

Summary
p62/SQSTM1 (p62) is a scaffolding protein that facilitates the formation and degradation of ubiquitinated aggregates via its self-interaction and ubiquitin binding domains. The regulation of this process is unclear but may relate to the post-translational modification of p62. In the present study, we find that Keap1/Cullin3 ubiquitinates p62 at lysine 420 within its UBA domain. Substitution of lysine 420 with an arginine diminishes p62 sequestration and degradation activity similar to that seen when the UBA domain is deleted. Overexpression of Keap1/Cullin3 in p62-WT expressing cells increases ubiquitinated inclusion formation, p62’s association with LC3 and rescues proteotoxicity. This effect is not seen in cells expressing a mutant p62 that fails to interact with Keap1. Interestingly, p62 disease mutants have diminished or absent UBA domain ubiquitination. These data suggest that the ubiquitination of p62’s UBA domain at lysine 420 may regulate p62’s function and be disrupted in p62 associated disease.
Introduction
Autophagic degradation can be a selective process targeting distinct cargoes via autophagic receptor proteins (Rogov et al., 2014). p62/SQSTM1 (p62) is an autophagic receptor that interacts with ubiquitinated cargo via its ubiquitin association (UBA) domain and recruits them via its LC3-interacting motif into the growing autophagosome membrane (Bjorkoy et al., 2005; Komatsu et al., 2007; Pankiv et al., 2007). Autophagic degradation of ubiquitinated proteins requires p62 to sequester them into inclusion bodies. This property is mediated by p62’s UBA domain but also via a PB1 domain that facilitates homo-oligomerization (Bjorkoy et al., 2005; Ciuffa et al., 2015; Itakura and Mizushima, 2011).
The oligomerization of p62 may serve several different roles in both inclusion body formation and autophagy. For example, p62 oligomers may organize along with ubiquitinated proteins sequestering them into inclusions (Bjorkoy et al., 2005). In addition, p62 forms filamentous structures via its PB1 domain that may serve as a site for phagophore membrane development (Itakura and Mizushima, 2011). Interestingly, p62 filaments are fragmented in the presence of polyubiquitin (Ciuffa et al., 2015). This fragmentation is mediated via p62’s UBA domain and suggests that higher-ordered oligomeric p62 structures may dynamically change in the setting of ubiquitin.
Consistent with p62’s role in the handling of ubiquitinated protein aggregates, dominantly inherited missense or deletion mutations within p62’s UBA domain are associated with degenerative diseases including Paget’s disease of the bone (PDB), amyotrophic lateral sclerosis (ALS), fronto-temporal dementia and more recently inclusion body myopathy (Bucelli et al., 2015; Rea et al., 2014). The common pathogenic feature among these disparate tissues is the accumulation of p62 aggregates and ubiquitinated inclusions. How p62 mutations contribute to disease pathogenesis is unclear but may relate to diminished ubiquitin binding activity (Layfield et al., 2006).
The post-translational modification of p62 has been demonstrated to regulate its function. Phosphorylation at one of two different serines within p62’s UBA domain enhances its association with ubiquitinated proteins promoting sequestering activity and rescue from proteotoxic stress (Lim et al., 2015; Matsumoto et al., 2011). Ubiquitination of p62 also occurs. Mass spectrometry approaches have identified multiple ubiquitination sites on p62 that include lysine residues within the PB1 and UBA domains (Kim et al., 2011; Song et al., 2016).
Ubiquitination of p62 can also modulate its function. Recently, studies identified ubiquitin ligases that depending upon their site of ubiquitination inhibited or facilitated p62’s function (Heath et al., 2016; Jongsma et al., 2016; Pan et al., 2016). The E3 ubiquitin ligase TRIM21 ubiquitylates p62 at lysine 7 within it PB1 domain. This ubiquitination abrogates p62 oligomerization thus inhibiting p62’s sequestration activity (Pan et al., 2016). p62 is also ubiquitinated within its UBA domain by the E3 ligase RNF26 although the exact residue in the UBA domain was not determined. This ubiquitination event was proposed to enhance p62’s interaction with other ubiquitin adaptors such as, TOLLIP, thus facilitating vesicular cargo sorting (Jongsma et al., 2016). In addition, RNF166 ubiquitinates p62 at residues K91 and K189 (Heath et al., 2016). Interestingly, these events involve atypical ubiquitin chains that are K29- and K33-linked. RNF166 mediated ubiquitin ligase activity facilitates p62’s role in the xenophagic degradation of intracellular bacteria (Heath et al., 2016).
p62 also associates with E3 ligases to regulate cell signaling pathways. Keap1 is an E3 ligase adaptor that contains a BR-C, ttk and bab (BTB) domain at its N-terminus, which mediates interaction with Cullin3 (Cul3) (Furukawa and Xiong, 2005). One substrate of the Keap1/Cul3 complex is Nrf2. When Keap1 is destabilized by oxidative stress, Nrf2 stabilizes and translocates to the nucleus where it activates the expression of genes regulated by antioxidant response elements (Kobayashi et al., 2006). Several studies demonstrate that p62 binds to Keap1; sequestering it into aggregates under conditions of autophagic inhibition (Ichimura et al., 2013; Komatsu et al., 2010; Lau et al., 2010). This p62-Keap1 interaction titrates Keap1 away from Nrf2, stabilizing Nrf2 by decreasing Cul3 mediated ubiquitination and allowing Nrf2 mediated activation of the antioxidant response pathway.
In this study, we find that p62 is ubiquitinated at lysine 420 within its UBA domain. This ubiquitination is mediated by the Keap1/Cul3 E3 ligase complex. Moreover, mutation of lysine 420 or disease mutations within p62’s UBA domain affect its ubiquitination and diminish its sequestration activity. The ubiquitination of ubiquitin binding proteins such as p62 is emerging as an important regulatory mechanism for autophagic adaptor proteins.
Results
p62’s UBA domain is ubiquitinated at K420
p62 associates with ubiquitinated proteins in cells (Bjorkoy et al., 2005). We co-expressed HA-p62 and Flag-ubiquitin (Flag-Ub) in p62−/−mouse embryonic fibroblasts (MEFs) for 24 hours and then immunoprecipitated with an HA antibody. Both p62 and Flag-Ub were detected within the immunoprecipitate (Figure 1A). Interestingly, whereas Flag-Ub migrated as a high molecular weight (HMW) species in the cell lysate, it migrated at ~75kDa in the HA immunoprecipitant (Figure 1A). Moreover, an immunoblot of p62 from similar lysates demonstrated both a 65kDa band and several HMW species (Figure 1A). This data suggested that Flag-Ub was conjugated to p62. Treatment of cell lysates with the catalytic domain of ubiquitin specific protease 2 (Usp2cc) collapsed the HMW species above p62 and abolished the presence of Flag-Ub within the HA immunoprecipitant (Figure 1A). N-ethylmaleimide (NEM) increased the amount of ubiquitinated p62 and inhibited the effect of Usp2cc (Figure 1A).
Figure 1. p62 is ubiquitinated within its UBA domain.
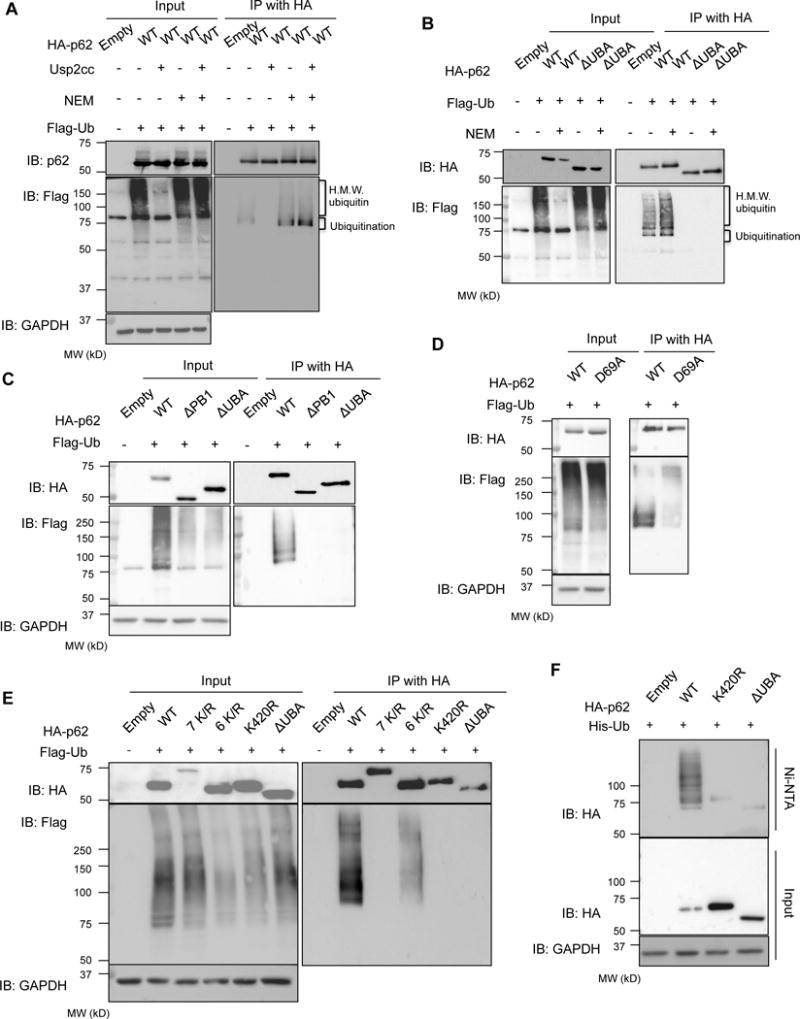
(A) anti-p62 and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62 and Flag-Ub. Some lysates were incubated with Usp2cc or NEM. (B) anti-p62 and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62-WT or HA-p62-ΔUBA and Flag-Ub. Some lysates were treated with NEM. (C) anti-HA and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62-WT, HA-p62-ΔPB1, or HA-p62-ΔUBA and Flag-Ub. (D) anti-HA and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62-WT or HA-p62-D69A along with Flag-Ub. (E) anti-HA and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA tagged p62-WT, p62-7K/R, p62-6K/R, p62-K420R or p62-ΔUBA and Flag-Ub. Anti-GAPDH serves as loading control for blots. (F) anti-HA immunoblots from p62−/−MEFs expressing HA, HA-p62-WT, HA-p62-K420R, or HA-p62-ΔUBA and histidine tagged ubiquitin (His-Ub). Anti-GAPDH serves as a loading control.
We co-expressed both Flag-Ub and a HA-p62 construct that lacks its UBA domain, p62-ΔUBA, and immunoprecipitated with an antibody to HA in p62−/−MEFs. p62-ΔUBA failed to co-immunoprecipitate any Flag-Ub even in the presence of NEM (Figure 1B). A similar experiment was performed that co-expressed Flag-Ub and a HA-p62 construct that lacks its N-terminal PB1 domain, p62-ΔPB1. Similar to p62-ΔUBA, p62-APB1 failed to co-immunoprecipitate any Flag-Ub (Figure 1C). To see if this related to a lack of a critical lysine residue such as K7 or K13 that have been previously reported to be ubiquitinated or related to a lack of p62 dimerization that is mediated via the PB1 domain, we performed the same experiment with HA-p62-D69A that fails to dimerize but maintains all N-terminal lysines (Figure 1D) (Pan et al., 2016; Song et al., 2016). Again, similar to p62-ΔPB1 no Flag-Ub was immunoprecipitated suggesting that the dimerization of p62 may be critical for the ubiquitination of p62’s C-terminal region.
To identify the principal lysine that is ubiquitinated on p62, we selected seven sites as determined by mass spectrometry analysis and previous proteomic studies (Kim et al., 2011; Mertins et al., 2013; Song et al., 2016; Wagner et al., 2011). We generated multiple HA-p62 mutant constructs with a lysine to arginine substitution (K/R; Figure S1). Notably, the HA tag does not contain a lysine. HA-p62 constructs were co-transfected with Flag-Ub into p62−/−MEFs for 24 hours and then immunoprecipitated with HA antibody in the presence of NEM. Mutation of all selected lysines, HA-p62-7K/R, abolished the ubiquitination of p62 as seen by the absence of Flag-Ub in the immunoprecipitant (Figure 1E). However, immunoprecipitation of p62 with mutations of 6 lysine residues (HA-p62-6K/R) that retains the C-terminal K420 residue demonstrates Flag-Ub in a pattern similar to p62-WT (Figure 1E). Changing the lysine at amino acid 420 to an arginine (HA-p62-K420R) diminished the amount of Flag-Ub in the immunoprecipitant similar to HA-p62-ΔUBA expression suggesting that the principal ubiquitinated lysine resides at K420 (Figure 1E).
To confirm that ubiquitin was indeed covalently attached to p62, we co-transfected HA-p62-WT, HA-p62-K420R or p62-ΔUBA with histidine tagged ubiquitin (His-Ub) into p62−/−MEFs for 24 hours and performed purification using a nickel charged affinity resin in the presence of 6M guanidine. Consistent with our results in RIPA buffer, only HMW HA-p62-WT was present in the affinity-purified lysate (Figure 1F).
p62’s UBA domain is ubiquitinated by Keap1/Cul3
Since p62 interacts with Keap1, an adaptor for the E3 ligase Cul3, we expressed HA-p62-WT, HA-p62-K420R and HA-p62-ΔUBA with Flag-Ub and Myc-Cul3 in p62−/−MEFs (Figure 2A) (Komatsu et al., 2010). After 24 hours, immunoblotting for p62 demonstrated an increase in ubiquitinated p62 in Myc-Cul3 expressing cells as evidenced by an increase in HMW p62 bands that were not present in HA-p62-K420R or HA-p62-ΔUBA expressing lysates (Figure 2A).
Figure 2. Cul3 complex ubiquitinates p62.
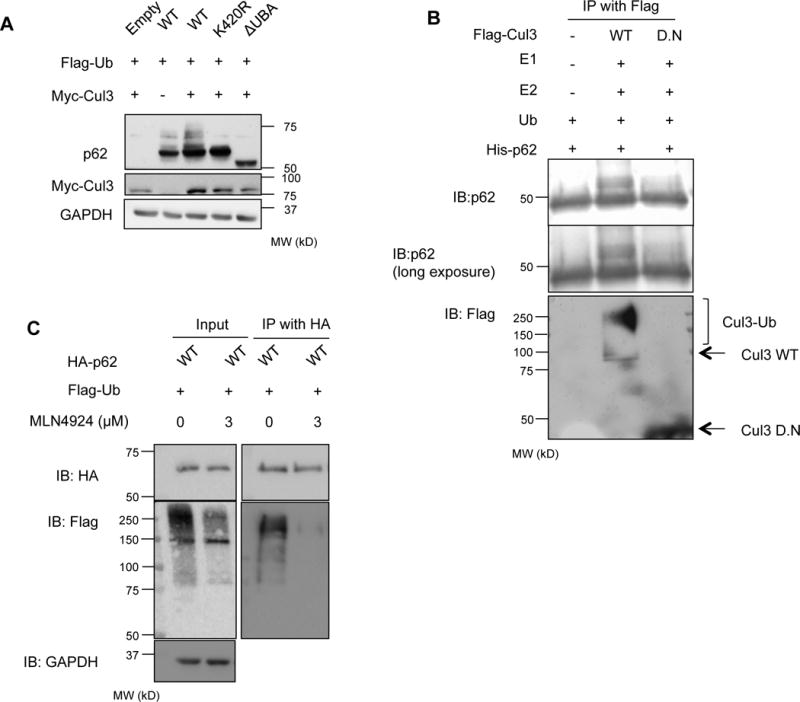
(A) anti-p62 and anti-myc immunoblots of lysates from p62−/−MEFs expressing HA tagged p62-WT, p62-K420R or p62-ΔUBA, Flag-Ub and myc-Cul3. (B) Anti-Flag and anti-p62 immunoblot of recombinant p62 (ΔN84) following in vitro ubiquitination with immunoprecipitated Flag-Cul3-WT or Cul3-DN from p62−/−MEFs. (C) anti-HA and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA tagged p62-WT and Flag-Ub. Some cells were treated with MLN4924. Anti-GAPDH serves as loading control for blots.
To further demonstrate that p62 could be ubiquitinated by Cul3 we transfected p62−/−MEFs with an expression construct containing a Flag tagged Cul3-WT or an inactive Cul3 that lacks its C-terminus (Cul3-DN) and immunoprecipitated Cul3 with an anti-Flag antibody. Purified His-p62 lacking the first 84 amino acids of p62 and thus does not contain two previously reported ubiquitination sites on p62 adjacent to the PB1 domain (K7 or K13) was incubated with ubiquitin, immunoprecipitated Flag-Cul3 and recombinant E1/E2 enzymes. Consistent with His-p62 ubiquitination there was an increase in its molecular weight when Flag-Cul3-WT and not Flag-Cul3-DN was included in the reaction (Figure 2B).
To function in ubiquitination reactions, Cul3 must be activated by, and bound to, NEDD8, in a process called neddylation (Singer et al., 1999). Cul3 activity can be inhibited using a nonspecific neddylation inhibitor, MLN4924 (Soucy et al., 2009). Treatment of p62−/−MEFs expressing HA-p62 and Flag-Ub with MLN4924 and subsequent immunoprecipitation of HA-p62 abolished p62 ubiquitination as assessed by Flag immunoblot (Figure 2C).
Since p62 associates with Keap1, an adaptor of the Cul3 ligase complex, we reasoned that p62 ubiquitination may be diminished in the absence of Keap1. We expressed HA-p62 and Flag-Ub in Keap1−/−MEFs with or without Myc-Keap1 for 24 hours and immunoprecipitated the lysates with an HA antibody. The amount of ubiquitinated p62 was increased when Myc-Keap1 was expressed in Keap1−/−MEFs (Figure 3A).
Figure 3. Keap1 modulates p62 ubiquitination.
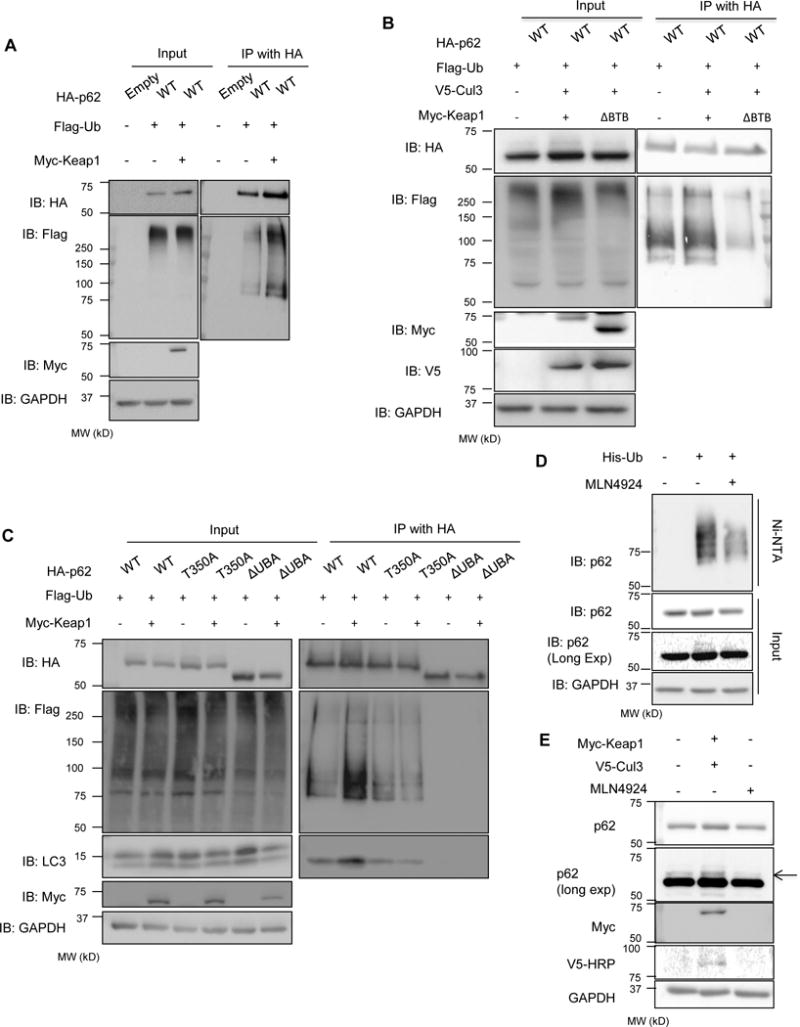
(A) Anti-HA and anti-Flag immunoblots of HA-immunoprecipitated lysates from Keap1−/−MEFs expressing HA-p62-WT, Flag-Ub and myc-Keap1. (B) Anti-HA and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62, Flag-Ub, V5-cul3 and myc-Keap1-WT or myc-Keap1-ΔBTB. (C) Anti-HA, anti-Flag, and LC3 immunoblots of HA-immunoprecipitated lysates from Keap1−/−MEFs expressing Flag-Ub, HA-p62-WT, HA-p62-T350A or HA-p62-ΔUBA with or without myc-Keap1. (D) anti-p62 immunoblot of nickel resin purified lysates from U20S cells expressing His-Ub and treated with DMSO or MLN4924. (E) Anti-p62 immunoblot of lysates from U20S cells with or without Myc-Keap1/V5-Cul3 expression or treated with MLN4924. Anti-GAPDH serves as loading control for blots.
Immunoprecipitation of HA-p62 in p62−/−MEFs expressing Flag-Ub, V5-Cul3 and Myc-Keap1 demonstrated more ubiquitinated p62 as compared with expression of HA-p62 and Flag-Ub alone (Figure 3B). This ubiquitination was abrogated when Myc-Keap1-ΔBTB that lacks its BTB domain, which is necessary for Cul3 association was expressed along with V5-Cul3 further suggesting that the Keap1/Cul3 complex mediates ubiquitination of p62 (Figure 3B).
p62 binds to Keap1 via its Keap1 interacting region (KIR). A single point mutation in p62, T350A, abolishes this interaction (Komatsu et al., 2010). To further establish that p62 ubiquitination is facilitated via Keap1, we expressed HA-p62-WT, HA-p62-T350A or HA-p62-ΔUBA along with Flag-Ub in Keap1−/−MEFs with or without Myc-Keap1 for 24 hours and immunoprecipitated the lysates with an HA antibody. Whereas Myc-Keap1 co-expression increased ubiquitinated HA-p62-WT as detected by Flag antibody, there was no augmentation of HA-p62-T350A when Myc-Keap1 was also expressed (Figure 3C).
Since the previous studies used an overexpressed human p62 construct in knockout mouse cells (see Figure S1B for level of overexpression as compared to control lines), we wanted to establish that endogenous p62 was indeed ubiquitinated. To do this, we expressed His-Ub in U20S cells treated with vehicle or MLN4924 for 24 hours and performed purification using a nickel charged affinity resin in the presence of 6M guanidine. Immunoblotting of the purified lysate with an antibody to p62 demonstrated a HMW smear in His-Ub transfected cells that was reduced when MLN4924 was added to the cells (Figure 3D). Similarly, we expressed Myc-Keap1 and V5-Cul3 in U20S cells or treated these cells with MLN4924. Lysates were then immunoblotted with a polyclonal antibody to p62. Myc-Keap1/V5-Cul3 co-expression increased the amount of a second band migrating above p62 and this band disappeared with MLN4924 treatment (Figure 3E).
UBA domain ubiquitination enhances p62’s sequestering activity
p62’s sequestering activity is required for ubiquitinated inclusion body formation in the setting of autophagic impairment and loss of p62 leads to a reduction of insoluble ubiquitinated proteins with a concomitant accumulation of soluble ubiquitinated proteins (Komatsu et al., 2007). To explore this function, we expressed control vector, HA-p62-WT, HA-p62-K420R and HA-p62-ΔUBA in p62−/−MEFs and fractionated cell lysates via ultracentrifugation (Figure 4A). In the absence of p62, ubiquitinated proteins remain in the supernatant and do not shift to the pelleted fraction. HA-p62-WT was enriched in the pelleted fraction along with HMW ubiquitinated proteins. In contrast, HA-p62-K420R and HA-p62-ΔUBA were enriched in the supernatant fraction and did not shift ubiquitinated proteins to the pelleted fraction.
Figure 4. Keap1/Cul3-mediated ubiquitination of p62 increases sequestering activity.
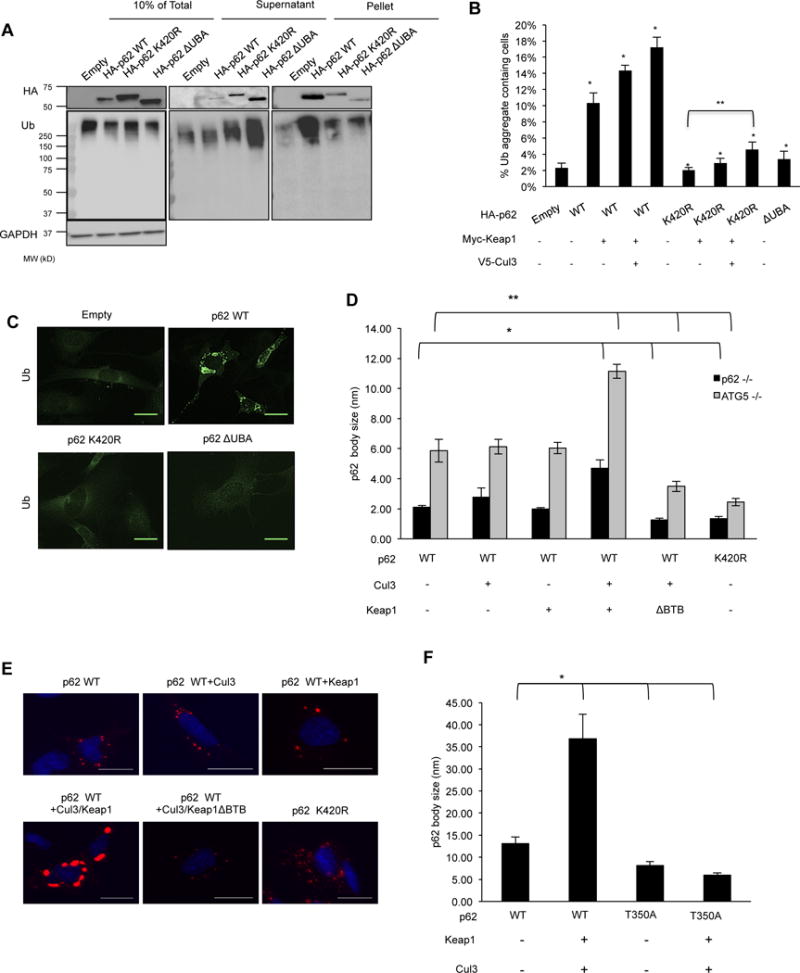
(A) Anti-HA and anti-ubiquitin immunoblots of cellular lysates (10% of total, supernatant and pellet) from p62−/−MEFs expressing empty HA vector, HA-p62-WT, HA-p62-K420R, or HA-p62-ΔUBA. (B) Quantitation of p62−/−MEFs expressing HA-p62-WT, HA-p62-K420R, HA-p62-ΔUBA or co-transfected with myc-Keap1 and myc-Keap1/V5-cul3 containing ubiquitinated inclusions. Representative data is pooled from four independent experiments. All data are represented as mean ± SEM. *p<0.05. (C) Anti-Ub immunostaining of p62−/−MEFs expressing empty vector, HA-p62-WT, K420R or ΔUBA. The scale bar indicates 1 μm. (D) Quantitation of p62 body size in p62−/− and ATG5−/− MEFs expressing mCherry tagged p62-WT or p62-K420R with myc-Keap1, myc-Keap1- ΔBTB and V5-Cul3. All data are represented as mean ± SEM. *p<0.05. (E) Representative images of ATG5−/−MEFs expressing Cherry-p62-WT or K420R along with Myc-Keap1/V5-Cul3. The scale bar indicates 1 μm. (F) Quantitation of p62 body size in MEFs expressing mCherry tagged p62-WT or p62-T350A with or without myc-Keap1/V5-cul3. All data are represented as mean ± SEM. *p<0.05.
Expression of HA-p62-WT in p62−/−MEFs generated multiple punctate ubiquitin positive inclusions throughout the cytoplasm. In contrast, HA-p62-ΔUBA and HA-p62-K420R had markedly fewer ubiquitin positive inclusions (Figure 4B–C). The co-expression of Myc-Keap1, V5-Cul3 or Myc-Keap1/V5-Cul3 increased the percentage of HA-p62-WT expressing cells containing ubiquitinated aggregates that did not occur in cells expressing HA-p62-K420R (Figure 4B and Figure S2). Similarly, we expressed mCherry-p62-WT and mCherry-p62-K420R in p62−/−MEFs or ATG5−/−MEFs and quantitated the average size of p62 bodies. mCherry-p62-WT formed larger p62 bodies and this was greater in ATG5−/−MEFs (Figure 4D–E). Co-expression of Myc-Keap1/V5-Cul3 further increased mCherry-p62-WT body size in both cell types but mCherry-p62-WT body size did not increase when Myc-Keap1-ΔBTB/V5-Cul3 was co-expressed. In control fibroblasts, co-expression of Myc-Keap1/V5-Cul3 increased mCherry-p62-WT body size but this augmentation did not occur with mCherry-p62-T350A expression (Figure 4F). Co-expression of Myc-Keap1/V5-Cul3 increased the size of endogenous p62 bodies, and MLN4924 treatment significantly decreased the size in U20S cells (Figure S3).
mCherry-p62-WT or p62-K420R was expressed in p62−/−MEFs and live cell imaging was performed to evaluate fluorescence recovery after photobleaching (FRAP) of individual puncta. The initial rapid phase of recovery has been suggested to correlate with p62’s sequestering activity (Matsumoto et al., 2011). The fluorescence exchange rate of mCherry-p62-K420R was more rapid than mCherry-p62-WT and co-expression of Myc-Keap1/V5-Cul3 decreased fluorescence recovery whereas co-expression of Myc-Keap1-ΔBTB/V5-Cul3 increased fluorescence recovery (Figure 5A–B). In a similar experiment, the co-expression of Myc-Keap1/V5-Cul3 failed to decrease the fluorescence recovery of mCherry-p62-T350A (Figure 5C). The differences in fluorescent recovery were not due to p62 presence within an autophagosome since an experiment performed in ATG5−/−MEFs gave similar results (Figure S4A–B).
Figure 5. p62 ubiquitination modulates its exchange rate.
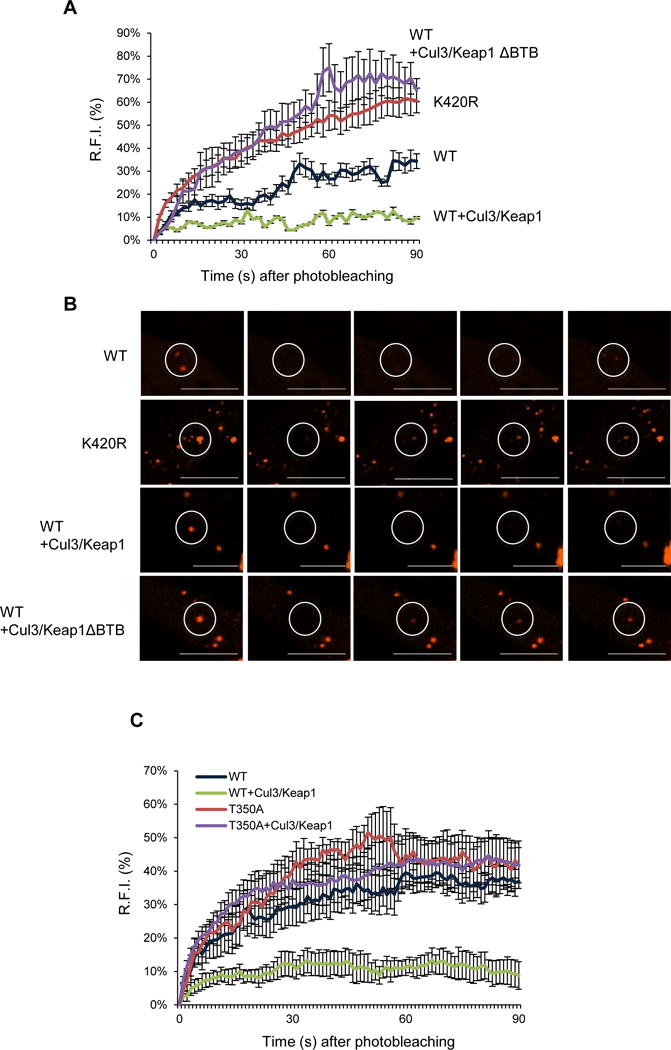
(A) The average RFI following photobleaching from p62−/−MEFs expressing mCherry-p62-WT or p62-K420R and V5-Cul3 with myc-Keap1-WT or myc-Keap1-ΔBTB. Representative data is pooled from four independent experiments. (B) Representative live cell images of mCherry-p62-WT or p62-K420R and V5-cul3 with myc-Keap1-WT or myc-Keap1-ΔBTB following photobleaching and fluorescent recovery. The circle indicates the photobleached area. The scale bars indicate 1 μm. (C) The average RFI following photobleaching from p62−/−MEFs expressing mCherry-p62-WT or p62-T350A and V5-Cul3 with myc-Keap1-WT. Representative data is pooled from three independent experiments.
UBA domain ubiquitination increases p62 degradation
We reasoned that the increased p62 exchange rate seen with mCherry-p62-K420R as compared with mCherry-p62-WT may reduce phagophore association with p62. To test this, we expressed HA-p62-WT or HA-p62-K420R with Flag-Ub in p62−/−MEFs for 24 hours. Cell lysates were immunoprecipitated with an anti-HA antibody and immunoblotted for HA and LC3B. HA-p62-K420R immunoprecipitants had less LC3BII as compared with HA-p62-WT (Figure 6A). Consistent with this, cell lysates immunoprecipitated with anti-HA from p62−/−MEFs expressing HA-p62-WT with Flag-Ub, Myc-Keap1 and V5-Cul3 in p62−/−MEFs had an increase in LC3BII association as observed on immunoblots probed with HA and LC3 (Figure 6B). Similarly, whereas Myc-Keap1/V5-Cul3 co-expression increased HA-p62-WT association with LC3B, this was not seen for HA-p62-T350A (Figure 3C).
Figure 6. Keap1/Cul3-mediated ubiquitination of p62 facilitates its role in autophagy.
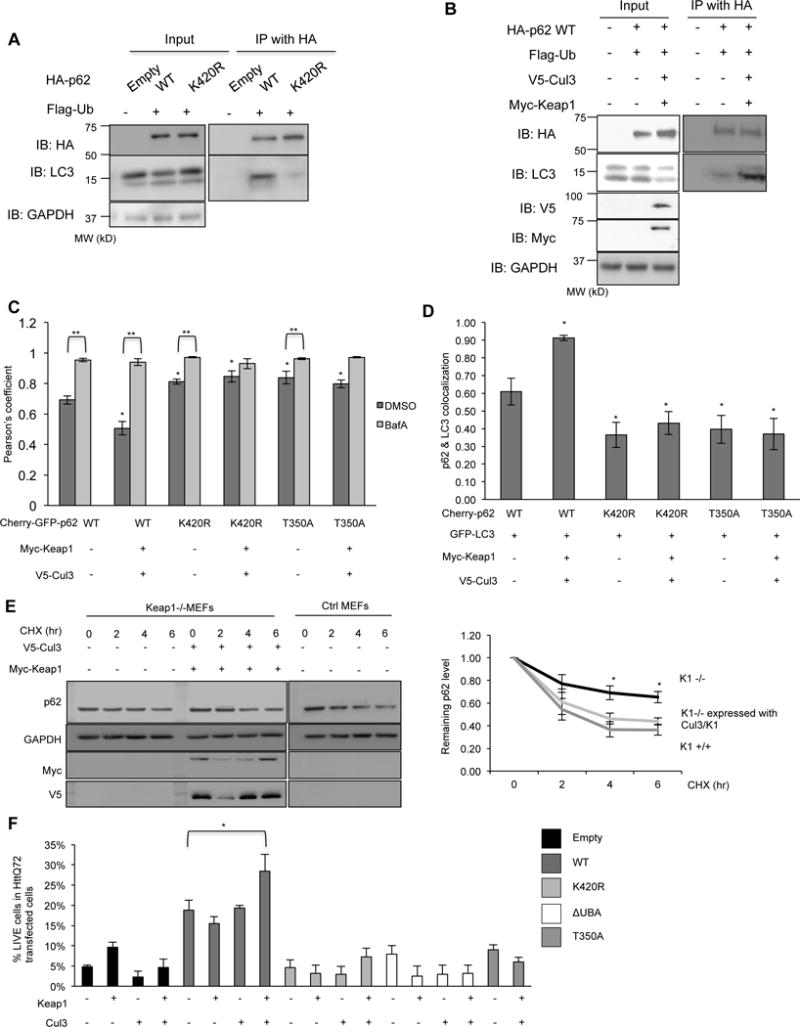
(A) Anti-HA and anti-LC3 immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62-WT or HA-p62-K420R and Flag-Ub. (B) Anti-HA and anti-LC3 immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62-WT and Flag-Ub with and without Myc-Keap1 and V5-Cul3. (C) Graph of the Pearson’s coefficient of co-localization of mCherry:GFP fluorescence in p62−/−MEFs expressing a dual fluorescent reporter mCherry-GFP fused to p62-WT, p62-K420R or p62-T350A with or without myc-Keap1 and V5-Cul3. Some cells were also with BafA. Representative data is pooled from three independent experiments. (D) Quantitation of the percent of mCherry-p62 bodies that co-localize with GFP-LC3 in p62−/−MEFs expressing mCherry fused p62-WT, p62-K420R or p62-T350A with or without Myc-Keap1 and V5-Cul3. Representative data is pooled from three independent experiments. (E) Anti-p62 immunoblot of lysates from control or Keap1−/−MEFs treated with cycloheximide for the indicated times. Some cells were also transfected with Myc-Keap1 and V5-Cul3. The percent of remaining p62 was quantified from three independent experiments and is represented graphically. (F) Quantitation of the percent of HttQ72-CFP positive p62−/−MEFs expressing HA-p62-WT, HA-p62-K420R, HA-p62-ΔUBA, T350A or co-expressing myc-Keap1 and V5-cul3. All data are represented as mean ± SEM. *p<0.05. Anti-GAPDH serves as loading control for blots. Representative data is pooled from three independent experiments.
To further explore the role of UBA domain ubiquitination by Keap1/Cul3 on p62’s degradation, we expressed tandem tagged mCherry-GFP-p62-WT in p62−/−MEFs for 24hours with and without BafilomycinA (BafA) (Figure 6C). When mCherry-GFP-p62-WT is in the cytosol as an inclusion body or within the non-acidic environment of an autophagosome, both mCherry and GFP fluorescence is present (Pankiv et al., 2007). However, when mCherry-GFP-p62-WT enters an acidic environment such as an autolysosome, GFP fluorescence quenches leaving only mCherry fluorescence. Consistent with this, the degree of mCherry:GFP co-localization was increased in BafA-treated cells as compared with untreated cells (Figure 6C and FigureS5A–B). In addition, co-expression of Myc-Keap1/V5-Cul3 with mCherry-GFP-p62-WT decreased the degree of mCherry:GFP co-localization suggesting that p62 ubiquitination enhances its autophagic degradation (Figure 6C). This effect was not seen in the presence of BafA.
The ratio of mCherry:GFP co-localization in p62−/−MEFs expressing mCherry-GFP-p62-K420R or mCherry-GFP-p62-T350A was significantly increased as compared to mCherry-GFP-p62-WT suggesting that fewer p62-K420R or p62-T350A puncta were within autolysosomes (Figure 6C). In contrast to mCherry-GFP-p62-WT puncta, there was not a decrease in mCherry:GFP co-localization with Myc-Keap1/V5-Cul3 expression in mCherry-GFP-p62-K420R or mCherry-GFP-p62-T350A expressing p62−/−MEFs (Figure 6C).
We reasoned that the increase in mCherry:GFP co-localization in p62−/−MEFs expressing mCherry-GFP-p62-K420R or mCherry-GFP-p62-T350A was due to its cytoplasmic accumulation in inclusion bodies rather than accumulation within autophagosomes. Consistent with this, p62−/−MEFs expressing mCherry-p62-K420R or mCherry-p62-T350A had a reduction in co-localization with co-expressed GFP-LC3B as compared to mCherry-p62-WT expressing p62−/−MEFs (Figure 6D). Moreover, expression of Myc-Keap1/V5-Cul3 increased the GFP-LC3B or endogenous LC3B co-localization with mCherry-p62-WT but failed to do so in mCherry-p62-K420R or mCherry-p62-T350A expressing p62−/−MEFs (Figure 6D and Figure S5C).
Further studies demonstrated that the stability of endogenous p62 was increased in Keap1−/−MEFs as compared with control MEFs when cell lysates were immunoblotted for p62 from cells treated with cycloheximide for varying times (Figure 6E). This increase in p62 stability was abrogated in Keap1−/−MEFs that were transfected with Myc-Keap1/V5-Cul3 expression constructs.
The expression of HttQ72-CFP in p62−/−MEFs is toxic with only ~5% of transfected cells remaining viable 24 hours post transfection (Figure 6F). This toxicity is rescued when HA-p62-WT is expressed but not by HA-p62-K420R, HA-p62-ΔUBA or HA-p62-T350A (Figure 6F). The expression of Myc-Keap1 and/or V5-Cul3 without HA-p62-WT in p62−/−MEFs did not significantly change HttQ72-CFP toxicity. However, the co-expression of Myc-Keap1/V5-Cul3 along with HA-p62-WT enhanced cell viability. This effect was not seen in the setting of HA-p62-K420R, HA-p62-ΔUBA or HA-p62-T350A (Figure 6F).
UBA domain ubiquitination in p62 is diminished with disease mutations
Some p62 disease mutations reside within its UBA domain (Figure 1C) (Rea et al., 2014). We expressed HA-p62-WT, p62-K420R and 5 different disease mutations, p62-A390X (equivalent to p62-ΔUBA used above), p62-P392L, p62-M404V, p62-G411S, and p62-G425R along with Flag-Ub in p62−/−MEFs (Figure 7A). Following 24 hours, we immunoprecipitated cell lysates with an HA antibody and immunoblotted for Flag and HA. Immunoprecipitants from HA-p62-WT but not HA-p62-K420R or HA-p62-A390X had Flag immunoreactive bands (Figure 7A). In addition, all expressed p62 disease mutants showed an absence or reduction in Flag immunoreactivity (Figure 7A). FRAP analysis of mCherry-p62-WT, p62-P392L, p62-M404V, p62-G411S or p62-G425R in p62−/−MEFs demonstrated that fluorescence recovery was more rapid with all disease mutants as compared with mCherry-p62-WT (Figure 7B and Figure S6). HA-p62-WT increased the number of live cells expressing HttQ72-CFP and this was augmented with co-expression of Keap1/Cul3 in p62−/−MEFs (Figure 7C). Whereas, expression of p62 disease mutants, with the exception of HA-p62-P392L, significantly decreased cell viability compared to HA-p62-WT. Co-expression of Keap1/Cul3 in p62 disease mutant-expressing cells reduced cell death.
Figure 7. p62 disease mutants have decreased ubiquitination and function.
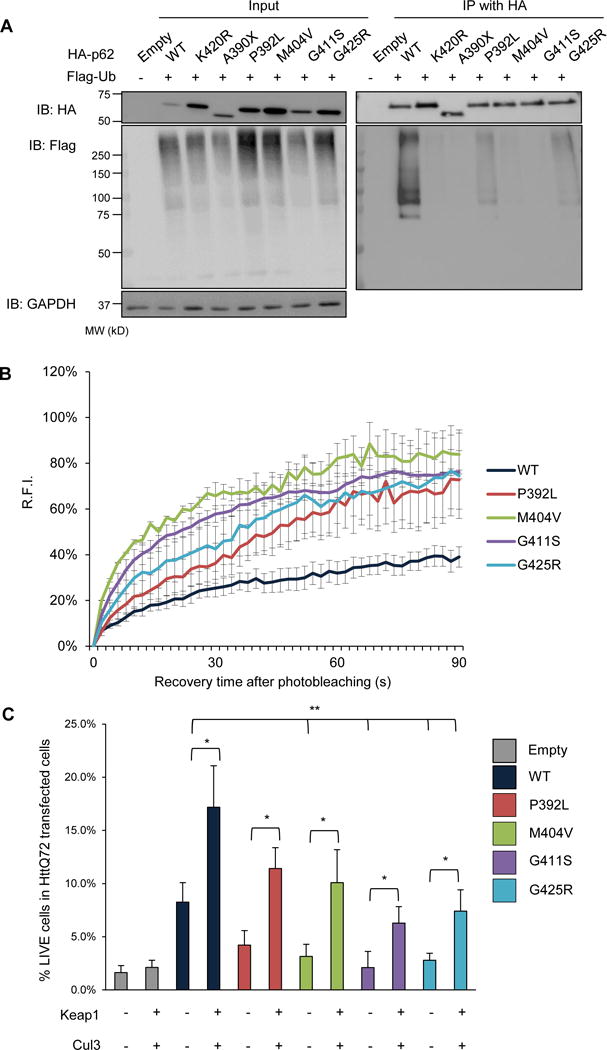
(A) Anti-HA and anti-Flag immunoblots of HA-immunoprecipitated lysates from p62−/−MEFs expressing HA-p62-WT, p62-K420R, p62-ΔUBA, p62-P392L, p62-M404V, p62-G411S, p62-G425R and Flag-Ub-WT. Anti-GAPDH is used as loading control. (B) The average RFI following photobleaching from p62−/−MEFs expressing mCherry-p62-WT, p62-P392L, p62-M404V, p62-G411S or p62-G425R. (C) Quantitation of the percent of HttQ72-CFP positive p62−/−MEFs expressing HA-p62-WT, p62-P392L, p62-M404V, p62-G411S, p62-G425R or co-expressing myc-Keap1 and V5-Cul3. All data are represented as mean ± SEM. *p<0.05. Representative data is pooled from three independent experiments.
Discussion
The present study finds that p62 is ubiquitinated at a highly conserved lysine within its UBA domain (K420) by Keap1/Cul3. Substitution of K420 to arginine, deletion of the UBA domain or mutation of the KIR domain in p62 decreases ubiquitinated inclusion formation, its LC3 interaction and cell viability. In contrast, Keap1/Cul3 expression can increase ubiquitinated inclusion formation, p62 inclusion body size and rescue cells from proteotoxic stress. Moreover, Keap1/Cul3 increases p62’s association with LC3B and its subsequent degradation. We propose that the ubiquitination of p62’s UBA domain by Keap1/Cul3 enhances its sequestration activity, subsequent phagophore association and degradation.
The molecular assembly of p62 oligomers is necessary for its function in autophagy. How this assembly is regulated is not fully understood. Recent studies support a model where the self-association of p62 occurs via its PB1 domain (Ciuffa et al., 2015). This interaction is a nidus for the extension of a filamentous p62 scaffold that allows the engulfing phagophore membrane to bind the LIR domain and degrade ubiquitinated proteins (Itakura and Mizushima, 2011). Deletion of the PB1 domain abrogates the self-association of p62 reducing p62’s sequestration activity and cellular function (Bjorkoy et al., 2005). Similarly, deletion of p62’s UBA domain impairs p62 body formation even in the setting of an intact PB1 domain. p62’s UBA domain can also self-associate as a dimer (Long et al., 2010). Phosphorylation of serines 403 and 409 within the UBA domain destabilize this dimer and regulates p62’s activity (Lim et al., 2015; Matsumoto et al., 2011). These studies suggest that p62’s molecular assembly is dependent upon both its PB1 and UBA domains.
The UBA domain of p62 has a shared interface that mediates dimerization and ubiquitin binding. Ubiquitin binding and UBA domain dimerization are mutually exclusive with dimerization “inactivating” ubiquitin binding (Long et al., 2010). Amino acid K420 resides at the C-terminus of helix 2. Structural studies have demonstrated that this region of p62’s UBA domain interacts with residues in loop 1. Indeed point mutations in loop 1 residues (E409K, G410K) or helix 2 (T419K) disrupt dimerization (Long et al., 2010). Whether ubiquitination of K420 disrupts dimerization allowing for increased ubiquitin binding is unclear. Alternatively, K420 ubiquitination may serve as means for increased oligomer formation via interaction of a p62 UBA domain with an adjacent p62’s ubiquitinated UBA domain or interactions with other proteins harboring ubiquitin-binding motifs.
More recently, it was demonstrated that polyubiquitin could fragment p62 filaments suggesting that the oligomeric structure of p62 was dynamic and could change with the addition of ubiquitinated cargo (Ciuffa et al., 2015). Our data extends this model and suggests that the ubiquitination status of p62’s UBA domain may alter p62’s oligomeric structure thus promoting inclusion body formation. This higher ordered p62 oligomer would then further promote p62’s sequestration activity, subsequent phagophore assembly and autophagic degradation. We suggest that by refining p62’s oligomeric structure via UBA domain ubiquitination, autophagic degradation and sequestration activity can be activated or inhibited locally within the p62 inclusion body.
The ubiquitination of autophagic adaptors is emerging as a means of regulating their function. Ubiquitination of optineurin by HACE1 increases its association with p62 enhancing autophagic flux and suppressing tumor growth (Liu et al., 2014). TRIM21 ubiquitinates p62 on its N-terminal PB1 domain inhibiting p62 oligomerization and sequestration activity (Pan et al., 2016). This event would antagonize Keap1/Cul3 mediated ubiquitination at the C-terminal UBA domain suggesting that the ubiquitination of p62 can both positively and negatively affect its sequestration function. Recently, the E3 ligase RPN26 was found to recruit and ubiquitinate p62’s UBA domain at the ER membrane (Jongsma et al., 2016). This event enhanced p62’s ability to capture a subset of endosomes via binding to ubiquitin binding domains on endocytic adaptors. Whether RPN26 ubiquitinates K420 or another lysine within the UBA domain is not established.
Previous studies have found that a Keap1 homodimer binds to two sites on Nrf2 facilitating Cul3 mediated ubiquitination. Oxidative stress destabilizes Keap1 and abrogates it interactions with Nrf2. Keap1 binds p62 at a single KIR domain. The affinity for this interaction becomes greater as p62 accumulates under conditions of impaired autophagy, sequestering Keap1 from Nrf2 leading to its stabilization (Jain et al., 2010; Komatsu et al., 2010; Lau et al., 2010). Similarly, Keap1 accumulates under conditions of impaired autophagy or in the setting p62 knockdown (Lau et al., 2013). Autophagic degradation of Keap1 through its interaction with p62 may help to maintain redox homeostasis (Watanabe et al., 2012). In fact, Keap1 itself enhances p62 mediated ubiquitin aggregate clearance (Fan et al., 2010). This may further serve to regulate Keap1 levels and its interactions with both Nrf2 and p62.
Our data suggests that Keap1/Cul3 can ubiquitinate p62 and enhance its autophagic activity. Only dimeric p62 is able to be ubiquitinated since p62 with a PB1 domain deletion or p62-D69A fail to be ubiquitinated (Figure 1C and 1D). This would suggest that a Keap1 homodimer binds to two high affinity KIR motifs on a p62 dimer leading to its ubiquitination. This model is similar to alternative models of Keap1 which suggests that a homodimer can associate with two Nrf2 molecules or with Nrf2 and a second protein with a KIR motif, such as PGAM5 which then targets this complex to the mitochondria (Lo and Hannink, 2006; Lo et al., 2006). Interestingly, loss of Keap1 stabilizes p62 whereas increased expression of Keap1/Cul3 increases p62 inclusion formation and subsequent degradation. Whether Keap1 is further degraded along with p62 is not known. However, Keap1/Cul3 mediated ubiquitination and subsequent enhancement of p62 degradation would be an efficient means by which Keap1 could decrease the levels of p62 thus releasing its antagonistic interaction.
Dominantly inherited mutations in p62 are associated with several degenerative diseases ranging from PDB to ALS (Rea et al., 2014). More recently, homozygous loss of function mutations in p62 were identified in patients with childhood onset neurodegeneration (Haack et al., 2016). Some p62 disease mutations reside within or lead to premature truncations of the UBA domain (Rea et al., 2014). One unifying feature for these mutations is variable degrees of impaired ubiquitin binding in vitro (Layfield et al., 2006). Our data finds that UBA domain associated p62 disease mutations also have a reduction in UBA domain ubiquitination. More recently, several p62 missense mutations associated with PDB and ALS have been identified within the Keap1 interacting region (Goode et al., 2016b; Wright et al., 2013). These mutations lead to a loss of Keap1 interaction and a reduction in Nrf2 signaling. It is intriguing to speculate that these mutations may also affect Keap1/Cul3 associated ubiquitination of p62’s UBA domain further unifying the pathogenic mechanism of p62 mutations. Alternatively, xenophagy associated phosphorylation of p62 at serine 349 can be disrupted by a point mutation within the UBA domain that diminishes its ubiquitin binding function (Ishimura et al., 2014). The phosphorylation of serine 349 on p62 then increases p62’s affinity for Keap1 (Ichimura et al., 2013). It is also conceivable that UBA domain associated disease mutations in p62 affect serine 349 phosphorylation abrogating Keap1/Cul3 mediated ubiquitination of the UBA domain.
The modulation of p62 activity via Keap1/Cul3 offers an attractive therapeutic target. Expression of Keap1/Cul3 increases p62’s sequestration activity. In the case of p62 associated disease mutations, Keap1/Cul3 expression restored p62’s cytoprotective role in polyglutamine toxicity. Whether Keap1/Cul3 mediated ubiquitination of p62 further enhances the sequestration of Keap1 resulting in the non-canonical activation of Nrf2, which may further enhance cell viability remains to be established. Indeed, activation of Nrf2 is cytoprotective in models of polyglutamine toxicity (Gan and Johnson, 2014). Our present data further connects p62-mediated autophagy with the Keap1-Nrf2 signaling pathway.
Experimental Procedures
Immunoprecipitation and Western blotting
Cells were harvested in NP-40 lysis buffers (50mM Tris pH7.7, 150mM NaCl, 1% NP-40) or RIPA buffers (50mM Tris pH 7.7, 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) with protease inhibitor cocktails (PIC; Sigma#P8340), 20mM NEM and phenylmethylsulfonyl fluoride (PMSF; Sigma#P7626) and washed with ice-chilled PBS. Lysates were centrifuged for 15mins at 4C°, 16,000g. The supernatants were collected. Some lysates were incubated with Usp2cc (1μg Usp2cc per 100μg cell lysates; R&D #075604). 200μg of cell lysates were pre-cleared for 1 hr with end-over-end rotator with 10μg of dynabeads M-270 Epoxy (Life Technologies #14302D) at 4°C. Samples were placed on a magnet and the supernatants were collected. The supernatants were incubated with antibody-conjugated Dynabeads on the rotator at 4°C for 2hrs. For antibody conjugation, dialyzed antibodies were coupled to Dynabeads M-270 Epoxy beads (Life Technologies #14311D) according to the manufacturer’s instruction. After 2hr incubation, the samples were placed on the magnet and washed three times with lysis buffer.
In the case of denaturing conditions, the assay was performed as previously described (Pan et al., 2016). Briefly, 10% of sample was kept as “input” samples. The rest of samples were mixed with equal volume of 6M guanidine buffer (6M guanidine-HCl, 100mM sodium phosphate, 600mM NaCl, 30mM imidazole, 0.05% Tween-20, pH 8.0). The mixed samples were pre-cleared for 1 hr on end-over-end rotator with 10μg of Dynabeads M-270 Epoxy at 4C°. The samples were placed on a magnetic stand and the supernatants were collected. The supernatants were incubated with HisPur Ni-NTA Magnetic Beads (Thermo Sci#88831) on the rotator for 2hrs at 4C°. The samples were placed on a magnetic stand and washed twice with 8M urea buffer (8M urea, 100mM sodium phosphate, 10mM Tris-Base, 50mM imidazole, pH 8.0). After all urea buffer completely removed, the samples containing beads were incubated with elution buffer (250mM imidazole, 100mM sodium phosphate, 600mM NaCl) for 30mins on a rotator. The samples were mixed with (β-mercaptoethanol containing sample buffer and boiled at 95°C for 5mins. The Western blot analysis was performed as previously described (Weihl et al., 2006).
In vitro ubiquitination assay
Recombinant E2 enzyme (UbcH5a/UBE2D1; #E2-616) and ubiquitylation kit containing E1 enzyme (#K-995) were purchased from Boston Biochem. Recombinant fragment of Histidine tagged human p62 (#p62-29505TH) containing amino acids 83-440 and was purchased from Biomart. Cells were transfected with Cul3 WT-Flag or Cul3-D.N.-Flag. 24hrs after, cells were harvested in RIPA buffer and immunoprecipitated with Flag-antibody-conjugated Dynabead for 2hrs at 4°C. The samples were washed three times with lysis buffer and incubated with E1, E2, ubiqutin and His-p62 (500ng) at 37°C for 1 hr. The samples were mixed with β-mercaptoethanol containing sample buffer and boiled at 95°C for 5mins.
Fractionation assay
p62−/−MEFs were grown in 10cm dishes at ~80% confluency. After three washes with ice-chilled PBS, cells were harvested in RIPA buffer with PIC, PMSF, and NEM. The cells were sonicated for 10s, two times at 4°C. 10% of sample was taken from the lysate as a ‘total’ fraction. The remaining sample was centrifuged at 100,000g for 30mins at 4°C. After centrifugation, the supernatant was transferred to a new tube and labeled as a ‘supernatant’ fraction. The rest of sample was resuspended in 200μl of 7M urea buffer (7M urea, 2M thiourea, 4% CHAPS, 30mM Tris, pH 8.5) and re-sonicated for 10s, two times at 4°C. The samples were re-centrifuged at 100,000g for 30mins at 4°C. The supernatant was transferred to a new tube and labeled as a ‘pellet’ fraction and the debris was discarded.
Cell viability assay
p62−/−MEFs grown at ~80% confluency were co-transfected with HttQ72-CFP and HA-p62. 24hr after the transfection, cells were washed three times with PBS. The assay was performed according to the manufacturer’s instruction (Life Technologies #L-3224).
Immunocytochemistry and fluorescence microscopy
Cells were grown on glass coverslips prior to transfection with plasmid constructs. 24hrs after the transfection, cells were washed three times with PBS, fixed in 4% PFA for 10mins and permeabilized with 0.1% Triton X-100 in PBS for 10mins. After washing three times with PBS, cells were blocked with 2% BSA in PBS for 30mins-1hr at room temperature (RT). Cells were stained with primary antibody at 4°C overnight followed by three-times washing with PBS. Cells were incubated with Alexa 555 or 488 Fluor-conjugated secondary antibody at RT for 1hr and mounted with Mowiol media containing DAPI. 10 random fields were taken with 20× objective equipped in a NIKON Eclipse 80i fluorescence microscopy. Co-transfected cells were counted as a total number of cells, and the cells containing ubiquitin-positive aggregates were counted using ImageJ software (NIH). The images were presented in pseudo-color. For Cherry-eGFP-p62 assay, p62−/−MEFs were transfected with Cherry-eGFP-p62-WT, K420R, or T350A along with or without Myc-Keap1/V5-Cul3 for 24hrs. Cells were washed two times with PBS and mounted with Mowiol media containing DAPI without fixation to avoid pH disruption (Goode et al., 2016a). Pearson’s coefficient between red and green channel was measured by ImageJ.
Fluorescence recovery after photobleaching
p62−/−MEFs were grown on 1.5 Glass bottom 35mm dishes (MatTek #P35G-1.5-10-C) and transfected with pDest-mCherry-p62 for 24hrs prior to imaging. Immediately before imaging, the medium was replaced with Phenol Red-free medium (Gibco #21063029) containing 10% FBS, 50 μg/mL P/S, 1% sodium pyruvate, and 1% non-essential amino acid. The live cell samples were placed on a heated chamber at 37°C with 5% CO2. The imaging and photobleaching were performed with 40× oil objective using an NIKON A1Rsi confocal microscopy. Before bleaching, the images of pre-bleached p62 bodies were taken. 561nm laser was used to photobleach p62 bodies for 500ms. Immediately after the bleaching, the images were collected every 2s for a total of 5mins as a post-bleached sample. The fluorescence intensities of post-bleached p62 bodies were individually measured. In the meantime, the fluorescence intensities of non-bleached p62 bodies were measured as a control. An average of p62 bodies in different cells (n≥7 cells containing p62 bodies) was calculated. The relative florescence intensities were calculated as the average fluorescence intensities of photobleached p62 bodies divided by the average fluorescence intensities of pre-bleached samples.
Cycloheximide assay
Keap1−/− or control MEFs was transfected at ~80% confluency with empty vector or Myc-Keap1/V5-Cul3. 24hrs after the transfection, cells were treated with 10μg/mL cycloheximide for varying time.
Statistical analysis
A two-tailed Student’s t-test was performed. For all tests, p-values<0.05 were considered statistically significant. Data are represented as mean ± SEM.
Supplementary Material
Acknowledgments
Funding included NIH AG031867 (C.C.W.), NIH AG042095 (C.C.W.), Muscular Dystrophy Association (C.C.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Y.L. and C.C.W designed and wrote the study. Y.L. conducted most of experiments with support from S.K.P. and A.L.K. T.F.C. reviewed the paper. B.R. offered reagents.
References
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM, Evila A, Hackman P, Udd B, Harms MB, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 2015;85:665–674. doi: 10.1212/WNL.0000000000001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuffa R, Lamark T, Tarafder AK, Guesdon A, Rybina S, Hagen WJ, Johansen T, Sachse C. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015;11:748–758. doi: 10.1016/j.celrep.2015.03.062. [DOI] [PubMed] [Google Scholar]
- Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, Zhu M, Zhong Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy. 2010;6:614–621. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Johnson JA. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim Biophys Acta. 2014;1842:1208–1218. doi: 10.1016/j.bbadis.2013.12.011. [DOI] [PubMed] [Google Scholar]
- Goode A, Butler K, Long J, Cavey J, Scott D, Shaw B, Sollenberger J, Gell C, Johansen T, Oldham NJ, et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy. 2016a;12:1094–1104. doi: 10.1080/15548627.2016.1170257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode A, Rea S, Sultana M, Shaw B, Searle MS, Layfield R. ALS-FTLD associated mutations of SQSTM1 impact on Keap1-Nrf2 signalling. Mol Cell Neurosci. 2016b;76:52–58. doi: 10.1016/j.mcn.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Ignatius E, Calvo-Garrido J, Iuso A, Isohanni P, Maffezzini C, Lonnqvist T, Suomalainen A, Gorza M, Kremer LS, et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am J Hum Genet. 2016;99:735–743. doi: 10.1016/j.ajhg.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath RJ, Goel G, Baxt LA, Rush JS, Mohanan V, Paulus GL, Jani V, Lassen KG, Xavier RJ. RNF166 Determines Recruitment of Adaptor Proteins during Antibacterial Autophagy. Cell Rep. 2016;17:2183–2194. doi: 10.1016/j.celrep.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Ishimura R, Tanaka K, Komatsu M. Dissection of the role of p62/Sqstm1 in activation of Nrf2 during xenophagy. FEBS Lett. 2014;588:822–828. doi: 10.1016/j.febslet.2014.01.045. [DOI] [PubMed] [Google Scholar]
- Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongsma ML, Berlin I, Wijdeven RH, Janssen L, Janssen GM, Garstka MA, Janssen H, Mensink M, van Veelen PA, Spaapen RM, et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell. 2016;166:152–166. doi: 10.1016/j.cell.2016.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol. 2013;33:2436–2446. doi: 10.1128/MCB.01748-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield R, Cavey JR, Najat D, Long J, Sheppard PW, Ralston SH, Searle MS. p62 mutations, ubiquitin recognition and Paget’s disease of bone. Biochem Soc Trans. 2006;34:735–737. doi: 10.1042/BST0340735. [DOI] [PubMed] [Google Scholar]
- Lim J, Lachenmayer ML, Wu S, Liu W, Kundu M, Wang R, Komatsu M, Oh YJ, Zhao Y, Yue Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11:e1004987. doi: 10.1371/journal.pgen.1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, Xu X, Wang H, Yang M, Liu X, et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. 2014;26:106–120. doi: 10.1016/j.ccr.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem. 2006;281:37893–37903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Garner TP, Pandya MJ, Craven CJ, Chen P, Shaw B, Williamson MP, Layfield R, Searle MS. Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF-kappaB signalling. J Mol Biol. 2010;396:178–194. doi: 10.1016/j.jmb.2009.11.032. [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, Yang B, Chen JS, Catanzaro JM, Du C, et al. TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol Cell. 2016;61:720–733. doi: 10.1016/j.molcel.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations–bridging Paget disease of bone and ALS/FTLD. Exp Cell Res. 2014;325:27–37. doi: 10.1016/j.yexcr.2014.01.020. [DOI] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167–178. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999;13:2375–2387. doi: 10.1101/gad.13.18.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Li S, Wu H, Gao R, Rao G, Wang D, Chen Z, Ma B, Wang H, Sui N, et al. Parkin promotes proteasomal degradation of p62: implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson’s disease. Protein Cell. 2016;7:114–129. doi: 10.1007/s13238-015-0230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10:M111013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Tatebe H, Taguchi K, Endo Y, Tokuda T, Mizuno T, Nakagawa M, Tanaka M. p62/SQSTM1-dependent autophagy of Lewy body-like alpha-synuclein inclusions. PLoS One. 2012;7:e52868. doi: 10.1371/journal.pone.0052868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Dalal S, Pestronk A, Hanson PI. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum Mol Genet. 2006;15:189–199. doi: 10.1093/hmg/ddi426. [DOI] [PubMed] [Google Scholar]
- Wright T, Rea SL, Goode A, Bennett AJ, Ratajczak T, Long JE, Searle MS, Goldring CE, Park BK, Copple IM, et al. The S349T mutation of SQSTM1 links Keap1/Nrf2 signalling to Paget’s disease of bone. Bone. 2013;52:699–706. doi: 10.1016/j.bone.2012.10.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.