

Abstract
Traumatic brain injury (TBI) disrupts the blood–brain barrier (BBB) and reduces cerebral glucose uptake. Vascular endothelial growth factor (VEGF) is believed to play a key role in TBI, and COG1410 has demonstrated neuroprotective activity in several models of TBI. However, the effects of COG1410 on VEGF and glucose metabolism following TBI are unknown. The current study aimed to investigate the expression of VEGF and glucose metabolism effects in C57BL/6J male mice subjected to experimental TBI. The results showed that controlled cortical impact (CCI)-induced vestibulomotor deficits were accompanied by increases in brain edema and the expression of VEGF, with a decrease in cerebral glucose uptake. COG1410 treatment significantly improved vestibulomotor deficits and glucose uptake and produced decreases in VEGF in the pericontusion and ipsilateral hemisphere of injury, as well as in brain edema and neuronal degeneration compared with the control group. These data support that COG1410 may have potential as an effective drug therapy for TBI.
Keywords: : brain edema, COG1410, metabolism, TBI, VEGF
Introduction
Traumatic brain injury (TBI) is the leading cause of death worldwide.1 Brain edema following TBI is the main determinant of adverse outcomes in survivors.2,3 Over the past several decades, efforts have been made to determine the pathophysiology of TBI4 and to develop drug therapies for TBI,5 but until now, there have been no effective neuroprotective drugs demonstrated in human clinical trials.6
Apolipoprotein E (apoE, protein; APOE, gene) is responsible for the transportation of lipids and the mediation of cholesterol in the central nervous system.7 Our previous research demonstrated that the APOEɛ4 allele was an adverse prognostic factor of TBI.8 However, apoE2 and apoE3 proteins act as neuroprotective factors, whereas apoE deficiency is related to motor and cognitive dysfunction after brain injury9,10 In addition, the administration of the exogenous apoE-mimetic peptide modifies inflammatory responses through binding with low-density lipoprotein (LDL) receptor-related protein (LRP) and improves motor dysfunction.11–15 Other studies have demonstrated that apoE-deficient mice have an increased expression of vascular endothelial growth factor (VEGF),16 which is believed to be related to blood–brain barrier (BBB) disruption after TBI,17 and to the increase in glucose utilization in astrocytes.18 COG1410, a mimetic peptide derived from the apoE receptor-binding region, not only retains most of the protective properties of holo-apoE protein, but also overcomes the deficiency in crossing the BBB. However, the effects of COG1410 on the expression of VEGF and on cerebral glucose metabolism after controlled cortical impact (CCI) have not been reported.
We hypothesized that COG1410 exerts neuroprotective effects on cerebral glucose metabolism and vestibulomotor function in an anti-VEGF manner. To investigate this hypothesis, vestibulomotor function, brain edema, VEGF, angiogenesis, neuronal degeneration and cerebral glucose uptake were assessed in a mouse model of CCI.
Methods
All procedures were approved by the Affiliated Hospital of Southwest Medical University and were consistent with the Laboratory Animal Regulations for the care and use of animals.
Animals
Adult (8–12 weeks) C57BL/6J male mice (Laboratory Animal Center of Chongqing Medical University, Chongqing, China) weighing 18–25 g were used. The mice were divided in four groups: the control group, the sham-operated group, the vehicle group, and the COG1410-treated group. The control group comprised native mice.
Experimental TBI: CCI
The method for creating the experimental CCI used in this study has been described previously.4 Briefly, the mice were anesthetized with 3.5% chloral hydrate (500 mg/kg), shaved, and placed in a stereotaxic frame. Using sterile techniques, a midline incision was made, and the scalp was reflected to expose the skull. A 3 mm right lateral craniotomy was performed with a motorized drill that was centered at 2.7 mm lateral from the midline and 3 mm anterior to lambda to allow the removal of bone without disrupting the dura. An impactor with a 2.0 mm diameter rod tip was driven at a velocity of 3 m/sec with a dwell time of 100 ms and an impact depth of 1.0 mm, which produced a moderate contusion in the right cortex causing pronounced behavioral deficits, but no mortality. After injury, a polyvinylidene fluoride skull cap was secured over the craniotomy, and the skin incision was sutured. The anesthetized mice were wrapped in a blanket (37°C) until they recovered, and were able to freely ambulate.
Administration of COG1410
COG1410 was provided by Cognosci (Research Triangle Park, NC) at a purity of 95% and was dissolved in a sterile 0.9% saline solution at a concentration of 0.1 mg/mL before administration. The mice were randomly divided into two groups: the COG1410-treated group received COG1410 (1 mg/kg), and the control group received 0.9% saline vehicle. COG1410 or vehicle was administered within 30 min of injury and then every 24 h via tail vein infusion.
Rotarod test
Motor function, particularly vestibulomotor function, was evaluated by a rotarod test (TME, Chengdu, China) according to Hamm and coworkers.19,20 Briefly, the day before injury, the mice underwent two consecutive conditioning trials at a constant rotational speed of 16 rpm for 1 min (interval = 5 min), followed by three additional trials with an accelerating rotational speed (starting at 0 rpm and accelerating by 3 rpm every 10 sec until the rotating speed reached 30 rpm). The average time to fall from the rotating cylinder in the latter three trials was recorded as the baseline of latency. At 1, 3, and 7 days after injury, the mice underwent three consecutive accelerating tests as described (interval = 15 min). The average time to fall from the rotating cylinder was recorded as the result of the rotarod test.
BBB permeability
Evans blue (EB) dye was used to measure the permeability of the BBB according to Kaya.21 Briefly, the mice were euthanized at 1, 3, or 7 days after injury. The cerebellum and olfactory brain were removed, and the injured right hemisphere was mechanically homogenized using formamide at a ratio of 1 g tissue to 2 mL formamide. The solution was warmed to 50°C for 72 h. The extract was ultracentrifuged (Thermo, USA) at a speed of 60,000 rpm for 45 min at 4°C. Absorbance of the EB in the high speed supernatant and in the homogenates was measured at 620 nm. The concentration of dye in the samples was calculated from a standard curve of EB in formamide. The ratio of EB absorbance in brain tissue in the high speed supernatant was calculated as a measure of BBB permeability.
Brain water content
The dry-wet method was used to determine brain water content.22 The mice were euthanized at 1, 3, or 7 days after injury. The cerebellum and olfactory brain were removed, and the injured right hemisphere was weighed to assess its wet weight. The injured right hemisphere was then dried for 24 h at 100°C to determine its dry weight. The brain water content was used to evaluate brain edema, which was obtained by the following calculation:
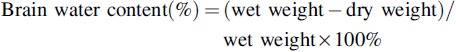 |
Enzyme-linked immunosorbent assay (ELISA)
ELISA was used to measure the expression of VEGF. The mice were anesthetized with an overdose of 3.5% chloral hydrate (1000 mg/kg) and then killed by decapitation. Cerebellum and olfactory brain were removed and the injured right hemisphere was weighed, then homogenized in phosphate-buffered saline (PBS) (1:9 weight/volume dilution in PBS, pH = 7.4), and centrifuged (4°C, 3,000 rpm for 20 min). Supernatant was diluted 10 times with sample diluent for ELISA assays. The protein content of each homogenate sample was measured with a BCA kit (Beyotime, Haimen, China). The results were normalized to total protein levels per sample.
Immunohistochemistry
The mice were deeply anesthetized with an overdose of 3.5% chloral hydrate (1000 mg/kg) and perfused intracardially with ice-cold PBS (pH = 7.4). After the mice were killed, the brains were carefully removed, fixed in 4% paraformaldehyde for 24 h, dehydrated, and embedded. Coronal sections (4 μ) were cut with a vibratome. The sections were dewaxed in xylene and rehydrated in a descending ethanol series. The sections were subsequently incubated in 7.5% H2O2 followed by incubation in normal goat serum for 20 min. The sections were incubated with rabbit anti-vWF (1:100, Bioss, Beijing, China) and detected with a Rat IgG ABC Kit (1:500, Vector Labs, Burlingame, CA). Additional sections were incubated with rabbit anti-degraded myelin basic protein (1:1000, Millipore, Billerica, USA) primary antibodies at 4°C overnight. Antibody binding was detected using a biotinylated secondary antibody and peroxidase-conjugated streptavidin. Visualization was performed with diaminobenzidine (DAB). After being counterstained with hematoxylin for 3 min, the sections were dehydrated in an ascending ethanol series and cover-slipped for microscopic viewing and quantification.
Immunohistochemical reactions for vWF were interpreted independently by two authors using the same microscope (E200, Nikon, Japan).23 The average microvessel number of five high-power fields ( × 200) was recorded as the microvessel density (MVD). National Institutes of Health (NIH) ImageJ software was used to determine the ipsilateral hemisphere immunoglobulin G (IgG) staining density and the total area of IgG staining.24
For some experiments, the mice were killed and the brains were carefully removed, fixed in 4% paraformaldehyde for 24 h, and equilibrated in 30% sucrose. Serial 50 μ thick coronal slices were cut on a freezing microtome. Silver staining was performed on a third set of sections to visualize the degenerating neuronal elements in the brain. Sections were processed with the FD NeuroSilverKit II (FD Neuro Technologies, Ellicott City, MD).4 NIH ImageJ software was used to measure the optical density of silver staining.
Micro-PET/CT Scan
An Inveon pre-clinical PET/CT scanner (Inveon MM gantry, Siemens, Germany) was used to detect cerebral glucose uptake at 1 day before injury and 1, 3, and 7 days after injury. The mice were deprived of food for 24 h prior to the PET scans to ensure stable plasma glucose levels. After being anesthetized with 3.5% chloral hydrate (500 mg/kg), they were injected with Fluorodeoxyglucose (18F) ([18F]FDG) through the tail vein and returned to their cages for a 30 min uptake period in a dark and quiet environment (T = 20°C). They were then positioned in the scanning bed. Imaging started with a CT scan for attenuation correction and localization of the lesion, which was immediately followed by a PET scan.25 The cerebral glucose uptake ratio was evaluated by standardized uptake value (SUV):
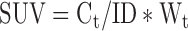 |
Where Ct (MBq/cc) is the decay-corrected activity concentration in the region of interest, ID (mCi) is the injected dose (the difference between prior to injection and after injection), and Wt (kg) is the weight of the mice.26
Statistical analysis
All data were analyzed using SPSS 13.0 (Statistical Product and Service Solutions, New York, NY). The results are presented as the mean ± standard error of the mean (SEM) of five animals. A two way ANOVA was used to analyze the EB, brain water content, IgG staining, VEGF, MVD, silver staining and SUV-glucose data. A repeated measure was used to analyze the rotarod test data. Pearson correlation analysis was used to analyze the relationship between vestibulomotor function and glucose uptake. A p value of <0.05 was considered to be a statistically significant difference when comparing parameters.
Results
COG1410 improved vestibulomotor function after CCI
The sham-operated group performed more poorly than did the control group at 1 day after injury (n = 5 for each group, p < 0.05); however, no significant differences were observed between the control group and the sham-operated group at 3 and 7 days after injury (p > 0.05). CCI-induced vestibulomotor deficits were significantly different from those of the sham-operated group (p < 0.05). The administration of COG1410 after the CCI-induced TBI significantly reduced vestibulomotor deficits compared with the vehicle group over the 7 day testing period (p < 0.05). Further, vestibulomotor deficits reached a maximum for the groups treated with saline and COG1410 at 1 day after injury; however, the COG1410-treated group returned to baseline (p > 0.05) whereas the vehicle group was still below baseline (p < 0.05) at 7 days after injury (Fig. 1).
FIG. 1.

COG1410 improved vestibulomotor performance on the rotarod test after controlled cortical impact (CCI). Mice administered COG1410 significantly increased their rotarod latencies compared with the vehicle group at 1, 3, and 7 days post-CCI (mean ± standard error of the mean, n = 5 per group, *p < 0.05, COG1410-treated group vs. vehicle group).
COG1410 reduced BBB disruption and brain water content
No significant difference was observed in EB dye extravasation, IgG leakage, or brain water content between the control group and the sham-operated group over the 7 day testing period (n = 5 for each group, p > 0.05). CCI induced increases in EB dye extravasation, IgG leakage, and brain water content compared with the sham-operated group (p < 0.05), and administration of COG1410 significantly reduced EB dye extravasation, IgG leakage, and brain water content compared with the vehicle group at 1 and 3 days after injury. However, there was no significant difference in EB dye and IgG leakage between the vehicle group and the COG1410-treated group at 7 days after injury. Further, the brain water content of the COG1410-treated group returned to baseline (p > 0.05), whereas that of the vehicle group did not return to baseline (p < 0.05) at 7 days after injury (Fig. 2).
FIG. 2.

(A) COG1410 reduced Evans blue (EB) dye extravasation after controlled cortical impact (CCI). The results showed that EB dye extravasation into the brain parenchyma was significantly reduced by the administration of COG1410 at 1 and 3 days after injury, and returned to baseline at 7 days after injury in the vehicle and COG1410-treated groups. (B) COG1410 reduced immunoglobulin G (IgG) leakage after CCI. The results showed that IgG leakage was significantly reduced by the administration of COG1410 at 1 and 3 days after injury and returned to baseline at 7 days after injury in the COG1410-treated and vehicle groups. (C) COG1410 reduced brain water content after CCI. The results showed that the brain water content was significantly reduced by the administration of COG1410 at 1 and 3 days after injury and returned to baseline at 7 days after injury in the COG1410-treated group. (D) IgG staining. (mean ± standard error of the mean, n = 5 per group, *p < 0.05, COG1410-treated group vs. vehicle group).
COG1410 suppressed the expression of VEGF after CCI
Surgery on the scalp did not increase the expression of VEGF in the brain tissue (n = 5 for each group, p > 0.05). However, CCI induced an increase in the expression of VEGF compared with the sham-operated group (p < 0.05). The administration of COG1410 significantly reduced the expression of VEGF over the 7 day testing period compared with the vehicle group (p < 0.05, Fig. 3).
FIG. 3.

COG1410 suppressed the expression of vascular endothelial growth factor (VEGF) after controlled cortical impact (CCI). The results showed that the expression of VEGF was significantly suppressed by the administration of COG1410 at 1, 3, and 7 days after injury in the COG1410-treated group (mean ± standard error of the mean, n = 5 per group, *p < 0.05, COG1410-treated group vs. vehicle group).
COG1410 suppressed excessive angiogenesis after CCI
There was no significant difference in MVD between the control and sham-operated groups over the 7 day testing period (n = 5 for each group, p > 0.05). An increase in MVD was observed at 3 and 7 days after injury in the CCI group compared with the sham-operated group (p < 0.05). Administration of COG1410 significantly suppressed excessive angiogenesis compared with the vehicle group at 3 and 7 days after injury (p < 0.05). Further, there was a conspicuous difference in morphology between neovascularization after CCI and normal microvessels in the control group (Fig. 4).
FIG. 4.

(A) COG1410 suppressed excessive angiogenesis after controlled cortical impact (CCI). The results showed that there was a significant increase in microvessel density (MVD) after CCI compared with the sham-operated group, and MVD was significantly suppressed by the administration of COG1410 at 3 and 7 days after injury in the COG1410-treated group. (B) There was a conspicuous difference in morphology between neovascularization after CCI and normal microvessels in the control group. The neovascularization was line-like or bud-like, but the normal microvessels were ring shaped ( × 400) (mean ± standard error of the mean, n = 5 per group, *p < 0.05, COG1410-treated group vs. vehicle group).
COG1410 reduced neurodegeneration
No significant difference was observed in the density of silver staining between the sham-operated group and the control group (n = 5 for each group, p > 0.05). CCI caused widespread silver-staining abnormalities compared with the sham-operated group (p < 0.05), and the silver-staining density reached its peak at 3 days after injury, as determined by densitometric methods. The administration of COG1410 significantly reduced the density of staining in the ipsilateral hemisphere of injury over the 7 day testing period compared with the vehicle group (p < 0.05, Fig. 5).
FIG. 5.

(A) COG1410 reduced the neurodegeneration in the ipsilateral hemisphere of injury after controlled cortical impact (CCI). The results showed that neurodegeneration was significantly reduced by the administration of COG1410 at 1, 3, and 7 days after injury in the COG1410-treated group. (B) Silver staining (mean ± standard error of the mean, n = 5 per group, *p < 0.05, COG1410-treated group vs. vehicle group).
COG1410 increased brain glucose uptake after CCI
The sham operation did not affect cerebral glucose uptake (n = 5 for each group, p > 0.05). However, there was a transient reduction of SUV in the pericontusion and ipsilateral hemisphere of injury. It reached a minimum at 1 day after injury and then increased and returned to baseline at 7 days after injury compared with the sham-operated group (p < 0.05). COG1410 increased the SUV of the pericontusion and ipsilateral hemisphere of injury compared with the vehicle group at 1 and 3 days after injury (p < 0.05). However, there was no statistically significant difference between the vehicle group and the COG1410-treated group at 7 days after injury (p > 0.05, Fig. 6).
FIG. 6.

COG1410 increased the glucose uptake of the ipsilateral hemisphere of injury and the pericontusion after controlled cortical impact (CCI). Mice treated with COG1410 had significantly increased standardized uptake value (SUV) in the ipsilateral hemisphere and pericontusion compared with the vehicle group at 1 and 3 days after injury (mean ± standard error of the mean, n = 5 per group, *p < 0.05, COG1410-treated group vs. vehicle group). Color image is available online at www.liebertpub.com/neu
Correlation analysis of SUV and the rotarod test
Cerebral glucose is believed to a reliable method for evaluating cognitive deficits and motor dysfunction after TBI.25,27 However, the relationship between cerebral glucose uptake and vestibulomotor function has not been known. In the current study, we performed a Pearson correlation analysis and determined that there was a close relationship between vestibulomotor function and cerebral glucose uptake (R = 0.828, R2 = 0.686, p < 0.05, Fig. 7). Higher cerebral glucose uptake was strongly associated with the recovery of vestibulomotor dysfunction after TBI.
FIG. 7.

Pearson correlation analysis was used to analyze the relationship between vestibulomotor function and glucose uptake in the ipsilateral hemisphere of injury. There was a close relationship between motor function and cerebral glucose metabolism (R = 0. 828, R2 = 0.686, p < 0.05).
Discussion
In summary, the moderate experimental CCI resulted in lower cerebral glucose uptake and vestibulomotor deficits and was associated the upregulation of VEGF and BBB disruption. COG1410 promoted recovery from vestibulomotor deficits and glucose uptake in the ipsilateral hemisphere and pericontusion of injury coupled with alleviating the excessive expression of VEGF and the BBB disruption. Further, cerebral glucose uptake exhibited a strongly positive correlation with vestibulomotor function. These results suggest that the therapeutic efficiency of COG1410 may be associated with the modulation of VEGF.
Previous findings have shown that there was an increase in the expression of VEGF after TBI.28 However, the role of VEGF in TBI is still being debated. Angiogenesis induced by the upregulation of VEGF improved subacute cerebral ischemia in the penumbra.29 However, the upregulation of VEGF activated matrix metalloproteinases (MMPs), which damaged the basilar membrane and disrupted the BBB.17 Further study demonstrated that there was a significant increase in the expression of VEGF in apoE-deficient mice,16 and the latest clinical research indicated that the elevation of VEGF exhibited an adverse effect from 4 to 14 days, whereas it had an advantageous effect from 14 to 21 days after TBI.30 Therefore, we hypothesized that administration of exogenous apoE mimetic peptide-COG1410 alleviated the BBB disruption via suppressing the excessive expression of VEGF as a new therapeutic target of TBI. Our experimental findings were consistent with the observation that CCI contributes to the expression of VEGF, and that a higher expression of VEGF increases BBB permeability and aggravates brain edema in the acute stage of injury. Therefore, from these perspectives, suppressing the excessive expression of VEGF is necessary and has therapeutic significance in the acute stage.
Angiogenesis is one of the predominant pathological processes after TBI and is believed to be a favorable factor in the subacute stage.31 In our study, we found that CCI induced angiogenesis and that COG1410 treatment decreased BBB permeability and brain edema while improving vestibulomotor deficits by partially suppressing angiogenesis in the acute stage. These outcomes might result from a structurally defective neovascularization32 and excessive cerebral perfusion pressure (CPP), which is increased by excessive angiogenesis, exacerbates extravasation of components, and aggravates brain edema.25 Johanson and coworkers reported that VEGF not only enhanced angiogenesis, but also could increase permeability and place a patient at risk of hemorrhaging in the early stage.33 In addition, numerous studies have demonstrated that the neovascularization resulted in submacular hemorrhage, and that anti-angiogenesis or anti-VEGF was useful for the submacular hemorrhage.34,35 Micro-PET/CT was applied to verify whether more angiogenesis led to higher cerebral glucose uptake. However, more MVD did not increase the SUV in the vehicle group compared with the COG1410-treated group. From this perspective, we believe that suppressing the excessive expression of VEGF and angiogenesis in the acute stage is necessary, and that COG1410 displays neuroprotective effects that are strongly associated with relieving brain edema.
In addition, our findings are consistent with study findings that BBB permeability was typically biphasic after CCI,36 and that COG1410 decreases vasogenic brain edema in the acute stage, as was shown in our previous research.37 Barzó and coworkers reported that vasogenic cerebral edema plays a major role in the acute stage after brain injury, and that cytotoxic cerebral edema becomes dominant at 1 week after injury.38 In our study, we found that BBB permeability nearly returned to baseline in both the vehicle and COG1410-treated groups at 7 days after injury; however, brain water content did not return to baseline in the vehicle group. These findings might mean that COG1410 decreases not only vasogenic cerebral edema, but also cytotoxic cerebral edema. Finally, there was a remarkable difference in morphology between the line-like and bud-like neovascularizations and the ring-shaped normal microvessels, which might result from the lack of smooth muscle function in neovascularization.32
APOE has been shown to be associated with the expression of VEGF, and apoE deficiency increases the expression of VEGF.16 Our research found that COG1410 reduced the expression of VEGF, and Gao and coworkers reported that anti-VEGF treatment attenuated the disruption of the BBB through the Toll-like receptor 4 (TLR4)/nuclear factor-κB (NF-κB) signaling pathway in the acute stage after TBI.39 Therefore, we hypothesize that COG1410 attenuates BBB disruption by reducing the expression of VEGF. In addition, Bell and coworkers reported that apoE reduced BBB permeability via the cyclophilin A (CypA)-NF-κB pathway through binding with the apoE receptor LRP-1.40 As COG1410 is a peptide derived from the receptor-binding domain of apoE, it may theoretically reduce BBB permeability through the abovementioned pathway; however, the mechanism requires further study.
The evaluation of cerebral glucose metabolism with PET with [18F]FDG is believed to be an effective and reliable imaging technique for studying brain metabolism. However, PET is a functional imaging modality and lacks precise anatomical localization. By providing anatomical localization and glucose metabolism imaging, micro-PET/CT is believed to be the best technique for studying traumatic penumbra.41 Micro-PET/CT was applied to test the cerebral glucose metabolism after suppressing the expression of VEGF. The present study demonstrated that CCI induced glucose hypometabolism and that the administration of COG1410 markedly increased cerebral glucose uptake not only in the penumbra, but also in the ipsilateral hemisphere. These findings are comparable to the glucose hypometabolism that has been reported in rat and human TBI studies.42 However, we did not find hypermetabolism after CCI, because the first testing time was 1 day after injury in our study, and hypermetabolism is usually observed several hours after injury in moderate to severe brain injuries.43 In addition, the change of cerebral glucose metabolism could be detected before the serology changes and clinical symptoms, and the PET/CT was applied to diagnose Alzheimer's disease (AD) in the earliest stages.44 In the current study, the cerebral glucose uptake value returning to the baseline occurred prior to the vestibulomotor function returning back to normal, and the cerebral glucose uptake value exhibited a strongly positive correlation with vestibulomotor function. Therefore, the cerebral glucose uptake value could be used as an important reference standard for judging the prognosis of TBI as described in the article.
Traditionally, ischemia after neurotrauma could be improved or overcome by angiogenesis in the subacute stage.29 However, increased angiogenesis not only aggravated brain edema but also decreased the pericontusion and ipsilateral hemisphere glucose uptake in the acute stage in our study. These outcomes might result from deepening brain edema, which leads to neuron death, and, eventually, to hypometabolism.25 To verify this hypothesis, silver staining was used in our study, and we found that there was less neuronal degeneration in the ipsilateral hemisphere after COG1410 treatment. From this perspective, we speculate that COG1410 might exert a neuroprotective effect by reducing brain edema and increasing neuron survival.
In addition, insulin was the most important endocrine factor for glucose uptake and utilization.45 Agudo and coworkers46 reported that the upregulation of VEGF leading to β-cell failure resulted from aggravating the inflammatory process, after which the glucose metabolism disorder was initiated and anti-VEGF treatment enhanced the glucose uptake.47,48 COG1410 suppressed the expression of VEGF in the current study, and it might reduce the β-cell failure to promote glucose uptake. Moreover, glucose transporter −4 (Glut-4) was the primary transporter of glucose transporting glucose into the cells, and the expression of Glut-4 was regulated by APOE.44,49 As COG1410 is a peptide derived from the receptor-binding domain of apoE, it might regulate the expression of Glut-4 to affect glucose uptake. Finally, Berkel and coworkers50 believed that the increased endothelial surface area contributed to higher accumulation of [18F]FDG supporting the idea that functionality of vessels is more critical for SUVs than MVD. Therefore, from these perspectives, suppression the expression of VEGF could increase the cerebral glucose uptake rather than decreasing the glucose uptake.
There are several limitations in the current study. First, a precise analysis of the accurate anatomical locations was not possible using micro-PET/CT. Co-registered technology, such as micro-PET/MRI, is necessary for further study. Second, because there are APOE polymorphisms in human beings, APOEɛ2, APOEɛ3, and APOEɛ4 transgenic mice need to be used to analyze the effect of the different apoE-protein isoforms on VEGF, angiogenesis, and cerebral glucose uptake. Finally, the effect of COG1410 on VEGF, angiogenesis, and cerebral glucose uptake in the subacute or chronic stage is worthy studying.
Conclusion
In conclusion, these findings support a neuroprotective effect of COG1410 by relieving the BBB disruption and brain edema, which promotes cerebral glucose uptake and vestibulomotor function recovery in the acute stage following CCI. Our current findings and the results of other studies suggest that COG1410 is a promising pre-clinical therapeutic agent for the treatment of TBI.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81371319, 81000528), the Program for New Century Excellent Talents in University (NCET-12-1057), and the Foundation for Outstanding Youth Academic Technology Leaders of Sichuan Province (2014JQ0022). We thank Cognosci Inc. for providing COG1410.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Johnstone V.P., Wright D.K., Wong K., O'Brien T.J., Rajan R., and Shultz S.R. (2015). Experimental traumatic brain injury results in long-term recovery of functional responsiveness in sensory cortex but persisting structural changes and sensorimotor, cognitive, and emotional deficits. J. Neurotrauma 32, 1333–1346 [DOI] [PubMed] [Google Scholar]
- 2.Terpolilli N.A., Kim S.W., Thal S.C., Kuebler W.M., and Plesnila N. (2013). Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 33, 311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vavilala M.S., Kernic M.A., Wang J., Kannan N., Mink R.B., Wainwright M.S., Groner J.I., Bell M.J., Giza C.C., Zatzick D.F., Ellenbogen R.G., Boyle L.N., Mitchell P.H., and Rivara F.P. (2014). Acute care clinical indicators associated with discharge outcomes in children with severe traumatic brain injury. Crit. Care Med. 42, 2258–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang Y., and Brody D.L. (2012). Administration of COG1410 reduces axonal amyloid precursor protein immunoreactivity and microglial activation after controlled cortical impact in mice. J. Neurotrauma 29, 2332–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mountney A., Bramlett H.M., Dixon C.E., Mondello S., Dietrich W.D., Wang K.K., Caudle K.L., Empey P., Poloyac S.M., Hayes R.L., Povlishock J., Tortella F.C., Kochanek P.M., and Shear D.A. (2016). Simvastatin treatment in traumatic brain injury: operation brain trauma therapy. J. Neurotrauma 33, 567–580 [DOI] [PubMed] [Google Scholar]
- 6.Schumacher M., Denier C., Oudinet J.P., Adams D., and Guennoun R. (2015). Progesterone neuroprotection: the background of clinical trial failure. J. Steroid Biochem. Mol. Biol. 160, 53–66 [DOI] [PubMed] [Google Scholar]
- 7.Phillips M.C. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 66, 616–623 [DOI] [PubMed] [Google Scholar]
- 8.Jiang Y., Sun X., Gui L., Xia Y., Tang W., Cao Y., and Gu Y. (2007). Correlation between APOE −491AA promoter in epsilon4 carriers and clinical deterioration in early stage of traumatic brain injury. J. Neurotrauma 24, 1802–1810 [DOI] [PubMed] [Google Scholar]
- 9.Huang Y., Liu X.Q., Wyss–Coray T., Brecht W.J., Sanan D.A., Mahley R.W.(2001). Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proceedings of the National Academy of Sciences of the United States of America 98(15):8838–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Namjoshi D.R., Martin G., Donkin J., Wilkinson A., Stukas S., Fan J., Carr M., Tabarestani S., Wuerth K., Hancock R.E., and Wellington C.L. (2013). The liver X receptor agonist GW3965 improves recovery from mild repetitive traumatic brain injury in mice partly through apolipoprotein E. PLoS One 8, e53529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufman N.A., Beare J.E., Tan A.A., Vitek M.P., McKenna S.E., Hoane M.R. (2010). COG1410, an apolipoprotein E-based peptide, improves cognitive performance and reduces cortical loss following moderate fluid percussion injury in the rat. Behav. Brain Res. 214, 395–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laskowitz D.T., Thekdi A.D., Thekdi S.D., Han S.K., Myers J.K., Pizzo S.V., and Bennett E.R. (2001). Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp. Neurol. 167, 74–85 [DOI] [PubMed] [Google Scholar]
- 13.Laskowitz D.T., Fillit H., Yeung N., Toku K., and Vitek M.P. (2006). Apolipoprotein E-derived peptides reduce CNS inflammation: implications for therapy of neurological disease. Acta Neurol. Scand. Suppl. 185, 15–20 [DOI] [PubMed] [Google Scholar]
- 14.Pocivavsek A., Burns M.P, and Rebeck G.W. (2009). Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N–terminal kinase. Glia 57, 444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pocivavsek A., Mikhailenko I., Strickland D.K., and Rebeck G.W. (2009). Microglial low-density lipoprotein receptor-related protein 1 modulates c-Jun N-terminal kinase activation. J. Neuroimmunol. 214, 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez–Robredo P., Sadaba L.M., Salinas–Alaman A., Recalde S., Rodriguez J.A., and Garcia–Layana A. (2013). Effect of lutein and antioxidant supplementation on VEGF expression, MMP-2 activity, and ultrastructural alterations in apolipoprotein E-deficient mouse. Oxid. Med. Cell. Longev. 2013, 213505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bauer A.T., Burgers H.F., Rabie T., and Marti H.H. (2010). Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J. Cereb. Blood Flow Metab. 30, 837–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allaman I., Fiumelli H., Magistretti P.J., and Martin J.L. (2011). Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology 216, 75–84 [DOI] [PubMed] [Google Scholar]
- 19.Hamm R.J., Pike B.R., O'dell D.M., Lyeth B.G., and Jenkins L.W. (1994). The Rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J. Neurotrauma 11, 187–203 [DOI] [PubMed] [Google Scholar]
- 20.Wang H., Durham L., Dawson H., Song P., Warner D.S., Sullivan P.M., Vitek M.P., and Laskowitz D.T. (2007). An apolipoprotein E-based therapeutic improves outcome and reduces Alzheimer's disease pathology following closed head injury: evidence of pharmacogenomic interaction. Neuroscience 144, 1324–1333 [DOI] [PubMed] [Google Scholar]
- 21.Kaya M., and Ahishali B. (2011). Assessment of permeability in barrier type of endothelium in brain using tracers: Evans blue, sodium fluorescein, and horseradish peroxidase. Methods Mol. Biol. 763, 369–382 [DOI] [PubMed] [Google Scholar]
- 22.Liew H.K., Pang C.Y., Hsu C.W., Wang M.J., Li T.Y., Peng H.F., Kuo J.S., and Wang J.Y. (2012). Systemic administration of urocortin after intracerebral hemorrhage reduces neurological deficits and neuroinflammation in rats. J. Neuroinflammation 9, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosomtwi A., Jiang Q., Ding G.L., Zhang L., Zhang Z. G., Lu M., Ewing J.R., and Chopp M. (2008). Quantitative evaluation of microvascular density after stroke in rats using MRI. J. Cereb. Blood Flow Metab. 28, 1978–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell K.L., Berman N.E., Gregg P.R., and Levant B. (2014). Fish oil improves motor function, limits blood–brain barrier disruption, and reduces Mmp9 gene expression in a rat model of juvenile traumatic brain injury. Prostaglandins Leukot. Essent. Fatty Acids 90, 5–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J., Gu L., Feng D.F., Ding F., Zhu G., and Rong J. (2012). Exploring temporospatial changes in glucose metabolic disorder, learning, and memory dysfunction in a rat model of diffuse axonal injury. J. Neurotrauma 29, 2635–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byrnes K.R., Wilson C.M., Brabazon F., von Leden R., Jurgens J.S., Oakes T.R., and Selwyn R.G. (2014). FDG-PET imaging in mild traumatic brain injury: a critical review. Front. Neuroenergetics 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato T., Nakayama N., Yasokawa Y., et al. (2007). Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma 24, 919–926 [DOI] [PubMed] [Google Scholar]
- 28.Thau–Zuchman O., Shohami E., Alexandrovich A.G., and Leker R.R. (2012). Subacute treatment with vascular endothelial growth factor after traumatic brain injury increases angiogenesis and gliogenesis. Neuroscience 202, 334–341 [DOI] [PubMed] [Google Scholar]
- 29.Ulbrich C., Zendedel A., Habib P., Kipp M., Beyer C., and Dang J. (2012). Long-term cerebral cortex protection and behavioral stabilization by gonadal steroid hormones after transient focal hypoxia. J. Steroid Biochem. Mol. Biol. 131, 10–16 [DOI] [PubMed] [Google Scholar]
- 30.Li M., Jia Q., Chen T., Zhao Z., Chen J., and Zhang J. (2016). The role of vascular endothelial growth factor and vascular endothelial growth inhibitor in clinical outcome of traumatic brain injury. Clin. Neurol. Neurosurg. 144, 7–13 [DOI] [PubMed] [Google Scholar]
- 31.Li L., Chopp M., Ding G.L., Qu C.S., Li Q.J., Lu M., Wang S., Nejad–Davarani S.P., Mahmood A., and Jiang Q. (2012). MRI measurement of angiogenesis and the therapeutic effect of acute marrow stromal cell administration on traumatic brain injury. J. Cereb. Blood Flow Metab. 32, 2023–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Damianovich M., Hout Siloni G., Barshack I., Simansky D.A., Kidron D., Dar E., Avivi C., and Onn A. (2013). Structural basis for hyperpermeability of tumor vessels in advanced lung adenocarcinoma complicated by pleural effusion. Clin. Lung Cancer 14, 688–698 [DOI] [PubMed] [Google Scholar]
- 33.Johanson C., Stopa E., Baird A., and Sharma H. (2011). Traumatic brain injury and recovery mechanisms: peptide modulation of periventricular neurogenic regions by the choroid plexus-CSF nexus. J. Neural Transm. 118, 115–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimopoulos S., Leitritz M.A., Ziemssen F., Voykov B., Bartz–Schmidt K. U., and Gelisken F. (2015). Submacular predominantly hemorrhagic choroidal neovascularization: resolution of bleedings under anti-VEGF therapy. Clin. Ophthalmol. 9, 1537–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin J.Y., Choi H.J., Chung B., Choi M., Lee J., and Byeon S.H. (2016). Anti-vascular endothelial growth factor with gas for submacular hemorrhage. Optom. Vis. Sci. 93, 173–180 [DOI] [PubMed] [Google Scholar]
- 36.Baskaya M.K., Rao A.M., Dogan A., Donaldson D., and Dempsey R.J. (1997). The biphasic opening of the blood–brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 226, 33–36 [DOI] [PubMed] [Google Scholar]
- 37.Cao F., Jiang Y., Wu Y., Zhong J., Liu J., Qin X., Chen L., Vitek M.P., Li F., Xu L., and Sun X. (2016). Apolipoprotein E-mimetic COG1410 reduces acute vasogenic edema following traumatic brain injury. J. Neurotrauma 33, 175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barzó P., Marmarou A., Fatouros P., Hayasaki K., and Corwin F. (1997). Contribution of vasogenic and cellular edema to traumatic brain swelling measured by diffusion-weighted imaging. J. Neurosurg. 87, 900–907 [DOI] [PubMed] [Google Scholar]
- 39.Gao W., Zhao Z., Yu G., Zhou Z., Zhou Y., Hu T., Jiang R., and Zhang J. (2015). VEGI attenuates the inflammatory injury and disruption of blood–brain barrier partly by suppressing the TLR4/NF-kappaB signaling pathway in experimental traumatic brain injury. Brain Res. 1622, 230–239 [DOI] [PubMed] [Google Scholar]
- 40.Bell R.D., Winkler E.A., Singh I., Sagare A.P., Deane R., Wu Z., Holtzman D.M., Betsholtz C., Armulik A., Sallstrom J., Berk B.C., Zlokovic B.V. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K., Liu B., and Ma J. (2014). Research progress in traumatic brain penumbra. Chin. Med. J. 127, 1964–1968 [PubMed] [Google Scholar]
- 42.Selwyn R., Hockenbury N., Jaiswal S., Mathur S., Armstrong R.C., and Byrnes K.R. (2013). Mild traumatic brain injury results in depressed cerebral glucose uptake: An (18)FDG PET study. J. Neurotrauma 30, 1943–1953 [DOI] [PubMed] [Google Scholar]
- 43.Foley N., Marshall S., Pikul J., Salter K., and Teasell R. (2008). Hypermetabolism following moderate to severe traumatic acute brain injury: a systematic review. J. Neurotrauma 25, 1415–1431 [DOI] [PubMed] [Google Scholar]
- 44.Keeney J.T., Ibrahimi S., and Zhao L. (2015). Human ApoE isoforms differentially modulate glucose and amyloid metabolic pathways in female brain: evidence of the mechanism of neuroprotection by ApoE2 and implications for Alzheimer's disease prevention and early intervention. J. Alzheimers Dis. 48, 411–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iliadis F., Kadoglou N., and Didangelos T. (2011) Insulin and the heart. Diabetes Res. Clin. Pract. 93, Suppl. 1, S86–91 [DOI] [PubMed] [Google Scholar]
- 46.Agudo J., Ayuso E., Jimenez V., Casellas A., Mallol C., Salavert A., Tafuro S., Obach M., Ruzo A., Moya M., Pujol A., Bosch F. (2012). Vascular endothelial growth factor–mediated islet hypervascularization and inflammation contribute to progressive reduction of b-cell mass. Diabetes 61, 2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hagberg C.E., Mehlem A., Falkevall A., Muhl L., Fam B.C., Ortsater H., Scotney P., Nyqvist D., Samen E., Lu L., Stone–Elander S., Proietto J., Andrikopoulos S., Sjoholm A., Nash A., and Eriksson U. (2012). Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature 490, 426–430 [DOI] [PubMed] [Google Scholar]
- 48.Wei K., Piecewicz S.M., McGinnis L.M., Taniguchi C.M., Wiegand S.J., Anderson K., Chan C., Mulligan K.X., Kuo D., Yuan J., Vallon M., Morton L.C., Lefai E., Simon M.C., Maher J.J., Mithieux G., Rajas F., Annes J.P., McGuinness O.P., Thurston G., Giaccia A.J., and Kuo C.J. (2013). A liver Hif-2alpha-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by VEGF inhibition. Nat. Med. 19, 1331–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foster L.J., and Klip A. (2000). Mechanism and regulation of GLUT-4 vesicle fusion in muscle and fat cells. Am. J. Physiol. Cell Physiol. 279, 877–890 [DOI] [PubMed] [Google Scholar]
- 50.van Berkel A., Rao J.U., Kusters B., Demir T., Visser E., Mensenkamp A. R., van der Laak J.A., Oosterwijk E., Lenders J.W., Sweep F.C., Wevers R.A., Hermus A.R., Langenhuijsen J.F., Kunst D.P., Pacak K., Gotthardt M., and Timmers H.J. (2014). Correlation between in vivo 18F-FDG PET and immunohistochemical markers of glucose uptake and metabolism in pheochromocytoma and paraganglioma. J. Nucl. Med. 55, 1253–1259 [DOI] [PubMed] [Google Scholar]