme precursors, only the spontaneous oxidation product of coproporphyrinogen (coproporphyrin) was observed to accumulate under any condition tested. Under aerobic conditions, WT S. aureus produced a small amount of coproporphyrin (0.45 ± 0.06 nmol/g), and hemB had none above the limits of detection. Pellets of ΔhemQ cells, by contrast, accumulated large amounts of coproporphyrin as follows: 2.5 ± 0.5 nmol/g, a greater than 5-fold increase relative to WT. Significantly smaller quantities of coproporphyrin were observed in aerobically inoculated cultures that were subsequently sealed, allowed to become anaerobic, and grown to saturation. ΔhemQ and WT strains produced 0.5 ± 0.08 and 0.09 ± 0.01 nmol/g of coproporphyrin, respectively. Control experiments with cultures grown in light and dark conditions suggested that the observed growth deficits for ΔhemQ were not due to coproporphyrin-mediated phototoxicity. (Data not shown here but are reported in Ref. 24.)
Recombinant SaHemQ Purifies with Little Heme Bound
When prepared under conditions that would give a fully heme-incorporated DaCld (7), the S. aureus homolog purified at high yields (15 mg/liter culture) but with minimal heme bound (∼0.1–0.2 heme molecules per protein monomer), consistent with prior characterizations of the HemQ proteins from M. tuberculosis and B. subtilis (15). The as-isolated protein was incubated overnight in the presence of 1–2 eq of hemin (from 1 mm stock in 0.1 m NaOH) at 4 °C to increase its heme content while avoiding precipitation and nonspecific association of heme with the protein. Following gel filtration, samples contained ∼0.5–0.6 heme per monomer. Heme-reconstituted and as-isolated holo-HemQ had overlapping UV-visible spectra and behaved identically in the measurements described below. Protein concentrations herein refer to the concentration of heme-containing monomer unless otherwise specified.
UV-visible Titrations
The affinity of apo-HemQ for heme was determined by UV-visible spectroscopic titration. To distinguish between free heme and the bound species, parallel titrations were carried out for a no-protein control and a sample containing 2 μm of freshly isolated apo-HemQ. The difference of the two spectra at the holo-HemQ Soret maximum was plotted as a function of the concentration of heme added. (Data not shown here but are reported in Ref. 24.) The data fit well to an equilibrium binding isotherm, yielding KD = 0.72 ± 0.2 μm.
Fluorescence Quenching
Apo-HemQ fluoresces from ∼300 to 350 nm when excited at 280 nm. The fluorescence emission was quenched with the addition of heme or metal-free porphyrin and was thus used as a signal in determining binding affinities for both heme and protoporphyrin IX (Fig. 5). Fluorescence quenching yielded a heme/protein KD of 1.68 ± 0.16 μm, which is similar to that found using UV-visible. The affinity of apo-HemQ for protoporphyrin IX is only slightly lower at KD = 2.2 ± 0.1 μm.
FIGURE 5.
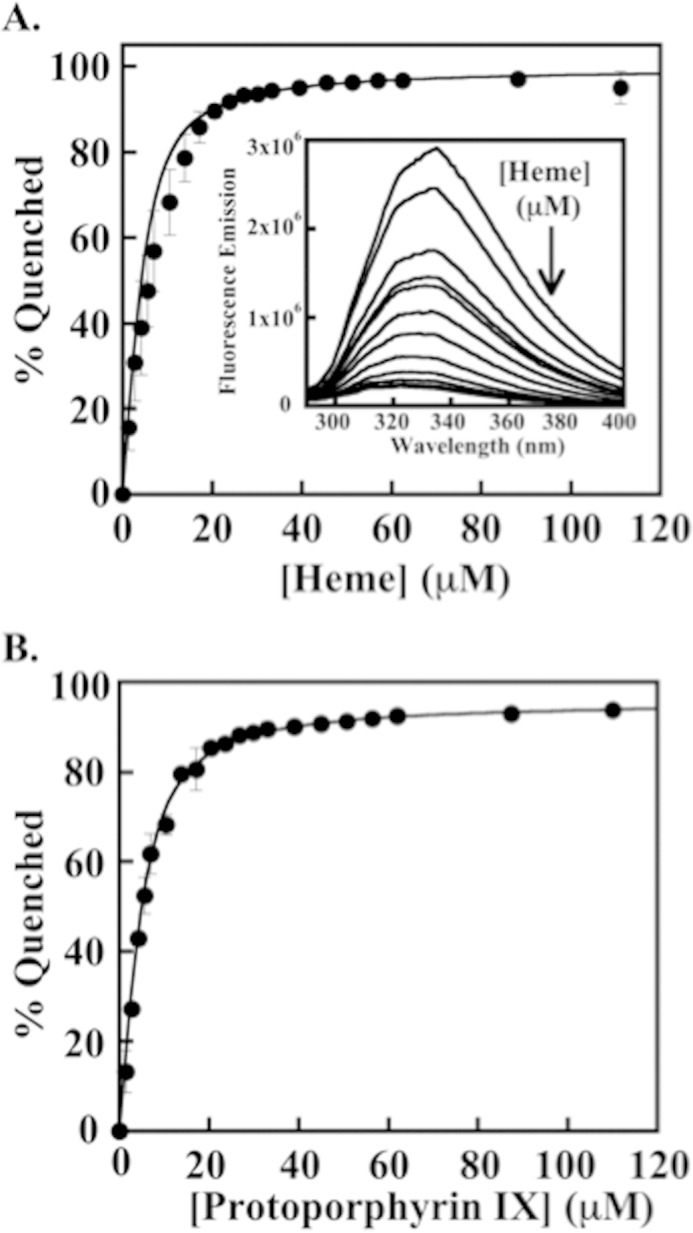
Equilibrium binding of apo-HemQ and heme/protoporphyrin IX measured by fluorescence quenching. A, binding isotherm for the quenching of apo-HemQ (∼5 μm) in 0.1 m potassium phosphate buffer, pH 6.8, by heme. The percent of quenching observed is plotted as a function of heme added and fit to a quadratic binding equation taking into account the concentration of protein (see “Experimental Procedures”). A Kd = 1.68 ± 0.16 μm was determined for heme. The inset shows quenching of apo-HemQ fluorescence over λ = 300–400 nm by addition of heme (excitation λ = 284 nm; 5 μm in 0.1 m potassium phosphate, pH 6.8, 25 °C). Heme was added from 1.4 to 111 μm, and spectra were collected after several minutes of incubation to allow equilibrium to be reached. B, binding isotherm for PPIX. The binding of PPIX was monitored in the same fashion as heme using a stock solution of PPIX (1 mm) dissolved in DMSO and diluted into 0.1 m potassium phosphate, pH 6.8. A Kd = 2.21 ± 0.1 μm was determined from the fit to the data.
Heme affinities for various proteins are listed in Table 1. The HemQ homolog from M. tuberculosis bound heme with a much weaker estimated KD of 30–40 μm (15). Titrations of heme into this protein did not produce a clear binding isotherm, leading to the estimate. Notably, the protein was expressed with a His tag attached, which in some instances has been shown to influence the reconstitution of weakly heme binding bacterial chaperone proteins with their cofactors (37, 38). It is therefore possible that some of the protein-based experiments from the prior study (15) may have been carried out with protein with heme not properly bound. Even without a His tag, handling of the SaHemQ likewise afforded experimental challenges due to its low affinity for heme (see below).
TABLE 1.
Heme and porphyrin affinities for S. aureus HemQ and other heme-binding proteins
| Protein, organism | Ligand (method used) | Kd | pH | Ref. |
|---|---|---|---|---|
| nm | ||||
| HemQ S. aureus | Heme (UV-visible) | 720 (0.21) | 6.8 | This work |
| Heme (fluorescence) | 1680 (160) | 6.8 | ||
| Protoporphyrin IX (fluorescence) | 2210 (100) | 6.8 | ||
| HemQ M. tuberculosis | Heme (UV-visible) | 3–4 × 104 | 8.0 | 15 |
| YfeX E. coli | Heme | 3.9 (1.6) | 8.0 | 41 |
| Protoporphyrin IX | 4.8 (2.8) | 8.0 | ||
| EfeB E. coli | Heme to apo-EfeB (ITC)a | 29.6 (4.3) | 8.0 | 41 |
| Heme to PPIX-EfeB (ITC) | 214 (50) | 8.0 | ||
| BtDyp Bacteroides theatiotaomicron | Heme (ITC) | 158 | 7.6 | 43 |
| TyrA Shewanella oneidensis | Heme (ITC) | 50 | 7.6 | |
| IsdC S. aureus | Heme (UV-visible) | 340 (120) | 6.0 | 44 |
| IsdG S. aureus | Heme (UV-visible) | 5,000 (1,500) | 7.5 | 14 |
| PhuS P. aeruginosa | Heme (Fluorescence) | 180 (10) | 7.8 | 29 |
| Myoglobin sperm whale | Heme (UV, CD, fluorescence) | 0.88 × 10−4 | 7.0 | 73 |
a ITC is isothermal titration calorimetry.
SaHemQ is a member of the Cld, DyP, and EfeB family proteins, which share a common structural superfamily (1). Members, including DaCld, YfeX, EfeB, and a well studied representative DyP, bind heme and/or PPIX with relatively high affinity, with KD values in the nanomolar range (39–41). Such values are expected for an enzyme/cofactor pair. Despite its strong sequence homology with DaCld and its likely close structural similarity, SaHemQ does not bind heme with strong affinity.
Proteins involved in heme acquisition, trafficking, metabolism, or homeostasis, including IsdC and IsdG, tend to bind heme in the low micromolar range (14, 42). The former protein's function is to acquire heme extracellularly and transport it into the cell; the latter functions as a heme oxygenase. The heme transport/chaperone protein PhuS (Pseudomonas aeruginosa) binds heme with a KD of 0.18 μm (29). The relatively low affinity is thought to be important for its role in delivering heme to heme oxygenase for degradation. With respect to its heme affinity, SaHemQ is most akin to the heme-binding proteins that do not use the heme as a catalytic cofactor. Notably, IsdG/I proteins are also part of the Cld, DyP, and EfeB structural superfamily and have close homologs to SaHemQ (see below).
The similar affinity of SaHemQ for iron-free protoporphyrin IX (PPIX) suggests that the protein is not selective for heme over porphyrin despite the presence of an iron-ligating histidine residue in the protein pocket, and it could in principle bind a heme precursor in vivo. Moreover, it suggests that substitution of the iron-ligating histidine for a nonligating residue is not certain to abrogate heme binding by SaHemQ.
UV-visible Spectroscopy Indicates a High Spin 5-Coordinate Heme
The Soret peak for holo-HemQ in 0.1 m potassium phosphate buffer at pH 6.8 has a maximum at 406 nm (ϵ406 = 37 mm−1cm−1), with broad visible bands near 510 (Q-band) and 630 nm (charge transfer). A shoulder in the Soret near 350 nm was present in some purifications of the protein and is consistent with the presence of a small amount of free heme. Dialysis of such preparations against 1 mm EDTA was sufficient to remove the 350 nm band. Protein samples with all heme stably even if sub-stoichiometrically bound were used for the experiments shown in Fig. 6 (described below) to be certain that the effects observed were due to stably bound heme.
FIGURE 6.
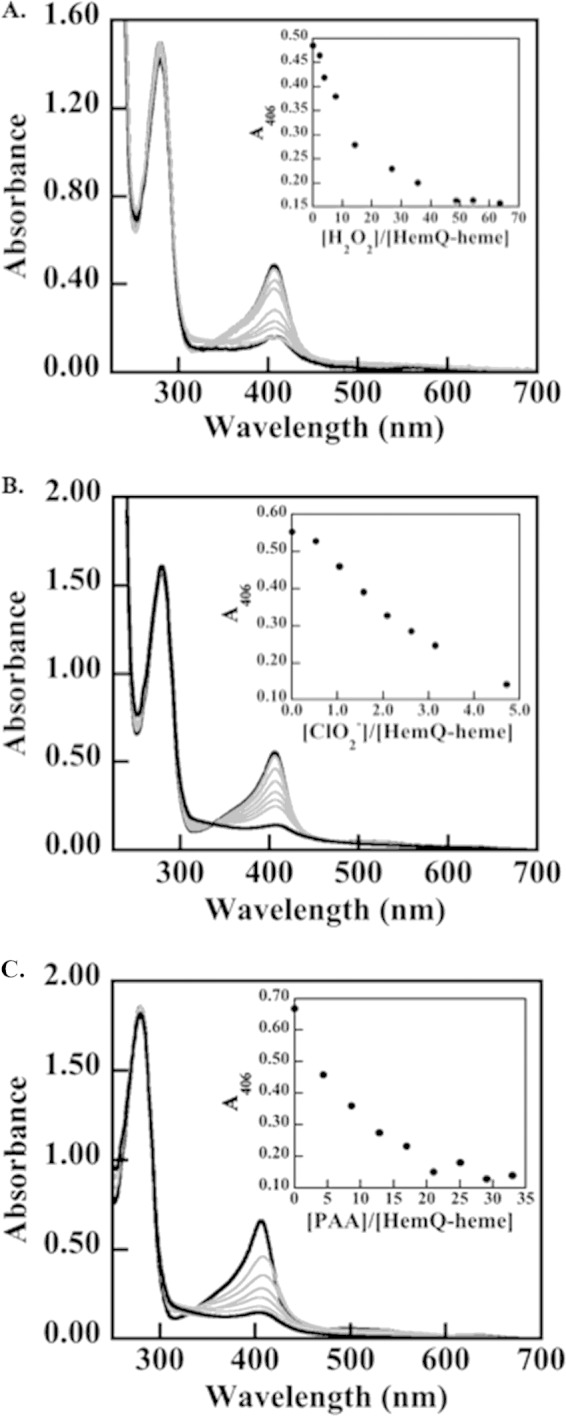
Heme in HemQ degrades in the presence of relatively small numbers of equivalents of H2O2, chlorite, or PAA. The most dramatic effects are observed for the low pKa/anionic oxidants PAA and chlorite. A 30 μm solution of HemQ (heme-containing monomer) in 20 mm potassium phosphate buffer (pH 6.8, 25 °C) was titrated with aliquots of oxidant. UV-visible spectra were measured until the solution spectra stopped changing, and the final spectra recorded after equilibrium was reached. Spectra were adjusted for volume changes. A, titration with H2O2. B, titration with chlorite. C, titration with PAA. The protein absorbance at 280 nm is shown along with the heme Soret band at 406 nm. Notably, addition of oxidant leads to heme destruction without evidence for aggregation or precipitation of protein. Free heme has an absorbance maximum near 380 nm. Spectra at the beginning and end of the titrations are shown as dark black lines, and spectra measured following the addition of oxidant are shown in gray.
Spectral peaks are summarized in Table 1 with comparisons to relevant related heme proteins. The band energies observed for SaHemQ are similar to those for five-coordinate high spin species and particularly for structurally related DaCld and dye decoloring peroxidases (DyP proteins) in their acidic forms (43). The ferrous species was generated via titrimetric addition of dithionite, shifting the Soret to 433 nm and yielding a visible peak at 558 nm.
pH Titration of Holo-SaHemQ Indicates a Wide Range of pH Stability and No Alkaline Transition
DaCld and most peroxidases undergo spectral transitions in the UV-visible range with pH (6, 44). At the high and low pH extremes, loss of the heme chromophore is associated with dissociation of the heme from the protein as it denatures. Within the range of pH over which the protein is stable, an alkaline transition may be observed, corresponding to the conversion of a 5-coordinate, typically high spin heme to a 6-coordinate, low spin ferric-hydroxy heme. The position of the pKa for this transition is related to the electronic structure of the distal pocket enclosing the heme's open coordination position. For heme peroxidases such as HRP (distal His/Arg), the transition occurs with a pKa above 11 (45). For DaCld (distal Arg-183), it shifts to pH 8.7 (6). In WT IsdG from S. aureus, this transition is particularly low, occurring at pH 7.1 (46). The ruffled heme cofactor of IsdG and the hydrophobicity of the distal pocket are expected to contribute to the unusual stability of the ferric-hydroxy species in this protein (47).
HemQ at pH 6.8 was diluted into buffers over a range of pH values, and changes in its UV-visible spectrum were examined. (Data are available in Ref. 24.) At pH values lower than 5.5, the Soret band rapidly diminishes, indicative of heme dissociation from the protein. By contrast, DaCld denatures with a midpoint pH of 4.8, and heme peroxidases can be stable at as low as pH 3–4 (6, 43). At higher pH values, no reversible transitions are observed. Rather, the Soret and protein peaks slightly diminish, suggestive of the precipitation of a small amount of protein (≤10% of the total) as the pH is raised to 10. This alkaline behavior is similar to that identified in DaCld R183Q, in which no alkaline transition occurs and loss of heme binding begins around pH 9 (3). This mutant has a distal pocket composition very similar to that predicted by sequence analysis for HemQ. It remains in a high spin mixed 5-/6-coordinate (presumably water bound) form at all values of pH for which the protein is stable.
Circular Dichroism Indicates Intact Secondary Structures for Holo- and Apo-HemQ
CD spectra were measured for both the freshly isolated apo- and reconstituted holo-forms of HemQ. (Data are available in Ref. 24.) The apoprotein has a peak maximum at 227 nm that shifts to 234 nm with the addition of heme. The percentages of secondary structure for the apo- and holoproteins were determined by fitting the data (K2D3 software). The results were summarized and compared with the known secondary structure composition of WT DaCld. The apoprotein appears to contain ∼17% α-helix, 30% β-sheet, and 40–50% turns/strands. The holoprotein contains slightly less α-helix (5–11%), more β-sheet (37–38%), and roughly the same amount of turns and strands (>50%). The similarity in spectra suggests that major rearrangements of secondary structure do not occur upon heme binding. Instead, the small shift in spectral peak energies and the changes in α-helix/β-sheet percentages imply subtle secondary structural changes upon the binding of heme as observed with cofactor binding in other proteins (48).
Apparent Oligomerization States for Holo- and Apo-HemQ Differ
The predicted molecular weight of HemQ based on amino acid sequence is 29.4 kDa, and the molecular weight of the purified protein according to MALDI-TOF MS analysis is 29.30 kDa. To determine the native molecular weight and the oligomeric state of the enzyme, analytical gel filtration was carried out for the apo- and reconstituted holoproteins. Retention times were derived from chromatograms of individual column runs of the protein standards and both the apo- and holoproteins. A plot of the linear relationship between the molecular weight and the elution time of each globular protein standard was generated (data not shown) and used to approximate the molecular weights of the unknowns. Apo-HemQ elutes near the column's void volume and gives an estimated molecular mass of 1200 kDa, suggesting a very high order multimer, a nonglobular protein structure, or a highly disordered but soluble protein. By contrast, holo-HemQ has an apparent molecular mass of ∼187 kDa, consistent with a homohexamer. Analytical gel filtration carried out in a similar fashion for DaCld suggested a homotetramer in solution; however, crystallography showed the functional protein to be a pentamer (4, 7). Other Cld-family proteins have exhibited a similar level of discrepancy between gel filtration estimates and crystallographic measurements of oligomerization states (11, 49, 50). Aside from a subset of Clds having short monomer sequences (< 200 amino acids) (10) and the Cld from Azospirae oryzae, which appears to be a crystallographic hexamer but a pentamer via mass spectrometry, all Clds examined to date are pentamers in the solid state (11, 51).
HemQ Exhibits Low Enzymatic Activities toward Chlorite and Peroxide
Holo-HemQ was examined for chlorite dismutase, peroxidase, and catalase reactivity under steady state conditions. No chlorite decomposition was detected up to 20 mm chlorite at 25 °C. By contrast, the R183Q mutant of DaCld exhibits reduced but still clearly observable chlorite decomposition activity (3). This mutant is expected to have a similar active site geometry to the SaHemQ.
At pH 6.8, the enzyme exhibits relatively weak peroxidase activity with the reductant ABTS as follows: Km(H2O2) = 3.4 ± 0.4 mm; kcat = 1.6 ± 0.1 s−1. Using a saturating amount of hydrogen peroxide (15 mm) and varying the concentration of ABTS, a Km(ABTS) of 30 ± 8 μm and a kcat of 1.8 ± 0.2 s−1 were measured. At higher concentrations of ABTS (>0.2 mm), steady diminution of the measured initial rate with increasing [ABTS] was observed, suggesting substrate inhibition. The measured kcat/Km(H2O2) of 4.8 × 102 m−1s−1 is roughly 4 orders of magnitude lower than in a canonical peroxidase (4.2 × 106 m−1s−1 for HRP-C with H2O2/ABTS at pH 7.0) (52).
Using PAA as the oxidant, by contrast, a much lower Km of 247 ± 106 μm (PAA) and higher kcat of 52 ± 9 s−1 were observed, with considerable loss of activity at higher [PAA]. Loss of activity is likely due to heme oxidation. The resulting specificity constant for the acidic oxidant PAA is several orders of magnitude higher than for H2O2, kcat/Km(PAA) = 2.1 × 105 m−1 s−1. The enhanced reactivity with PAA relative to H2O2, also observed for DaCld (53), may be a result of the lower pKa oxidant not requiring base-catalyzed removal of a proton for complex formation with the ferric heme.
HemQ was also studied as a catalase via the production of O2 from H2O2 under near anaerobic conditions. Initial rates of O2 production follow Michaelis-Menten-like behavior as a function of H2O2 concentration, yielding Km(H2O2) = 2.6 ± 0.3 mm, kcat = 5.2 ± 0.2 s−1, and kcat/Km(H2O2) = 3.4 × 103 (pH 6.8, 25 °C). The high Km value for the catalase reaction is similar to that measured for the peroxidase reaction and again is consistent with the lack of an active site base. The measured kcat/Km(H2O2) is about 3 orders of magnitude lower than values for typical catalases, which tend to be high, kcat/Km ≥106 m−1 s−1 (54). HemQ from M. tuberculosis and B. subtilis exhibited catalase activity, pH 8, with slightly higher turnover numbers than the S. aureus protein at pH 6.8 (15). Faster reactivity may be due to the use of higher pH conditions. Low peroxidase and catalase activities of the S. aureus HemQ, in conjunction with its weak association with heme, suggest that a catalytic role with H2O2 is unlikely in vivo.
Transient Reactions with Oxidants Yield No Observable Intermediates
Spectral changes were monitored following rapid mixing of SaHemQ with PAA (10 eq) and H2O2 (>100 eq), respectively, at pH 6.8. No characteristic spectra for iron-oxygen intermediates (ferryl, ferric-hydroperoxy, etc.) were detected, due to low or no formation of such species or their rapid decay. Concentrations of oxidant ranging from 1 to 100 eq were assayed (data not shown) to detect potential reaction intermediates. No intermediates were observed under any conditions. Instead, the reaction of holo-HemQ with <5 eq of PAA results in a slow decrease in the intensity of the entire UV-visible spectrum, consistent with heme degradation. Following reaction with even very low eq of H2O2 (<5) the Soret band shifts from 403 to 407 nm prior to bleaching. Production of verdoheme, a product of H2O2-mediated heme lysis in catalases, peroxidases, and heme oxygenase (52, 55, 56) is not clearly observed.
PAA has a pKa near 8 and therefore does not require base catalysis for rapid reaction with ferric heme. The lack of an intermediate following reaction with PAA contrasts with prior measurements with the DaCld and its R183Q mutant, which is expected to resemble SaHemQ in the identity of the critical distal residue (53). Following mixing with <10 eq of PAA, this mutant forms an apparent ferryl (Fe(IV)=O) exchange coupled to a porphyrin radical cation (compound I). Hence, the lack of SaHemQ reactivity with PAA cannot be attributed to the lack of an active site base or to the unavailability of a positively charged Arg residue in the active site.
Susceptibility of the Heme to Oxidation
Because transient kinetics showed partial heme destruction even after addition of less than 5 eq of oxidant, the chromophore was titrated with H2O2 and chlorite to quantify its stability in the presence of these oxidants. Measuring either residual activity after incubation with increasing oxidizing equivalents or optically titrating DaCld gave similar estimates of total turnovers prior to complete loss of heme cofactor as follows: ∼17,000 turnovers with chlorite and 500 turnovers with H2O2 (7). HRP is inactivated after ∼600 turnovers in the presence of excess H2O2 and without reductant (57). By contrast, the S. aureus HemQ porphyrin spectrum is 90% abolished after addition of only ∼49 eq of H2O2 (Fig. 6). This indicates significant redox susceptibility of the bound heme. The same level of heme destruction is observed even more quickly in the presence of peracetic acid (21 eq) or chlorite (5 eq). The small number of turnovers sustained by the heme suggests that a primary role in detoxifying these oxidants is unlikely. At the same time, the reaction of the protein with each of these oxidants results in apparent heme scission (disappearance of the Soret absorbance due to the heme) without significant protein denaturation or aggregation (no disruption of the protein's absorbance band at 280 nm).
Because of HemQ's structural and sequence similarities to the heme oxygenase protein IsdG (see below), we assessed the stability of the HemQ-bound heme in the presence of excess O2, reducing agent, catalase, and superoxide dismutase. The antioxidant enzymes were added to distinguish between direct Fe(II)/O2-mediated heme decomposition and the reaction between the heme and Fe(II)-generated diffusible H2O2 (29). Prior studies have used excess ascorbate (up to 1000 eq) as the reductant (29, 58). Under these conditions, a small shift in the HemQ heme spectrum, possibly due to binding of the ascorbate, was observed. If a stronger thiolate-based reductant such as dithionite is used (59), no diminution of the heme Soret and visible absorbance bands occurs.
Network Analysis Shows Closest Sequence Relationships with Structurally Similar Monooxygenases
A network analysis was used to assess the diversity of proteins with sequence and/or structural relationships to the SaHemQ. Network analyses use algorithms similar to BLAST, comparing and connecting not just sequence pairs but concurrently defining relationships among whole groups. As output, thousands of individual protein sequences can be displayed as nodes/dots. A line is drawn between two sequences if the statistical significance of the similarity score between them is better than a defined E-value cutoff. Sequences with common biochemical functions form clusters in these analyses. As the level of stringency (E-value) is raised, sequences that were connected or grouped become subdivided into smaller clusters, revealing finer shades of functional differences. Hence, the use of large numbers of sequences in a network analysis allows for a larger scale visualization of sequence relationships than could otherwise be achieved with conventional sequence alignments.
A number of conclusions were drawn. First, proteins from the same UniProt clan as the S. aureus HemQ are biochemically diverse, coming from 11 different functional classes. Database-derived functional annotations are frequently erroneous; however, the probable functions of clusters of sequences highlighted here (Clds, DyPs, IsdGs, and antibiotic biosynthesis-associated monooxygenases) could be validated in each case using biochemically characterized sequences as landmarks. Second, even at a relatively low level of stringency, the DyP proteins (Fig. 7) form a distinct cluster that is unconnected from the remaining sequences, reflecting their high degree of sequence divergence from the Clds/HemQs despite their strong structural similarity. Third, overlaying available structures from at least 10 functionally divergent clusters from Fig. 7 clearly demonstrates the retention of the protein domain fold across this broad selection of sequences. (See Fig. 8 for examples.) Fourth, of the available structures, only the DyPs and chlorite-degrading Clds contain stably bound heme, although all contain a structurally similar cavity for binding heme and/or substrate. Finally, the antibiotic biosynthesis monooxygenases (ABMOs) and IsdG/IsdI proteins are the most closely related on a sequence level to the Clds/HemQs, with a few sequences from these classes clustering with Clds even at very high stringency (E-value cutoff = 10−7; data not shown).
FIGURE 7.

Network analysis of Cld/HemQ sequences and their homologs shows the highest levels of similarities to IsdG/IsdI and cofactor-independent monooxygenases. Important functional clusters of proteins are as labeled. Individual protein sequences are represented by dots colored according to their Uniprot-designated function as follows: chlorite dismutases (pink); antibiotic biosynthesis monooxygenases (dark blue); IsdG/IsdI proteins (cyan); DyP and EfeB proteins (red); uncharacterized proteins (gray). Left, network was generated at low stringency, with an E-value cutoff at 1e−5. DyP and EfeB proteins, the closest structural relatives of the Clds/HemQ proteins, remain unconnected to them on a sequence level even at the lowest stringency sampled. Right, network was generated at a higher stringency (E-value cutoff at 1e−3). The most closely related sequences to the Clds/HemQ proteins (pink cluster, lower right) are indicated by the many lines connecting this cluster to the antibiotic biosynthesis monooxygenase group. IsdG/IsdI protein sequences are interspersed with this group.
FIGURE 8.

Cld homologs of S. aureus HemQ share strong sequence and structural similarities with IsdG and antibiotics associated monooxygenase proteins. A, DaCld like the Cld from G. stearothermophilus (GsCld, Protein Data Bank code 1T0T) forms a homopentamer. GsCld crystallizes as the apoprotein. B, structural overlay of GsCld (magenta) and DaCld (blue, Protein Data Bank code 3Q08) monomers shows the location of the DaCld heme (rendered as sticks) and illustrates the overall similarity of the two proteins. C, structural overlay of GsCld (magenta) and the S. aureus IsdG protein homodimer (cyan, Protein Data Bank code 2ZD0 (18)) illustrates the similarity of the two protein structures. IsdG-bound hemes (one per monomer) are rendered as sticks. D, overlay of the heme-binding domain of DaCld (blue) and the IsdG monomer (cyan) shows the location of the heme in each. E, overlay of the DaCld monomer (blue) and the biochemically characterized ABMO protein SnoaB (green, Protein Data Bank code 3KNG (19)). Figure was generated using Chimera.
Apo-HemQ Does Not Catalyze Coproporphyrinogen Oxidation
The ΔhemQ strain of S. aureus accumulates coproporphyrin. Moreover, HemQs/Clds have very close sequence and close structural relationships with antibiotic biosynthesis-associated monooxygenases, a class of proteins that includes coproporphyrinogen oxidase (CPO, encoded by hemF), which catalyzes the cofactor-free oxidation of coproporphyrinogen (60–62). Under reaction conditions that successfully demonstrated CPO activity for the enzyme from E. coli (30), no conversion of the starting material was observed for the SaHemQ. These results are consistent with prior work using heme-containing HemQ proteins from M. tuberculosis and B. subtilis (15).
Equilibrium Ligand Affinities for the Heme Iron Illustrate the Distal Pocket's Hydrophobicity
The equilibrium affinities of ligands for the heme iron provide an indirect measure of the electronic structure of the distal pocket environment. Affinities of cyanide, imidazole, and fluoride for holo-HemQ were measured at pH 6.8 via UV-visible titration. (Data are not shown. See Ref. 24.) The HemQ-CN complex Soret band intensifies and shifts to 420 nm with a shoulder at 370 nm and a visible peak at 550 nm. An equilibrium-binding constant (Kd = 8.9 ± 0.87 μm) was determined by fitting Equation 1 to ΔA (425 nm) versus [KCN]. DaCld residue Arg-183 has been shown to interact with bound CO and to stabilize bound anions (6). Holo-HemQ has a relatively high affinity for CN− compared with the R183Q mutant of the HemQ homolog DaCld (3), although sequence comparisons suggest the two have a similar set of amino acids lining the distal pocket.
The HemQ-imidazole Soret band shifts to 414 nm with α and β bands at 570 and 530 nm and loss of the charge transfer band at 630 nm. The Kd values for imidazole at pH 6.8 are 1.0 ± 0.1 mm. In DaCld WT and Arg-183 mutants, including R183Q, by contrast, the affinity for imidazole is in the micromolar range, again suggesting very different factors controlling imidazole stabilization in SaHemQ. Finally, no spectral changes were observed with addition of fluoride up to 60 mm. The protein has a low affinity for monoanions much like DaCldR183Q or DaCldR183A and distinct from WT DaCld (3).
DISCUSSION
Genome sequencing and allied systems-biology techniques have identified protein families that have yet to find firm chemical or biological functions. Such functional assignments are necessarily complex and often require a confluence of experimental approaches. The Cld or C-family proteins are widespread in unicellular life, but their roles are unclear. Although the first characterized Clds are known to detoxify chlorite via an unusual heme-dependent, O2-generating process, the majority of the many organisms that contain a cld gene have no need for such a reaction, which, moreover, may have recently evolved.
Genetic context points strongly toward a role for cld genes in heme metabolism in Actinobacteria, where the gene is co-operonic with hem genes encoding what appears to be a complete heme biosynthetic pathway (Scheme 1). Dailey et al. (15) experimentally confirmed a functional association with heme biosynthesis using strains of E. coli in which the native protoporphyrinogen oxidase (PPO) and ferrochelatase (Fc) genes (hemG and hemH) were inactivated. The genes, either alone or together, were not complemented by functionally equivalent hemY/hemH gene pairs from two different Gram-positive bacteria from two separate phyla, B. subtilis and M. tuberculosis (15). However, including the hemQ gene along with hemY/hemH allowed for full complementation of the single and double knock-outs. Similar results were obtained in vitro, where all three proteins were necessary for processing the PPO substrate protoporphyrinogen to heme, the final product of Fc, and where HemQ appeared to have a stimulatory effect specifically on PPO activity. These results highlight some of the diversity in heme biosynthesis pathways, particularly at the final steps. In addition to possessing a hemQ gene, Gram-positive bacteria, for example, possess a PPO and Fc that are soluble in the cytoplasm (63). Membrane association of PPO and Fc in eukaryotes is believed to regulate proper coupling of toxic PPIX and Fe(II) and could serve an analogous role in Gram-negative bacteria (64). It is not clear how Gram-positive organisms enforce PPIX/Fe(II) coupling. At the same time, the Gram-positive Fc lacks the iron-sulfur cluster found in eukaryotic and Gram-negative versions of the enzyme.
Although the necessity of HemQ for PPO and Fc function was demonstrated by prior work, its actual role and in particular the role of heme binding to HemQ have not been ascertained. HemQ has catalase activity when heme-bound but, as acknowledged here and previously, this activity is of a magnitude comparable with that found for heme-binding proteins that are not catalases (65). To address these questions, we generated a knock-out of the hemQ gene in an organism with a native hemQ gene, S. aureus, to study the role of hemQ in its biological context. We undertook in parallel a thorough biochemical and sequence-structural analysis of the SaHemQ protein, which is representative of a large sub-branch of the Cld-family tree (1), a focus of ongoing research in our group. Our results confirm prior studies linking members of this family to heme biosynthesis, demonstrate unusual biochemical properties for the protein, and suggest regulatory or combined regulatory-trafficking roles in heme metabolism for the gene product in S. aureus.
The small colony (<1 mm) phenotype of the ΔhemQ strain on solid media (Fig. 2) provides the first indication of a role in heme metabolism. The SCV phenotype has been observed in clinical S. aureus isolates for decades and has more recently been associated with defects in cellular respiration (66, 67). The best characterized SCVs include hemB-impaired strains and other auxotrophs for either heme or menadione (32, 35, 68). The latter molecule is a primary electron carrier in the respiratory electron transport chain. Heme auxotrophy is believed to exert its influence on colony size (and bacterial doubling time) via the loss of respiration-associated cytochromes and terminal reductases (69). We therefore compared the ΔhemQ and hemB growth phenotypes in the liquid phase under aerobic and anaerobic (fermentative) conditions, observing significantly slowed growth and attenuated final culture densities for the mutants specifically in the aerobic case (Fig. 3).
This same defect is indirectly observed via parallel decreases in cellular catalase activity and increases in peroxide susceptibility for both the ΔhemQ and hemB strains. The majority of cellular heme is known to be commandeered by catalase in the related Gram-positive organism, B. subtilis (36). These results and the restoration of growth via heme b supplementation in each case further indicate that the influence of either hemQ or hemB is not related either to biosynthesis or delivery of one particular heme b derivative (e.g. respiration-associated heme a and heme o) and that the deficits caused by each mutation influence heme production globally. Hence, we rule out a role for HemQ in heme trafficking to a particular enzymatic or cellular target in S. aureus and instead consider direct roles in heme biosynthesis or regulation.
The possibility that HemQ serves a direct biosynthetic role as an alternative coproporphyrinogen oxidase (CPO) at first seemed like a strong possibility, suggested by four lines of evidence. First, examination of the porphyrin content of ΔhemQ, hemB, and WT cultures grown under aerobic and fermentative conditions indicated substantial accrual of coproporphyrin (the spontaneous oxidation product of coproporphyrinogen) specifically by ΔhemQ under aerobic conditions (Fig. 4). However, inactivation of the PPO from B. subtilis, for example, results in accumulation of coproporphyrin (70). It was proposed that the ΔhemY cultures initially accumulate protoporphyrinogen, the substrate of PPO, which ultimately inhibits or dysregulates CPO. The ΔhemH (ferrochelatase) mutant from B. subtilis likewise accumulates coproporphyrin (71). A possible explanation offered was that the terminal enzymes of heme biosynthesis (CPO, PPO, and Fc) form a functional complex (see Scheme 1), and loss of any of the three results in an apparent CPO deficiency. Therefore, although coproporphyrin accumulation is consistent with CPO activity for HemQ, a coproporphyrin accumulation phenotype has been observed under other circumstances.
Second, despite being a facultative aerobe, S. aureus like all or most other members of its phylum (Firmicutes) does not have an annotated hemF gene encoding a CPO. Rather, these organisms contain a hemN gene encoding an S-adenosylmethionine and FeS cluster-dependent coproporphyrinogen dehydrogenase, which converts coproporphyrinogen III to protoporphyrinogen III in an O2-independent fashion. Because its cofactors are unstable in air, it is possible that HemN is likewise not stable aerobically in vivo. In a cyanobacterium possessing both hemF and hemN genes, for example, the latter was shown to be preferentially expressed under low O2/microaerophilic conditions (72). It is possible that hemQ could encode an O2-dependent CPO that is not related to hemF. Consistent with this hypothesis, ΔhemQ is only growth-restricted under aerobic conditions.
Finally, network analysis of HemQ sequences identifies these proteins as most closely related to two highly similar groups of monooxygenases as follows: IsdG/IsdI-like proteins, which catalyze a heme oxygenase reaction as part of the pathway used by S. aureus and other Gram-positive bacteria for nutritional uptake of heme iron (14), and ABMOs, cofactor-independent proteins that catalyze reactions on organic substrates capable of facile one-electron oxidations (62). Both the IsdG/IsdI and ABMO proteins share strong structural similarities with the Clds (Fig. 9), including the Cld from a member of the same phylum, Geobacillus stearothermophilus (59% identity with S. aureus HemQ). The CPOs encoded by hemF genes are also cofactor-independent monooxygenases (30), although from a different sequence/structural family from HemQ.
When we examined apo- and holo-SaHemQ for coproporphyrinogen oxidase activity, however, we found none, consistent with prior work (15). Furthermore, supplementation of ΔhemQ cultures with PPIX, the product of PPO and substrate for Fc, did not restore growth in an iron-rich medium. We therefore concluded that a simple biosynthetic role for HemQ as a CPO, at least independent of other secondary roles that it might serve in conjunction with other proteins, was unlikely.
With side-chain modification, trafficking, or direct enzymatic roles in heme biosynthesis ruled less likely, either an indirect role in biosynthesis, a regulatory function, or both remains possible. While weighing the possibility of such roles, we first noted that HemQ has close structural and evolutionary relationships with IsdG/IsdI proteins and ABMOs, proteins that use heme or porphyrins not as cofactors but as substrates for O2-mediated oxidation or oxygenation (Figs. 7 and 8). Consistent with these relationships, heme or equivalently PPIX binding by HemQ is relatively weak compared with what one would expect for a redox cofactor and closer to the range observed for proteins that either transport heme or use heme as a substrate (Table 1). We had likewise observed that the heme-free apoprotein appears to aggregate or unfold over minutes to hours at 4 °C. This observed instability is consistent with a recent comprehensive study of the stability of the holo- and apo-forms of the homologous Cld protein from Nitrospira defluvii (NdCld). This work showed, via a variety of thermochemical and spectroscopic methods, that the apo-form of the pentameric NdCld is substantially less stable than the holo- and that the pentameric oligomerization state assumed by the apoprotein is lost over time (74). We therefore hypothesized that auto-degradation of the heme in SaHemQ could be a signal that would lead to disassembly and inactivation of the “protein, alone or in complex with other enzymes.” This would in turn lead to termination of heme biosynthesis.
To test this hypothesis, we examined the ability of SaHemQ to facilitate oxidation or oxygenation of its own heme. SaHemQ does not catalyze heme oxygenation, consistent with this protein stably maintaining its heme under aerobic conditions even in the presence of reducing equivalents. However, we found the heme to be remarkably unstable in the presence of H2O2, almost completely degrading in <50 eq of the oxidant (Fig. 5). Although peroxidases likewise undergo oxidative degradation of their bound hemes in the absence of reducing substrates, they typically do so following exposure to several hundred equivalents (65); catalases survive many thousands of turnovers with peroxide. Even more striking, the heme completely degrades after reaction with as few as 5 eq of chlorite, indicating that the heme environment is profoundly different in HemQ than in close sequence relatives from the chlorite dismutase family that efficiently convert on the order of 20,000 eq of chlorite to Cl− and O2. Enhanced sensitivity to the anionic oxidants relative to H2O2 is consistent with the behavior of other Clds and likely due to HemQ's lack of an active site base. This level of redox sensitivity and the proximity of PPO as a continuous source of H2O2 suggest that heme degradation is essential to HemQ's role. However, we caution that we cannot exclude the possibility that some of these properties of HemQ could be a consequence of working under in vitro conditions with isolated HemQ in its heme-reconstituted form.
HemQ's conditional stability is reminiscent of the α-proteobacterial regulatory protein Irr (75). Irr represses heme biosynthesis by physically associating with Fc. When iron is insufficient, Irr binds protoporphyrin IX and inhibits ferrochelatase, in addition to serving further roles as a DNA-binding transcriptional regulator (76, 77). When iron is replete, Irr binds Fc-generated heme, which stimulates Irr's autodegradation. Release of the apo-Irr from Fc allows heme biosynthesis to proceed. Hence, Irr regulates heme biosynthesis at the protein level, preventing the production of redox-active and toxic PPIX when iron is not present and enforcing the coupling of PPIX to Fe(II) when it is.
The data here suggest that HemQ could by analogy serve to sense iron availability or redox status, as the protein's heme binding status and consequent stability respond to each of these conditions. For example, when iron is available for ferrochelatase, HemQ is expected to be heme-bound and therefore stable. Hence, heme binding to HemQ allows PPO and Fc to continue to produce PPIX and heme, respectively. When iron is not available, PPO would continue to make PPIX as well as H2O2, where the latter can lyse the heme in HemQ quickly, especially in the absence of reducing equivalents to support peroxidase chemistry. Without heme from Fc to replenish the lost cofactor, HemQ would denature and PPO/Fc activity would cease. This model has Fc periodically resupplying HemQ with heme.
Alternatively, HemQ could serve as a redox sensor, a sensor of heme status, or both. The acute sensitivity of HemQ's heme to the nonbiological anions PAA or chlorite suggests that other more biologically common anionic reduced oxygen species such as hypochlorite, peroxynitrite, or superoxide can also likely rapidly degrade the heme in HemQ. S. aureus has a source of superoxide in its membrane, where it maintains a reservoir of heme molecules that use the respiratory electron carrier menaquinone to cycle O2 to O2− (22). With sufficient or perhaps toxic levels of heme already in the membrane, the cell could communicate the need to shut down heme biosynthesis via the production of superoxide that would in turn deactivate HemQ. Alternatively, HemQ could serve as a general sensor of oxidative and nitrosative stress, for example, the kind induced by the human immune response. Redox stress is well known to lead to alterations in bacterial iron metabolism. HemQ could communicate such stress to the heme biosynthetic machinery. The association in B. subtilis between HemQ and the switch between aerobic/anaerobic lifestyles suggest that the shutdown of heme biosynthesis could be part of a coordinated response to stress (16, 17) that could ultimately lead to the adoption of the clinically intractable SCV phenotype. HemQ offers a means by which the SCV phenotype could be adopted reversibly in response to conditions in which slow, intracellular growth becomes favorable to S. aureus. These hypotheses, as well as the nature of the interaction between HemQ and the Fc and PPO enzymes, will be probed in vivo and in vitro as part of ongoing work.
CONCLUSIONS
The necessity of HemQ for heme metabolism is dramatically illustrated by the small colony phenotype of the ΔhemQ strain, which grows slowly and accumulates coproporphyrin under aerobic conditions. The HemQ protein behaves very differently from its homologs from perchlorate-respiring Proteobacteria such as DaCld. It has no detectable chlorite decomposing activity, limited peroxidase and catalase activities, and relatively low affinity for heme. The sensitivity of the heme toward redox degradation is consistent with the close sequence relationship between HemQ and heme-degrading IsdG/IsdI proteins. These data, together with documented associations of HemQ homologs with the final two catalytic steps of heme biosynthesis (15), suggest a novel role for HemQ in regulating heme production and/or homeostasis. A regulatory function for coupling Fe(II) availability to PPIX synthesis or for the termination of the heme biosynthetic process under conditions of redox stress (ultimately leading to adoption of the SCV phenotype) is both biologically exigent and consistent with the data. Similar roles for hemQ genes in a broad variety of medically important Gram-positive bacteria are possible.
Acknowledgments
We thank Garrett Moraski and Bennett Streit for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM090260 (to J. L. D.) and R01AI069233 and R01AI073843 (to E. P. S.).
E. P. Skaar, unpublished data.
- Cld
- chlorite dismutase
- SCV
- small-colony variant
- PAA
- peracetic acid
- MWCO
- molecular weight cutoff
- ABMO
- antibiotic-associated monooxygenase
- ABTS
- 2,2′-azino-bis[3-ethylbenzthiazoline-6-sulfonic acid
- TSB
- tryptic soy broth
- PPIX
- protoporphyrin IX
- CPO
- coproporphyrinogen oxidase
- PPO
- protoporphyrinogen oxidase
- Fc
- ferrochelatase.
REFERENCES
- 1. Goblirsch B., Kurker R. C., Streit B. R., Wilmot C. M., DuBois J. L. (2011) Chlorite dismutases, DyPs, and EfeB: 3 microbial heme enzyme families comprise the CDE structural superfamily. J. Mol. Biol. 408, 379–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coates J. D., Achenbach L. A. (2004) Microbial perchlorate reduction: Rocket-fuelled metabolism. Nat. Rev. Microbiol. 2, 569–580 [DOI] [PubMed] [Google Scholar]
- 3. Blanc B., Mayfield J. A., McDonald C. A., Lukat-Rodgers G. S., Rodgers K. R., DuBois J. L. (2012) Understanding how the distal environment directs reactivity in chlorite dismutase: Spectroscopy and reactivity of Arg183 mutants. Biochemistry 51, 1895–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goblirsch B. R., Streit B. R., DuBois J. L., Wilmot C. M. (2010) Structural features promoting dioxygen production by Dechloromonas aromatica chlorite dismutase. J. Biol. Inorg. Chem. 15, 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee A. Q., Streit B. R., Zdilla M. J., Abu-Omar M. M., DuBois J. L. (2008) Mechanism of and exquisite selectivity for O–O bond formation by the heme-dependent chlorite dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 15654–15659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Streit B. R., Blanc B., Lukat-Rodgers G. S., Rodgers K. R., DuBois J. L. (2010) How active-site protonation state influences the reactivity and ligation of the heme in chlorite dismutase. J. Am. Chem. Soc. 132, 5711–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Streit B. R., DuBois J. L. (2008) Chemical and steady state kinetic analyses of a heterologously expressed heme-dependent chlorite dismutase. Biochemistry 47, 5271–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kostan J., Sjöblom B., Maixner F., Mlynek G., Furtmüller P. G., Obinger C., Wagner M., Daims H., Djinović-Carugo K. (2010) Structural and functional characterisation of the chlorite dismutase from the nitrite-oxidizing bacterium “Candidatus Nitrospira defluvii”: Identification of a catalytically important amino acid residue. J. Struct. Biol. 172, 331–342 [DOI] [PubMed] [Google Scholar]
- 9. Maixner F., Wagner M., Lücker S., Pelletier E., Schmitz-Esser S., Hace K., Spieck E., Konrat R., Le Paslier D., Daims H. (2008) Environmental genomics reveals a functional chlorite dismutase in the nitrite-oxidizing bacterium “Candidatus Nitrospira defluvii”. Environ. Microbiol. 10, 3043–3056 [DOI] [PubMed] [Google Scholar]
- 10. Mlynek G., Sjöblom B., Kostan J., Füreder S., Maixner F., Gysel K., Furtmüller P. G., Obinger C., Wagner M., Daims H., Djinović-Carugo K. (2011) Unexpected diversity of chlorite dismutases: a catalytically efficient dimeric enzyme from Nitrobacter winogradsky. J. Bacteriol. 193, 2408–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Geus D. C., Thomassen E. A., Hagedoorn P.-L., Pannu N. S., van Duijn E., Abrahams J. P. (2009) Crystal structure of chlorite dismutase, a detoxifying enzyme producing molecular oxygen. J. Mol. Biol. 387, 192–206 [DOI] [PubMed] [Google Scholar]
- 12. Hagedoorn P. L., de Geus D. C., Hagen W. R. (2002) Spectroscopic characterization and ligand-binding properties of chlorite dismutase from the chlorate respiring bacterial strain GR-1. Eur. J. Biochem. 269, 4905–4911 [DOI] [PubMed] [Google Scholar]
- 13. Bab-Dinitz E., Shmuely H., Maupin-Furlow J., Eichler J., Shaanan B. (2006) Haloferax volcanii PitA: an example of functional interaction between the Pfam chlorite dismutase and antibiotic biosynthesis monooxygenase families? Bioinformatics 22, 671–675 [DOI] [PubMed] [Google Scholar]
- 14. Skaar E. P., Gaspar A. H., Schneewind O. (2004) IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J. Biol. Chem. 279, 436–443 [DOI] [PubMed] [Google Scholar]
- 15. Dailey T. A., Boynton T. O., Albetel A.-N., Gerdes S., Johnson M. K., Dailey H. A. (2010) Discovery and characterization of HemQ: an essential heme biosynthetic component. J. Biol. Chem. 285, 25978–25986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marino M., Hoffmann T., Schmid R., Möbitz H., Jahn D. (2000) Changes in protein synthesis during the adaptation of Bacillus subtilis to anaerobic growth conditions. Microbiology 146, 97–105 [DOI] [PubMed] [Google Scholar]
- 17. Ye R. W., Tao W., Bedzyk L., Young T., Chen M., Li L. (2000) Global gene expression profiles of Bacillus subtilis grown under anaerobic conditions. J. Bacteriol. 182, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee W. C., Reniere M. L., Skaar E. P., Murphy M. E. (2008) Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J. Biol. Chem. 283, 30957–30963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grocholski T., Koskiniemi H., Lindqvist Y., Mäntsälä P., Niemi J., Schneider G. (2010) Crystal structure of the cofactor-independent monooxygenase SnoaB from Streptomyces nogalater: Implications for the reaction mechanism. Biochemistry 49, 934–944 [DOI] [PubMed] [Google Scholar]
- 20. Erman J. E., Vitello L. B., Miller M. A., Shaw A., Brown K. A., Kraut J. (1993) Histidine-52 is a critical residue for rapid formation of cytochrome-C peroxidase compound-I. Biochemistry 32, 9798–9806 [DOI] [PubMed] [Google Scholar]
- 21. Duthie E. S., Lorenz L. L. (1952) Staphylococcal coagulase: mode of action and antigenicity. J. Gen. Microbiol. 6, 95–107 [DOI] [PubMed] [Google Scholar]
- 22. Wakeman C. A., Hammer N. D., Stauff D. L., Attia A. S., Anzaldi L. L., Dikalov S. I., Calcutt M. W., Skaar E. P. (2012) Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus. Mol. Microbiol. 86, 1376–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bae T., Schneewind O. (2006) Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58–63 [DOI] [PubMed] [Google Scholar]
- 24. Kurker R. C. (2013) Ph.D. thesis, Beyond O2: Biochemistry of chlorite dismutases from Klebsiella and Staphylococcus. University of Notre Dame, Notre Dame, IN [Google Scholar]
- 25. Woodward J. J., Martin N. I., Marletta M. A. (2007) An Escherichia coli expression-based method for heme substitution. Nat. Methods 4, 43–45 [DOI] [PubMed] [Google Scholar]
- 26. Berry E. A., Trumpower B. L. (1987) Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 161, 1–15 [DOI] [PubMed] [Google Scholar]
- 27. Kawakami N., Shoji O., Watanabe Y. (2012) Single-step reconstitution of apo-hemoproteins at the disruption stage of Escherichia coli cells. ChemBioChem 13, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 28. Louis-Jeune C., Andrade-Navarro M. A., Perez-Iratxeta C. (2011) Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 80, 374–381 [DOI] [PubMed] [Google Scholar]
- 29. Lansky I. B., Lukat-Rodgers G. S., Block D., Rodgers K. R., Ratliff M., Wilks A. (2006) The cytoplasmic heme-binding protein (PhuS) from the heme uptake system of Pseudomonas aeruginosa is an intracellular heme-trafficking protein to the δ-regioselective heme oxygenase. J. Biol. Chem. 281, 13652–13662 [DOI] [PubMed] [Google Scholar]
- 30. Breckau D., Mahlitz E., Sauerwald A., Layer G., Jahn D. (2003) Oxygen-dependent coproporphyrinogen III oxidase (HemF) from Escherichia coli is stimulated by manganese. J. Biol. Chem. 278, 46625–46631 [DOI] [PubMed] [Google Scholar]
- 31. Atkinson H. J., Morris J. H., Ferrin T. E., Babbitt P. C. (2009) Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One 4, e4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. von Eiff C., Heilmann C., Proctor R. A., Woltz C., Peters G., Götz F. (1997) A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J. Bacteriol. 179, 4706–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heinemann I. U., Jahn M., Jahn D. (2008) The biochemistry of heme biosynthesis. Arch. Biochem. Biophys. 474, 238–251 [DOI] [PubMed] [Google Scholar]
- 34. Sendi P., Proctor R. A. (2009) Staphylococcus aureus as an intracellular pathogen: the role of small colony variants. Trends Microbiol. 17, 54–58 [DOI] [PubMed] [Google Scholar]
- 35. Proctor R. A., von Eiff C., Kahl B. C., Becker K., McNamara P., Herrmann M., Peters G. (2006) Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4, 295–305 [DOI] [PubMed] [Google Scholar]
- 36. Faulkner M. J., Ma Z., Fuangthong M., Helmann J. D. (2012) Derepression of the Bacillus subtilis PerR peroxide stress response leads to iron deficiency. J. Bacteriol. 194, 1226–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arnesano F., Banci L., Barker P. D., Bertini I., Rosato A., Su X. C., Viezzoli M. S. (2002) Solution structure and characterization of the heme chaperone CcmE. Biochemistry 41, 13587–13594 [DOI] [PubMed] [Google Scholar]
- 38. Stevens J. M., Daltrop O., Higham C. W., Ferguson S. J. (2003) Interaction of heme with variants of the heme chaperone CcmE carrying active site mutations and a cleavable N-terminal His tag. J. Biol. Chem. 278, 20500–20506 [DOI] [PubMed] [Google Scholar]
- 39. Létoffé S., Heuck G., Delepelaire P., Lange N., Wandersman C. (2009) Bacteria capture iron from heme by keeping tetrapyrrole skeleton intact. Proc. Natl. Acad. Sci. U.S.A. 106, 11719–11724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu X., Du Q., Wang Z., Zhu D., Huang Y., Li N., Wei T., Xu S., Gu L. (2011) Crystal structure and biochemical features of EfeB/YcdB from Escherichia coli O157: ASP235 plays divergent roles in different enzyme-catalyzed processes. J. Biol. Chem. 286, 14922–14931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zubieta C., Joseph R., Krishna S. S., McMullan D., Kapoor M., Axelrod H. L., Miller M. D., Abdubek P., Acosta C., Astakhova T., Carlton D., Chiu H. J., Clayton T., Deller M. C., Duan L., Elias Y., Elsliger M. A., Feuerhelm J., Grzechnik S. K., Hale J., Han G. W., Jaroszewski L., Jin K. K., Klock H. E., Knuth M. W., Kozbial P., Kumar A., Marciano D., Morse A. T., Murphy K. D., Nigoghossian E., Okach L., Oommachen S., Reyes R., Rife C. L., Schimmel P., Trout C. V., van den Bedem H., Weekes D., White A., Xu Q., Hodgson K. O., Wooley J., Deacon A. M., Godzik A., Lesley S. A., Wilson I. A. (2007) Identification and structural characterization of heme binding in a novel dye-decolorizing peroxidase, TyrA. Proteins 69, 234–243 [DOI] [PubMed] [Google Scholar]
- 42. Villareal V. A., Pilpa R. M., Robson S. A., Fadeev E. A., Clubb R. T. (2008) The IsdC protein from Staphylococcus aureus uses a flexible binding pocket to capture heme. J. Biol. Chem. 283, 31591–31600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dunford H. B. (1999) Heme Peroxidases, Wiley-VCH, Hoboken, NJ [Google Scholar]
- 44. Vitello L. B., Erman J. E., Miller M. A., Mauro J. M., Kraut J. (1992) Effect of Asp-235–Asn substitution on the absorption-spectrum and hydrogen-peroxide reactivity of cytochrome-C peroxidase. Biochemistry 31, 11524–11535 [DOI] [PubMed] [Google Scholar]
- 45. Epstein N., Schejter A. (1972) On the nature of the alkaline ionization of horseradish peroxidase. FEBS Lett. 25, 46–48 [DOI] [PubMed] [Google Scholar]
- 46. Takayama S. J., Ukpabi G., Murphy M. E., Mauk A. G. (2011) Electronic properties of the highly ruffled heme bound to the heme degrading enzyme IsdI. Proc. Natl. Acad. Sci. U.S.A. 108, 13071–13076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Olea C., Jr., Kuriyan J., Marletta M. A. (2010) Modulating heme redox potential through protein-induced porphyrin distortion. J. Am. Chem. Soc. 132, 12794–12795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Anderson L., Palmer T., Price N. C., Bornemann Ss, Boxer D. H., Pau R. N. (1997) Characterisation of the molybdenum-responsive ModE regulatory protein and its binding to the promoter region of the modABCS (molybdenum transport) operon of Escherichia coli. Eur. J. Biochem. 246, 119–126 [DOI] [PubMed] [Google Scholar]
- 49. Stenklo K., Thorell H. D., Bergius H., Aasa R., Nilsson T. (2001) Chlorite dismutase from Ideonella dechloratans. J. Biol. Inorg. Chem. 6, 601–607 [DOI] [PubMed] [Google Scholar]
- 50. Mehboob F., Wolterink A. F., Vermeulen A. J., Jiang B., Hagedoorn P.-L., Stams A. J., Kengen S. W. (2009) Purification and characterization of a chlorite dismutase from Pseudomonas chloritidismutans. FEMS Microbiol. Lett. 293, 115–121 [DOI] [PubMed] [Google Scholar]
- 51. Ebihara A., Okamoto A., Kousumi Y., Yamamoto H., Masui R., Ueyama N., Yokoyama S., Kuramitsu S. (2005) Structure-based functional identification of a novel heme-binding protein from Thermus thermophilus HB8. J. Struct. Funct. Genomics 6, 21–32 [DOI] [PubMed] [Google Scholar]
- 52. Rodriguez-Lopez J. N., Hernández-Ruiz J., Garcia-Cánovas F., Thorneley R. N., Acosta M., Arnao M. B. (1997) The inactivation and catalytic pathways of horseradish peroxidase with m-chloroperoxybenzoic acid-A spectrophotometric and transient kinetic study. J. Biol. Chem. 272, 5469–5476 [DOI] [PubMed] [Google Scholar]
- 53. Mayfield J. A., Blanc B., Rodgers K. R., Lukat-Rodgers G. S., DuBois J. L. (2013) Peroxidase-type reactions suggest a heterolytic/nucleophilic O-O joining mechanism in the heme-dependent chlorite dismutase. Biochemistry, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chelikani P., Fita I., Loewen P. C. (2004) Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 61, 192–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilks A., Torpey J., Ortiz de Montellano P. R. (1994) Heme oxygenase (HO-1). Evidence for electrophilic oxygen addition to the porphyrin ring in the formation of α-meso-hydroxyheme. J. Biol. Chem. 269, 29553–29556 [PubMed] [Google Scholar]
- 56. Andreoletti P., Mouesca J. M., Gouet P., Jaquinod M., Capeillère-Blandin C., Jouve H. M. (2009) Verdoheme formation in Proteus mirabilis catalase. Biochim. Biophys. Acta 1790, 741–753 [DOI] [PubMed] [Google Scholar]
- 57. Arnao M. B., Acosta M., del Río J. A., García-Cánovas F. (1990) Inactivation of peroxidase by hydrogen peroxide and its protection by a reductant agent. Biochim. Biophys. Acta 1038, 85–89 [DOI] [PubMed] [Google Scholar]
- 58. Wu R., Skaar E. P., Zhang R., Joachimiak G., Gornicki P., Schneewind O., Joachimiak A. (2005) Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J. Biol. Chem. 280, 2840–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mayhew S. G. (1978) The redox potential of dithionite and SO; from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur. J. Biochem. 85, 535–547 [DOI] [PubMed] [Google Scholar]
- 60. Layer G., Pierik A. J., Trost M., Rigby S. E., Leech H. K., Grage K., Breckau D., Astner I., Jänsch L., Heathcote P., Warren M. J., Heinz D. W., Jahn D. (2006) The substrate radical of Escherichia coli oxygen-independent coproporphyrinogen III oxidase HemN. J. Biol. Chem. 281, 15727–15734 [DOI] [PubMed] [Google Scholar]
- 61. Rand K., Noll C., Schiebel H. M., Kemken D., Dülcks T., Kalesse M., Heinz D. W., Layer G. (2010) The oxygen-independent coproporphyrinogen III oxidase HemN utilizes harderoporphyrinogen as a reaction intermediate during conversion of coproporphyrinogen III to protoporphyrinogen IX. Biol. Chem. 391, 55–63 [DOI] [PubMed] [Google Scholar]
- 62. Fetzner S., Steiner R. A. (2010) Cofactor-independent oxidases and oxygenases. Appl. Microbiol. Biotechnol. 86, 791–804 [DOI] [PubMed] [Google Scholar]
- 63. Panek H., O'Brian M. R. (2002) A whole genome view of prokaryotic haem biosynthesis. Microbiology 148, 2273–2282 [DOI] [PubMed] [Google Scholar]
- 64. Boynton T. O., Daugherty L. E., Dailey T. A., Dailey H. A. (2009) Identification of Escherichia coli HemG as a novel, menadione-dependent flavodoxin with protoporphyrinogen oxidase activity. Biochemistry 48, 6705–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vlasits J., Jakopitsch C., Bernroitner M., Zamocky M., Furtmüller P. G., Obinger C. (2010) Mechanisms of catalase activity of heme peroxidases. Arch. Biochem. Biophys. 500, 74–81 [DOI] [PubMed] [Google Scholar]
- 66. Swingle E. L. (1935) Studies on small colony variants of Staphylococcus aureus. J. Bacteriol. 29, 467–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Proctor R. A., van Langevelde P., Kristjansson M., Maslow J. N., Arbeit R. D. (1995) Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin. Infect. Dis. 20, 95–102 [DOI] [PubMed] [Google Scholar]
- 68. Kohler C., von Eiff C., Liebeke M., McNamara P. J., Lalk M., Proctor R. A., Hecker M., Engelmann S. (2008) A defect in menadione biosynthesis induces global changes in gene expression in Staphylococcus aureus RID C-1631-2010 RID C-1618-2010. J. Bacteriol. 190, 6351–6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Möbius K., Arias-Cartin R., Breckau D., Hännig A.-L., Riedmann K., Biedendieck R., Schröder S., Becher D., Magalon A., Moser J., Jahn M., Jahn D. (2010) Heme biosynthesis is coupled to electron transport chains for energy generation. Proc. Natl. Acad. Sci. U.S.A. 107, 10436–10441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hansson M., Hederstedt L. (1994) Bacillus subtilis HemY is a peripheral membrane protein essential for protoheme IX synthesis which can oxidize coproporphyrinogen III and protoporphyrinogen IX. J. Bacteriol. 176, 5962–5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Olsson U., Billberg A., Sjovall S., Sjo S. (2002) In vivo and in vitro studies of Bacillus subtilis ferrochelatase mutants suggest substrate channeling in the heme biosynthesis pathway. J. Bacteriol. 184, 4108–4124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aoki R., Takeda T., Omata T., Ihara K., Fujita Y. (2012) MarR-type transcriptional regulator ChlR activates expression of tetrapyrrole biosynthesis genes in response to low-oxygen conditions in cyanobacteria. J. Biol. Chem. 287, 13500–13507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Culbertson D. S., Olson J. S. (2010) Role of heme in the unfolding and assembly of myoglobin. Appl. Microbiol. Biotechnol. 49, 6051–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hofbauer S., Gysel K., Mlynek G., Kostan J., Hagmüller A., Daims H., Furtmüller P. G., Djinov́ic-Carugo K., Obinger C. (2012) Impact of subunit and oligomeric structure on the thermal and conformational stability of chlorite dismutases. Biochim. Biophys. Acta 1824, 1031–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Qi Z., O'Brian M. R. (2002) Interaction between the bacterial iron response regulator and ferrochelatase mediates genetic control of heme biosynthesis. Mol. Cell 9, 155–162 [DOI] [PubMed] [Google Scholar]
- 76. Qi Z., Hamza I., O'Brian M. R. (1999) Heme is an effector molecule for iron-dependent degradation of the bacterial iron response regulator (Irr) protein. Proc. Natl. Acad. Sci. U.S.A. 96, 13056–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yang J., Ishimori K., O'Brian M. R. (2005) Two heme binding sites are involved in the regulated degradation of the bacterial iron response regulator (Irr) protein. J. Biol. Chem. 280, 7671–7676 [DOI] [PubMed] [Google Scholar]