

Abstract
The selective clearance of organelles by autophagy is critical for the regulation of cellular homeostasis in organisms from yeast to humans. Removal of damaged organelles clears the cell of potentially toxic byproducts and enables reuse of organelle components for bioenergetics. Thus, defects in organelle clearance may be detrimental to the health of the cells, contributing to cancer, neurodegeneration and inflammatory diseases. Organelle-specific autophagy can clear mitochondria, peroxisomes, lysosomes, endoplasmic reticulum, chloroplasts and the nucleus. Here, we review our understanding of the mechanisms that regulate the clearance of organelles by autophagy and highlight gaps in our knowledge of these processes.
eTOC
The selective clearance of organelles by autophagy is critical for the regulation of cellular homeostasis in organisms from yeast to humans. Defects in organelle clearance can be detrimental and contribute to cancer, neurodegeneration and inflammatory diseases. Anding and Baehrecke review the understanding of the regulation of organelles clearance by autophagy.
Introduction
Organelles are specialized structures within a cell that carryout specific functions that are critical for its function and survival. Cells modulate the number of organelles or degrade parts of organelles according to functional and energetic needs. Thus, it is crucial that overall organelle health and number be tightly regulated. Both surplus and dysfunctional organelles are cleared through an evolutionarily conserved, catabolic self-eating process called macroautophagy (hereafter autophagy), which is the focus of this review. Here we will discuss only the autophagic degradation of membrane-bound organelles.
Autophagy involves the sequestration of components of the cytoplasm into double membrane vesicles called autophagosomes. Autophagosomes then fuse with lysosomes (vacuoles in yeast and plants) to degrade their contents. This degradation process is critical to maintain cellular homeostasis, ridding the cell of either excess or damaged organelles, aggregated proteins, or pathogens. This recycling process also provides energy for the cell. Defects in autophagy have been linked to various human disorders, including cancer, neurodegeneration and inflammatory diseases (Mizushima et al., 2008; Ravikumar et al., 2010b). Autophagy is active at basal levels and this is thought to be important for the maintenance of cellular homeostasis, clearing the cell of damaged organelles or debris. Autophagy can also be activated by cellular stresses such as either starvation, hypoxia, or chemotherapeutic drugs, triggering a cell survival process that provides the cell with energy during times of energy depletion. In addition, programmed autophagy is triggered by cell signals during development, but how these signals integrate with core autophagy regulatory factors remains largely unclear. Although autophagy is most often thought of as a cell survival process, autophagy can also promote cell death in specific contexts, including during development (Anding and Baehrecke, 2015).
Autophagy was long considered to be a nonselective degradation pathway. This is likely because early genetic studies of autophagy were conducted under nutrient limiting conditions. However, recent research has revealed that autophagy is also critical for the selective degradation of specific cargos, such as organelles and proteins. In this review, we will summarize the recent findings on the selective autophagy of organelles. We will focus on both the stimuli that activate autophagy and the proteins critical for cargo recognition and clearance. In general, selective autophagy starts with a cellular signal that tags the organelle, often through ubiquitination, for destruction (Figure 1). Cargo recognition proteins then detect the cargo to be degraded and bring the cargo through either direct or indirect interaction with Atg proteins to the forming autophagosome membrane, known as the phagophore. Despite the common mechanisms involved in organelle clearance, the degradation cues and molecules used in selective autophagy are diverse and specific for each organelle involved. Having a thorough understanding of these pathways is crucial as defects in the selective autophagy of various organelles have been associated with conditions such as neurodegeration (Nixon, 2013), aging (Nordgren et al., 2013), and renal injury (Maejima et al., 2013). In addition, a detailed understanding of how cargoes are recognized may provide valuable information that can be used in the development of therapeutics to specifically target autophagy sub-routines for precision medicine.
Figure 1. Selective autophagy.
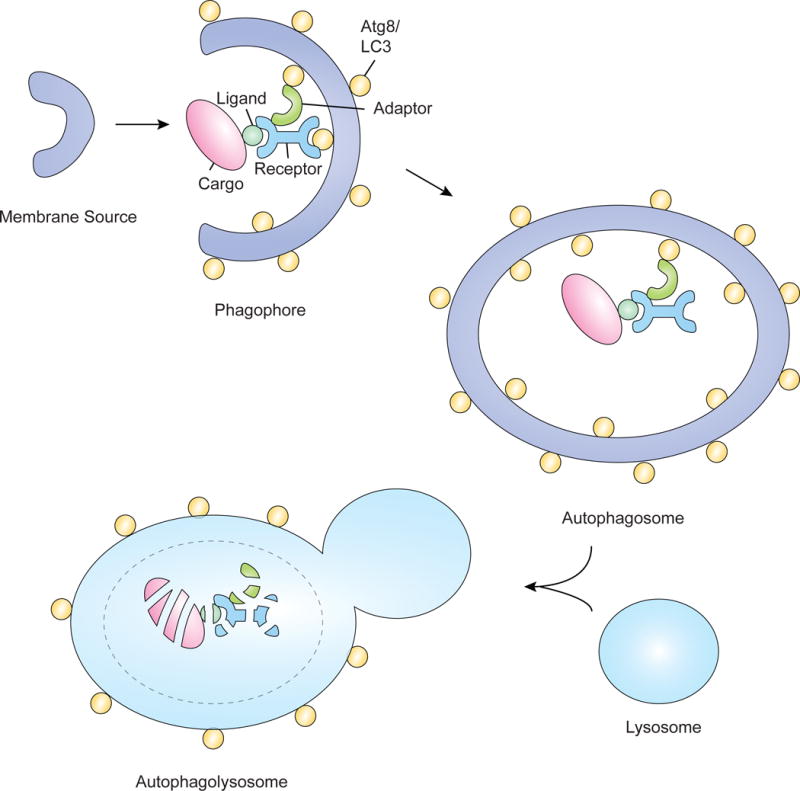
In selective autophagy, cargo, such as organelles, are degraded by the autophagolysosome. Cargo are often tagged with ligands such as ubiquitin, enabling them to interact with receptor proteins that can then interact with the autophagosomal membrane. This interaction occurs by binding to Atg proteins such as Atg8/LC3 or through interaction with adaptor proteins. After the cargo is brought to the phagophore, formation of the autophagosome occurs and this structure can then fuse with the lysosome to form the autophagolysosome, degrading the cargo.
Regulation of Autophagy
Upon induction of autophagy, proteins of the Atg1 complex, including Atg1, Atg13, Atg17, Atg29 and Atg31 in yeast, assemble at the pre-autophagosomal structure (PAS) to form the isolation membrane, also known as the phagophore (Kamada et al., 2000; Matsuura et al., 1997; Suzuki et al., 2001). In humans, this complex consists of ULK1/2, ATG13, and FIP200, which assemble with the unique ATG101 protein (Kamada et al., 2000). The formation of this complex triggers the initiation of autophagosome formation. In yeast, the PAS is likely to be assembled de novo (Kovács et al., 2007). In mammals, however, the origin of the autophagosomal membrane is debated. Though nucleation of the isolation membrane has been shown to occur at a site originating from the endoplasmic reticulum (ER) called the omegosome (Axe et al., 2008; Hayashi-Nishino et al., 2009), other sources of membrane have also been shown to contribute to autophagosome formation, including ER-Golgi intermediate compartments (Ge et al., 2013), ER-mitochondria junctions (Hamasaki et al., 2013), mitochondria (Hailey et al., 2010), Golgi-endosomal membranes (Ohashi and Munro, 2010) and the plasma membrane (Ravikumar et al., 2010a).
Following the initiation step of autophagy, the nucleation stage involves the class III phosphatidylinositol (PtdIns) 3-kinase (PI3K) complex consisting of Beclin-1 (Atg6 in yeast), VPS34 (class III PI3K), VPS15 (also called p150 in mammals or ird1 in Drosophila), and ATG14 (Itakura et al., 2008). This complex promotes local production of phosphatidyl-inositol 3-phosphate specific to autophagosomes and recruits effector proteins such as Atg18/WIPI1/2 to the PAS (Obara et al., 2008). Atg18 forms a complex with Atg2 that functions in autophagosome formation and controls the morphology of vesicles and PtdIns 3,5-bisphosphate homeostasis in complex with other proteins (Efe et al., 2007). Finally, the elongation step involves both the elongation and closure of the isolation membrane and is regulated by two ubiquitin-like conjugation systems. The first is the Atg12-Atg5-Atg16 complex, in which the E1-like Atg7 (Tanida et al., 1999) and the E2-like Atg10 (Shintani et al., 1999) conjugate the ubiquitin-like Atg12 to Atg5. This Atg12-Atg5 conjugate then interacts with Atg16 to function as the Atg12-Atg5-Atg16 complex (Mizushima et al., 1998). The second ubiquitin-like conjugation system functions to add a phosphatidylethanolamine (PE) lipid to Atg8 (LC3 in mammals). First, Atg8 is processed by the protease Atg4 (Poole et al., 2008). Atg8 is then conjugated to PE by Atg7 and the E2-like Atg3 enzymes. Evidence also exists that the Atg12-Atg5 complex produced in the first ubiquitin-like conjugation system possesses an E3-like activity for efficient lipidation of Atg8 (Kanki et al., 2009; Okamoto et al., 2009). Lipidation of Atg8/LC3 facilitates its interaction with the forming autophagosomal membrane which facilitates cargo recruitment to autophagosomes, and allows autophagosome maturation steps such as expansion and closure of the isolation membrane to occur (Geisler et al., 2010).
When initiation, elongation, and expansion have completed, the autophagosome closes and can fuse with lysosomes to form the autophagolysosome. Fusion of the autophagosome vesicle with either late endosomes or lysosomes involves a set of SNARE proteins including syntaxin-17 (STX17), SNAP29, and VAMP8 (Itakura et al., 2012; Takáts et al., 2013). The activity of the SNARE complex is controlled by ATG14 which directly binds to the STX17-SNAP29 binary complex on autophagosomes and promotes STX17-SNAP29-VAMP8-mediated autophagosome fusion (Diao et al., 2015). It was recently shown that EPG5 acts at this step as a tethering factor, allowing the assembly and stabilization of SNAREs and determining the fusion specificity of autophagosomes with late endosomes/lysosomes (Wang et al., 2016). Once fusion is complete, the autophagolysosome contents are degraded.
Selective Autophagy of Organelles
The selective autophagy of organelles is critical for the maintenance of cellular homeostasis both by maintaining organelle integrity and number in the context of varying environments and stresses. The clearance of organelles, known as organellophagy, differs from the bulk degradation process that occurs in starvation-induced autophagy, for example, in that it is a selective process involving the specific sequestration of cellular components. Though the types of organellophagy discussed in this review are diverse, they all involve initiation by a signal that induces downstream events triggering degradation cues for a specific target, molecules that tag the target as cargo to be degraded, and autophagy-related components that sequester and eliminate the cargo. Several organelles including mitochondria, peroxisomes, lysosomes, the endoplasmic reticulum, nucleus, and chloroplasts have all been identified as cargo that can be degraded via autophagy in various taxa.
Mitophagy
The selective degradation of mitochondria by autophagy is called mitophagy (Lemasters, 2005). Mitochondria are the powerhouses of the cell as they generate most of the cell’s adenosine triphosphate (ATP), which is used as a source of chemical energy. Mitochondria are also critical for processes such as programmed cell death (Green and Kroemer, 2004). Consequently, damage and subsequent dysfunction of mitochondria can lead to a wide range of disorders due to the impact on cellular metabolism and production of reactive oxygen species (ROS). Thus, clearance of damaged mitochondria is critical for the overall health of the cell. Additionally, because of their role in metabolism, mitochondria are thought to be cleared due to shifts in energy demands of the cell. The clearance of mitochondria, whether due to damage, shifting energy demands, or programmed maturation of the cell such as with the loss of mitochondria in reticulocytes, is carried out by autophagy.
Mitophagy can be triggered by various stimuli that result in mitochondrial damage, such as hypoxia (Bellot et al., 2009; Zhang et al., 2008), chemical uncouplers (Narendra et al., 2008), and ROS (Frank et al., 2012; Zhang et al., 2008) (Figure 2). Additionally, mitophagy can occur in response to developmentally programmed changes in the cell, such as in mammalian reticulocytes (Schweers et al., 2007) and the enterocyte cells of the Drosophila intestine midgut (Chang et al., 2013). Mitophagy is also critical for the elimination of paternal mitochondria from fertilized oocytes during C. elegans embryogenesis (Rawi et al., 2011)(Sato and Sato, 2011). The best-studied mitophagy pathway involves Pink1 and Parkin (Matsuda et al., 2010; Narendra et al., 2008; 2010a; Vives-Bauza et al., 2010), both genes known to be linked to familial Parkinson’s disease (Kitada et al., 1998; Valente et al., 2004). PINK1, a mitochondrial serine/threonine-protein kinase, is normally imported into the mitochondrial inner membrane where it is cleaved by PARL. When mitochondrial membrane potential has been compromised, PINK1 fails to be imported and accumulates on the surface of the mitochondria. This accumulated PINK1 phosphorylates various targets, including ubiquitin (Lazarou et al., 2015), and recruits and activates the E3 ubiquitin ligase, Parkin (Koyano et al., 2014). Parkin then acts as an amplifier of the PINK1-generated mitophagy signal, ubiquitinating mitochondrial surface proteins that can be recognized by cargo receptor proteins that bring the mitochondria to forming autophagosomes for degradation (Sarraf et al., 2013). Parkin-independent mitophagy can also occur. For example, loss of Drp1 in mouse cardiomyocytes led to increased mitochondrial ubiquitination and p62 mitochondrial targeting independent of Parkin (Kageyama et al., 2014). Additionally, upon mitophagy induction, AMBRA1 localizes to damaged mitochondria through LIR motif-dependent interactions with LC3, promoting both canonical PARKIN-dependent and –independent mitochondrial clearance (Strappazzon et al., 2015).
Figure 2. Mitophagy.
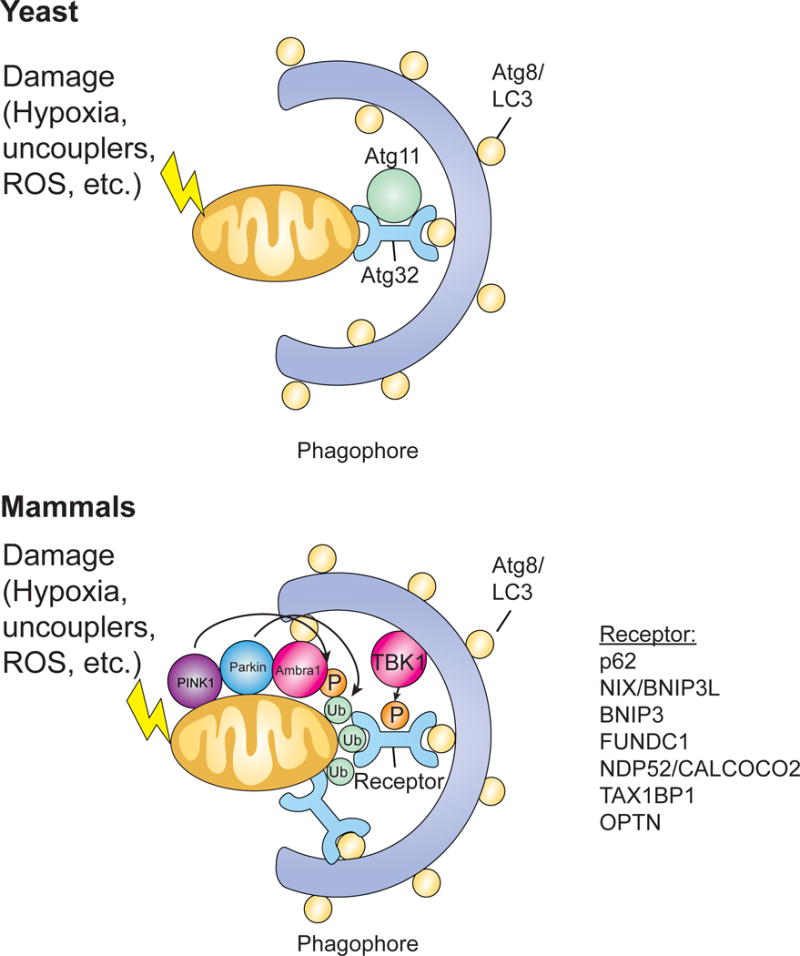
Mitophagy can be triggered by various stimuli, such as hypoxia, uncouplers, or ROS, that cause damage to mitochondria. In yeast, Atg32 acts as the mitophagy receptor, binding the adaptor protein Atg11 and interacting with Atg8 on the inner membrane of autophagosomes. In mammals, Pink1 phosphorylates various targets, including ubiquitin, and recruits Parkin which can then amplify the signal by ubiquitinating mitochondrial surface proteins. These ubiquitinated proteins can then be recognized by cargo receptor proteins that bring the mitochondria to forming autophagosomes for degradation. Mitochondrial surface receptor proteins can also recognize LC3 to facilitate recruitment to the phagophore. AMBRA1 also localizes to damaged mitochondria through LIR motif-dependent interactions with LC3, promoting both canonical PARKIN-dependent and –independent mitochondrial clearance. Additionally, TBK1 phosphorylates autophagy receptors to create a signal amplification loop in mitophagy.
PINK1 may also play a role in mitochondrial quality control through regulation of mitochondrial fission (Yang et al., 2008). In mammals, mitochondrial fusion is mediated by the fusion proteins mitofusin 1 (Mfn1), Mfn2, and optic atrophy 1 (OPA1), and fission is mediated by dynamin-related protein 1 (Drp1) (Youle and van der Bliek, 2012). In Drosophila, mutations in both Drp1 and PINK1 are lethal and over-expression of Drp1 can rescue flies deficient in either PINK1 or Parkin, implicating fission in mitophagy (Poole et al., 2008). Additionally, PINK1 has been shown to trigger displacement of protein kinase A (PKA) from A-kinase anchoring protein 1 (AKAP1) that is associated with the outer mitochondrial membrane, preventing phosphoinhibition of DRP1 and ensuring requisite fission of damaged mitochondria for organelle degradation (Pryde et al., 2016). AMPK has also been implicated in mitochondrial dynamics, as it is activated by mitochondrial stress and triggers mitochondrial fission, at least in part by phosphorylation of MFF (Toyama et al., 2016). AMPK is also known to phosphorylate and activate ULK1, thus providing a link between fission and the activation of mitophagy.
In the yeast S. cerevisiae, Atg32 acts as the mitophagy receptor (Kanki et al., 2009; Okamoto et al., 2009), binding the adaptor protein Atg11 and bringing the mitochondria to the vacuole, the equivalent of the animal lysosome. In addition to interacting with Atg11 through its coiled-coil domain, Atg32 also has an Atg8-interacting motif (AIM; also LC3 interacting region or LIR) near the N terminus that allows it to interact with Atg8 on the inner membrane of autophagosomes. In mammals, multiple receptors have been implicated in mitophagy, including p62/SQSTM1 (Geisler et al., 2010), NIX/BNIP3L (Novak et al., 2010; Zhang and Ney, 2008), BNIP3 (Zhang and Ney, 2009), FUNDC1 (Liu et al., 2012a), NDP52 (CALCOCO2), TAX1BP1 and optineurin (OPTN; (Lazarou et al., 2015)). Whether p62/SQSTM1 is necessary for the PINK1/Parkin mitophagy pathway is inconclusive, as the data are conflicting (Geisler et al., 2010; Lazarou et al., 2015; Narendra et al., 2010b; Okatsu et al., 2010). Lazarou et al. (2015) used genome editing to knock out five autophagy receptors (pentaKO) in HeLa cells and each receptor was re-expressed in pentaKOs to determine which receptors were necessary for mitophagy. Single, double, and triple knockout cell lines were also generated, and it was determined that while p62 and NBR1 are dispensable for Parkin-mediated mitophagy, OPTN and NDP52 are the primary, yet redundant receptors. In neuronal cells, cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy, with the interaction between LC3 and cardiolipin facilitated by LC3’s cardiolipin binding sites (Chu et al, 2013). Clearly the role of certain receptors in mitophagy is context dependent and more studies are needed to determine which receptors are involved in various conditions. Additionally, it was recently shown that Tank-binding kinase 1 (TBK1) phosphorylates autophagy receptors to create a signal amplification loop during mitophagy (Richter et al., 2016).
Once PINK1 accumulates on the mitochondrial surface and phosphorylates several targets, including ubiquitin, Parkin is recruited and amplifies the signal by ubiquitination of other mitochondrial surface-associated proteins. This then promotes the recruitment of ubiquitinbinding mitophagy receptors. p62 interacts with ubiquitin through its UBA domain, which interacts preferentially with K63-polyUb chains (Seibenhener et al., 2004). The UBAN domain of OPTN is essential for its interaction with ubiquitin and binds to a variety of ubiquitin chains, including K63- and M1-linked (Richter et al., 2016). NDP52 and TAX1BP1 utilize their ubiquitinbinding zinc finger (UBZ) domains to interact with ubiquitin and both of these proteins can bind M1-, K48- and K63-linked ubiquitin chains (Tumbarello et al., 2015; Wild et al., 2011). Other mitophagy receptor protiens, such as Nix, FUNDC1, BNIP3, and Atg32 are mitochondrial outer membrane proteins and, thus, do not require interaction with ubiquitin as they interact with the mitochondrion directly. Deubiquitinases (DUBs) such as USP30 have been shown to antagonize parkin-dependent mitophagy (Bingol et al., 2014). Overexpression of USP30 removes ubiquitin attached by Parkin onto mitochondrial proteins thereby blocking mitophagy, and reduction of USP30 leads to enhanced mitochondrial degradation in neurons.
In order to recruit mitochondria to the forming autophagosome for degradation, these receptor proteins must also facilitate interaction with the autophagosomal membrane. Each of these receptors contains LC3 interacting regions (LIRs) that facilitate their interaction with LC3-like molecules on the autophagosome. In yeast, Atg32 interacts with Atg8 indirectly through Atg11 and directly through its LIR in the N-terminal cytosolic domain (Kondo-Okamoto et al., 2012). In mammalian cells, the three outer mitochondrial membrane mitophagy receptors, Nix (Novak et al., 2010), FUNDC1 (Liu et al., 2012a), and BNIP3 (Hanna et al., 2012) all have a LIR in their cytosolic N-terminal domains that have been implicated in mitophagy. p62 (Ichimura et al., 2008; Noda et al., 2008; Pankiv et al., 2007) and OPTN (Wong and Holzbaur, 2014) also possess LIR motifs whereas TAX1BP1 and NDP52 both possess non-canonical LIR motifs (Rogov et al., 2014) that facilitate their interaction with LC3.
Finally, although mitophagy is traditionally thought to clear entire mitochondria, there are also mechanisms whereby mitochondrial fragments can be delivered to the lysosome directly through mitochondrial derived vesicles (MDVs) (Sugiura et al., 2014). Potentially any cargo could be included in these MDVs, provided they are first oxidized. Also, like mitophagy, this process is dependent on PINK1 and Parkin (McLelland et al., 2014). In addition, the SNARE syntaxin-17 and the homotypic fusion and vacuole protein sorting tethering complex is involved in the targeting of Parkin/PINK1-generated MDVs to endolysosomal compartments for turnover (McLelland et al., 2016).
While much is known about the initiation and mechanisms of mitophagy, there are still many remaining questions. For example, does phosphorylated ubiquitin activate ubiquitin ligases other than Parkin? Are other E3 ligases involved in Parkin-independent mitophagy? After recruitment of mitochondria to the forming autophagosomal membrane, what recruits other autophagy machinery components and initiates formation of the autophagosome around mitochondria? Also, is the machinery required for autophagosome formation in mitophagy similar to that used in starvation-induced autophagy? Finally, does a mechanism exist to exclude reasonably good mitochondria from this process, such as a mechanism involving USP30 or another DUB?
Pexophagy
Peroxisomes are small organelles involved in the catabolism of branched and long chain fatty acids and in the reduction of reactive oxygen species, specifically hydrogen peroxide, among other functions (Tripathi and Walker, 2016). Because of their important role in clearing toxic ROS, peroxisome homeostasis is critical, and this includes the formation of new peroxisomes as well as the removal of damaged peroxisomes. The degradation of peroxisomes by autophagy is known as pexophagy (Katarzyna and Suresh, 2016; Oku and Sakai, 2016). In the yeast Pichia pastoris, pexophagy is triggered by a shift in nutrient conditions, such as from methanol to glucose or ethanol media in which peroxisomal metabolism is not critical for viability and cell growth (Manjithaya et al., 2010; Oku and Sakai, 2016). Treatment of cultured cells or animals with hypolipidemic drugs, phthalate esters, or non-classical peroxisome proliferators, such as 4-phenylbutyrate (Till et al., 2012), in addition to amino acid starvation (Sargent et al., 2016), induces pexophagy (Figure 3).
Figure 3. Pexophagy.
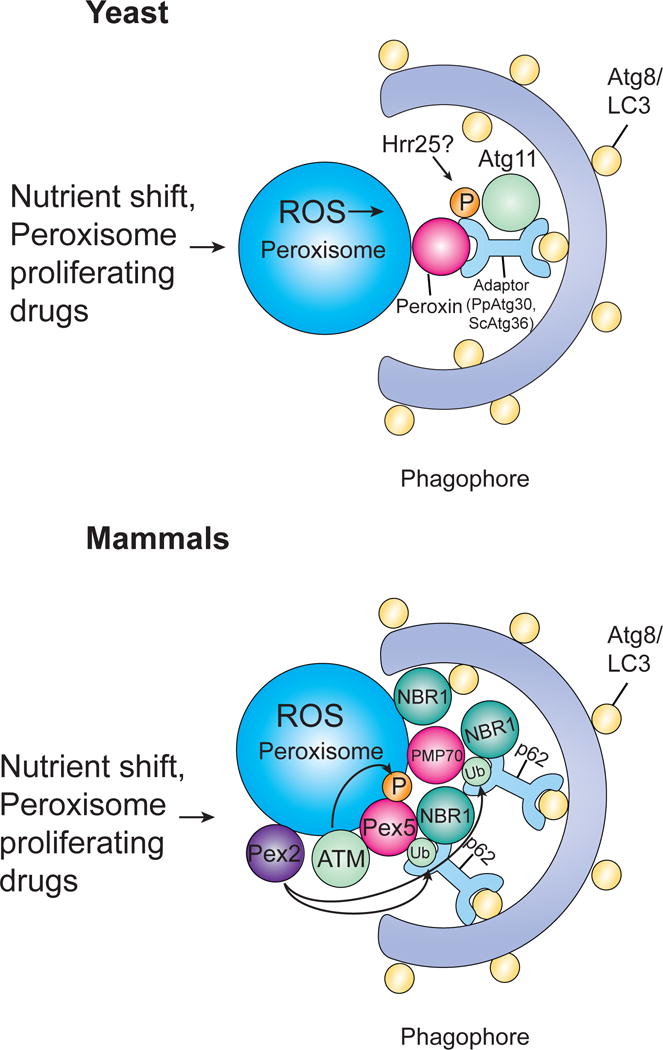
Pexophagy can be triggered by either a shift in nutrient conditions or peroxisome proliferating drugs. In yeast, Atg30 (P. pastoris) or Atg36 (S. cerevisiae) acts as the autophagy receptor, binding peroxisomes via their interaction with at least one peroxin and recruiting autophagic machinery by interacting with Atg proteins. Both Atg30 and Atg36 are phosphorylated (Atg36 by Hrr25), allowing them to interact with Atg11. In mammals, pexophagy has been shown to involve either NBR1 or p62. NBR1 binds peroxisomes through its JUBA domain which allows it to interact with phospholipids and ubiquitin. p62 supports and cooperates with NBR1 to bind ubiquitinated proteins. ATM kinase activates pexophagy in response to ROS by phosphorylating PEX5 leading to ubiquitination of PEX5 which can then be recognized by p62, targeting peroxisomes for pexophagy. PMP70 has also been implicatd in pexophagy and both PMP70 and Pex5 are ubiquitinated by Pex2.
As with other selective autophagy pathways, pexophagy requires specific cargo receptor(s) and/or adaptors. The first pexophagic adaptor protein to be identified in P. pastoris was Atg30 (Farré et al., 2008). In S. cerevisiae, Atg36 was found to be the adaptor protein for pexophagy (Motley et al., 2012). Although Atg30 and Atg36 possess limited protein sequence similarity, they share a conserved AIM and Atg11 binding site (Farré et al., 2013). Both Atg30 and Atg36 attach to peroxisomes via their interaction with at least one peroxin, such as Pex3, Pex5 or Pex14, and recruit autophagic machinery by interacting with Atg proteins. Both Atg30 and Atg36 are phosphorylated, allowing them to interact with Atg11 and, thus, assisting in the biogenesis of the autophagic membrane structure targeting the peroxisomes. Atg36 is phosphorylated by the kinase Hrr25 facilitating its interaction with the autophagosomal membrane (Tanaka et al., 2014). Mammals do not have Atg30 or Atg36 homologs, but instead use NBR1 and p62 as pexophagy adaptors (Katarzyna and Suresh, 2016). In COS-7 cells, pexophagy occurs in a p62-dependent manner (Kim et al., 2008) and down-regulation of either NBR1 or p62 has been shown to suppress degradation of peroxisomes in HeLa cells (Deosaran et al., 2013). An increase in ubiquitinated proteins on the surface of peroxisomes results in their targeting to autophagosomes for degradation (Kim et al., 2008). NBR1 binds to peroxisomes through its JUBA domain (a combination of a J and UBA domain), which binds to peroxisomes through interactions with ubiquitin (Deosaran et al., 2013). p62 supports and cooperates with NBR1 to bind ubiquitinated peroxisomes through its UBA domain and enhances the efficiency of degradation in NBR1-induced pexophagy (Deosaran et al., 2013; Yamashita et al., 2014; Zhang et al., 2015). Overexpression of PEX3 facilitates ubiquitination of peroxisomes and subsequent pexophagy, implicating it in this process (Yamashita et al., 2014). Additionally, the NBR1 LIR domain is necessary for pexophagy upon PEX3 overexpression. More recent studies have implicated the ubiquitination of mammalian PEX5 (Nordgren et al., 2015; Zhang et al., 2015) as well as PMP70 (Sargent et al., 2016) in pexophagy. In response to ROS, ATM kinase has also been implicated in the activation of pexophagy (Zhang et al., 2015). ATM that is activated by ROS phosphorylates PEX5, leading to monoubiquitination of peroxisome-localized PEX5 which can then be recognized by p62, targeting the peroxisomes for pexophagy. During amino acid starvation conditions, PEX2, but not PEX10 or PEX12, acts as the E3 ubiquitin ligase to selectively ubiquitinate peroxisomal membrane proteins to designate peroxisomes for autophagy-mediated degradation (Sargent et al., 2016).
Recent studies have provided insight into how pexophagy is triggered in mammalian cells, and how dysfunctional peroxisomes are recognized by the pexophagy machinery. Several questions remain to be addressed, however, and it will be interesting to compare differences in the mechanisms that regulate clearance of different organelles, such as peroxisomes and mitochondria. For example, how does the ubiquitination of PEX5 lead to pexophagy, as various residues can be ubiquitinated on PEX5 leading to different fates (Subramani, 2015). Also, though PMP70 is ubiquitinated by PEX2 during amino acid depletion conditions, it is unclear whether ubiquitination of PMP70 alone induces pexophagy. Additionally, other peroxisomal membrane proteins may also be ubiquitinated during amino acid depletion, as multiple ubiquitination bands were observed during PEX2 overexpression (Sargent et al., 2016). Is the ubiquitination of multiple proteins required for pexophagy?
Lysophagy
The lysosome is an acidic, membrane-bound organelle that is responsible for the degradation of unwanted intracellular materials. This degradation is carried out by a large variety of hydrolytic enzymes contained within the lysosome. Destabilization of the lysosome and the leaking of these enzymes into the cytoplasm is detrimental to the cell (Aits and Jäättelä, 2013). Additionally, if damaged lysosomes that lack digestive ability are not removed, the total number of intracellular lysosomes does not alter even if some of them become dysfunctional and, thus, cells are unable to maintain cellular homeostasis. Therefore, in order to protect the cell and maintain cellular homeostasis, the cell degrades damaged lysosomes by an autophagic process called lysophagy (Hung et al., 2013; Maejima et al., 2013).
Lysophagy can be induced by a variety of stimuli that lead to lysosomal damage (Figure 4). Experimentally, lysophagy can be initiated using photochemical internalization, a process that facilitates gene delivery through light-induced lysosome breakage (Hung et al., 2013). Several substances including mineral crystals such as silica and monosodium urate, bacterial or viral toxins, lipids, β-amyloid, lysosomotropic compounds, and proteases are also known to impair lysosomal membranes in vivo (Aits and Jäättelä, 2013; Maejima et al., 2013). Although studies of the mechanisms that control lysophagy are limited, it has been shown that upon lysosomal injury, galectin-3 and LC3 are recruited to the injured lysosome (Maejima et al., 2013). Interestingly, damaged lysosomal membranes are galectin-3-positive, ubiquitinated and co-localize with p62 in mouse embryonic fibroblasts (MEFs) (Maejima et al., 2013). In addition, a similar association of ubiquitin and p62 has been associated with damaged lysosomes in HeLa cells (Hung et al., 2013). These results suggest that ubiquitination and subsequent recruitment of the cargo adapter protein p62 are involved in this process.
Figure 4. Lysophagy.
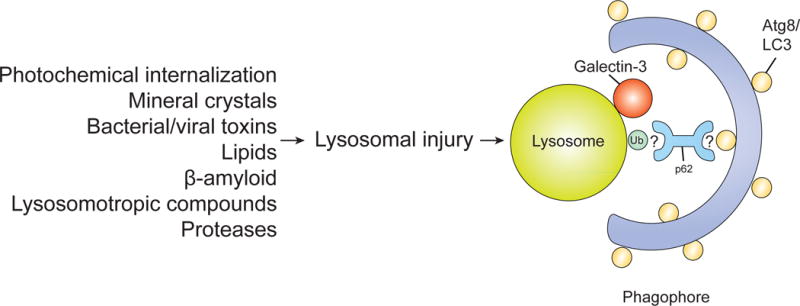
Lysophagy can be induced by a variety of stimuli that can lead to lysosome damage. Upon lysosomal injury, galectin-3 and LC3 are recruited to the injured lysosome. Lysosomal membranes are ubiquitinated and co-localize with p62, suggesting that ubiquitination and subsequent recruitment of p62 are involved in this process. However, the exact mechanisms of lysophagy regulation are still unclear.
Many questions about the molecular details of lysophagy remain. For example, what triggers the recruitment of galectin-3 and LC3 to the lysosomal membrane? Although ubiquitin and p62 are associated with damaged lysosomes, is p62 acting as either a classical cargo receptor protein, or in some other capacity such as a signaling scaffold protein? These early studies of lysophagy suggest similarities to mitophagy and the clearance of other organelles, and thus functional and biochemical analyses of the role of ubiquitin, ubiquitin receptors, and factors that modulate their activities should enhance our understanding of this process.
ERphagy (Reticulophagy)
The endoplasmic reticulum (ER) is a network of membranous tubules within the cytoplasm of a cell that is important in protein and lipid synthesis, as well as calcium storage. Both secretory and integral membrane proteins are folded and modified in the ER and homeostatic control of this process is critical for the overall health of the cell. Central to this control are the unfolded protein response (UPR) and ER-associated degradation (ERAD) pathways, which are activated upon the accumulation of unfolded proteins in the ER (Wang and Kaufman, 2016). The UPR is a signaling cascade aimed at both the elimination of misfolded proteins that accumulate in the organelle and increasing its folding capacity (Wang and Kaufman, 2012). In contrast, ERAD recognizes misfolded proteins and retrotranslocates these proteins into the cytoplasm where they are degraded by the ubiquitin-proteasome system (Ruggiano et al., 2014). Additionally, autophagy is activated upon ER stress and autophagosomes formed during this period of stress have been shown to contain portions of the ER (Schuck et al., 2014). This autophagy of the ER is known as ER-phagy or reticulophagy (Figure 5) and counterbalance ER expansion during the UPR, contributing to cell homeostasis.
Figure 5. ER-phagy/Reticulophagy.
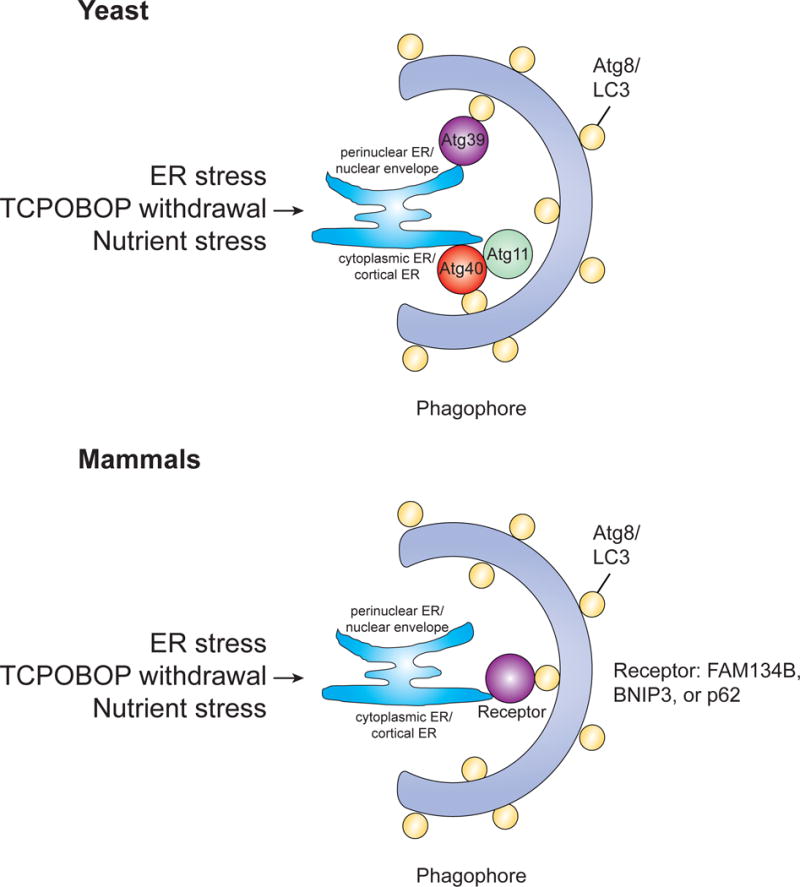
ER stress, TCPOBOP withdrawal, and nutrient stress are all stimuli that can lead to the induction of ER-phagy. In yeast, both Atg39 and Atg40 have been shown to mediate ER-phagy, localizing to distinct subdomains of the ER and interacting with Atg8. In mammals, the functional counterpart of Atg40 is FAM134B. Both BNIP3/Nix and p62 have also been implicated in ER-phagy.
ER stress is a clear inducer of ER-phagy (Bernales et al., 2006; Schuck et al., 2014), but other stimuli have also been shown to induce this process. In mammals, ER-phagy is used to remove excess hepatic ER after withdrawal of the hepatic mitogen TCPOBOP (1, 4-bis[2-(3, 5-dicholoropyridyloxy)] benzene) (Yang et al., 2016). Additionally, nutrient limiting conditions and rapamycin treatment induce selective autophagy of the ER (Mochida et al., 2015). As with other types of selective autophagy, ER-phagy is a receptor-mediated process, with receptor proteins playing a critical role in target selection. In the yeast S. cerevisiae, Atg39 and Atg40 are used to mediate ER-phagy, localizing to distinct subdomains of the ER and interacting with Atg8, facilitating the formation of autophagosomes (Mochida et al 2015).
The functional counterpart of Atg40 in mammals is the family with sequence similarity 134 member B (FAM134B) protein. FAM134B contains a conserved LIR motif and FAM134B positive ER fragments co-localize with LC3B. Additionally, whereas downregulation of FAM134B leads to ER swelling, overexpression of FAM134B leads to ER fragmentation and lysosomal degradation (Khaminets et al., 2015). Interestingly, this fragmentation of the ER prior to ER-phagy is reminiscent of mitochondrial fission prior to mitophagy. Disruption of FAM134B in mice was also shown to cause swelling of the ER and inhibit ER-phagy, indicating that FAM134B is indeed the ER-phagy receptor in mammals. The reticulon domain of FAM134B as well as its LIR motif are both required for ER-phagy. It was proposed that FAM134B promotes membrane remodeling and ER scission via the membrane-bending capacity of its reticulon domain, whereas the LIR motif acts in a canonical fashion to target ER to autophagosomes for degradation (Khaminets et al., 2015). Bnip3/Nix has also been associated with ER-phagy in HeLa cells, with ER-localized Bnip3 binding to LC3 through interaction with the Bnip3 LIR motif (Hanna et al., 2012). However, in this study ER-phagy was not completely attenuated when the Bnip3/LC3 interaction was disrupted, suggesting that Bnip3 was not the only autophagy receptor on the ER. Additionally, p62 has also been associated with autophagic removal of excess hepatic endoplasmic reticulum in mice (Yang et al., 2016).
Like with other organelle-specific selective autophagy pathways, many questions remain regarding the precise mechanisms underlying the process of ER-phagy. For example, though FAM134B has been suggested to promote membrane remodeling, it is still unknown how the ER is fragmented and sequestered by the phagophore and whether this event occurs prior to autophagosome formation. Additionally, since p62 has been implicated in ER-phagy, it would be interesting to determine if ubiquitin plays an important role in autophagic clearance of ER. Furthermore, studies have shown both Bnip3/Nix and p62 to be associated with ER-phagy, but further clarification on the exact cargo receptor(s) involved in this process is warranted.
Nucleophagy
The nucleus is the organelle containing the cells’ genetic material, and autophagic degradation of the nucleus is known as nucleophagy (Mochida et al., 2015). Because degradation of the nucleus would generally be detrimental to cells, nucleophagy for the most part involves only partial degradation of nuclear components (piecemeal micronucleophagy or PMN) (Figure 6). In S. cerevisiae, PMN specifically degrades non-essential nuclear components by pinching off portions of the nuclear envelope (NE) and the granular nucleolus enriched in pre-ribosomes, but excludes chromosomal DNA, nuclear pore complexes, and spindle pole bodies (Farré et al., 2009; Kraft et al., 2009; Kvam and Goldfarb, 2007; Millen et al., 2009; Roberts et al., 2003). This process was shown to require much of the core autophagy machinery (Krick et al., 2008). In PMN, a junction between the nucleus and the vacuole is generated by the interaction of two key proteins, Vac8p and Nvj1p. Upon initiation of PMN by a stimulus such as starvation, these junctions bulge into the vacuole and a piece of the nucleus buds off, releasing a vesicle into the vacuole where it is degraded by resident vacuolar hydrolases (Krick et al., 2008; Kvam and Goldfarb, 2007; Roberts et al., 2003).
Figure 6. Nucleophagy.
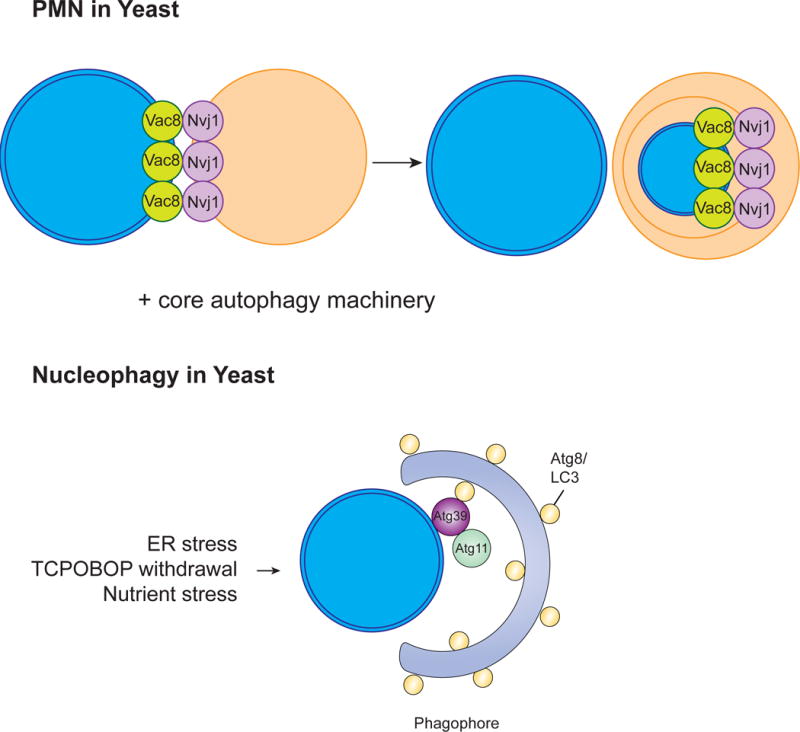
The clearance of nuclear contents by autophagy using either PMN or the involvement of the cargo receptor Atg39. In yeast, PMN involves the formation of a junction between the nucleus and the vacuole generated by two key proteins, Vac8 and Nvi1. These junctions bulge into the vacuole, a piece of the nucleus buds off, and this vesicle is released into the vacuole where it is degraded by vacuolar hydrolases. This process involves much of the core autophagy machinery. The nucleophagy receptor Atg39 was also shown to localize to the yeast perinuclear ER/nuclear envelope and lead to degradation of nuclear components. Less is known about the mechanism behind nucleophagy in mammals.
Nucleophagy can also be activated in yeast after prolonged periods of nitrogen starvation, a process termed late nucleophagy (Mijaljica et al., 2012). In contrast to PMN, late nucleophagy can occur in the absence of Nvj1p or Vac8p and doesn’t require Vps34 PtdIns(3)P-kinase complex components. In the filamentous fungus Aspergillus oryzae, autophagy mediates the degradation of the entire nucleus which appears to be important for hyphal tip growth through the recycling of nutrients that are generated by nuclear degradation (Shoji et al., 2010). Autophagy-mediated degradation of the nucleus also occurs in the rice blast fungus, Magnaporthe oryzae (Liu et al., 2012b). The nucleophagy receptor Atg39 was recently identified and localized to the yeast perinuclear ER/nuclear envelope where double membrane vesicles derived from this area contained intranuclear compartments (Mochida et al., 2015). Although macroautophagy of the nucleus has been reported in animals, including mammals, there are no obvious homologs to Atg39 in these species.
In mammalian nuclear envelopathies, disorders caused by mutations in the genes encoding nuclear envelope proteins (such as A-type lamins and emerin), a part of the nucleus is degraded by autophagy when nuclei are damaged and/or partially extruded into the cytoplasm (Park et al., 2009). The degradation of the entire nucleus was also reported to occur in cultured murine seminal vesicle epithelial cells through an autophagy process termed nucleophagy, but the mechanism that regulates this process remains unknown (Kovács et al., 2000). In keratinization, the differentiation of keratinocytes resulting in the formation of the stratum corneum, dissolution of the nucleus occurs through autophagy (Akinduro et al., 2016). Parakeratosis, the retention of nuclei in the stratum corneum, is a characteristic of psoriasis. Akinduro et al. (2016) showed that the autophagy proteins LC3, WIPI1, and ULK1 were decreased in the parakeratotic regions of epidermis taken from patients with psoriasis, suggesting that a failure of nucleophagy may contribute to its pathogenesis.
Unfortunately, because not much is known about the mechanisms that control nucleophagy in mammals and how selectivity is established, many questions remain. The functional homolog of yeast Atg39 in mammals has yet to be identified. Additionally, like with reticulophagy, it is still unclear how the nuclear membrane is fragmented and sequestered by the phagophore and whether this event occurs prior to autophagosome formation. In mammals, the autophagy proteins WIPI1 and ULK1 are required for nucleophagy during keratinocyte terminal differentiation in vitro (Akinduro et al., 2016), but what other proteins are involved in the regulation of nucleophagy? Are ubiquitin and other factors that are common to clearance of organelles by autophagy involved in nucleophagy?
Chlorophagy
Chloroplasts are the organelles of plants and photoautotrophs that perform photosynthesis and are central to plant metabolism. Because plants are immobile, the use and recycling of cellular components is critical for their survival and health in differing environments. During plant senescence, cellular macromolecules are degraded and their components are mobilized and reused. For example, the degradation of chloroplast proteins, such as Rubisco (ribulose-1,5-bisphosphate carboxylase/oxygenase), is a major source of nitrogen. Early studies proposed that the major pathway for chloroplast protein degradation in senescing leaves was through sequential degradation within the vacuole, a process now widely recognized as autophagy (Lin and Wittenbach, 1981; Wittenbach et al., 1982) (Figure 7).
Figure 7. Chlorophagy.
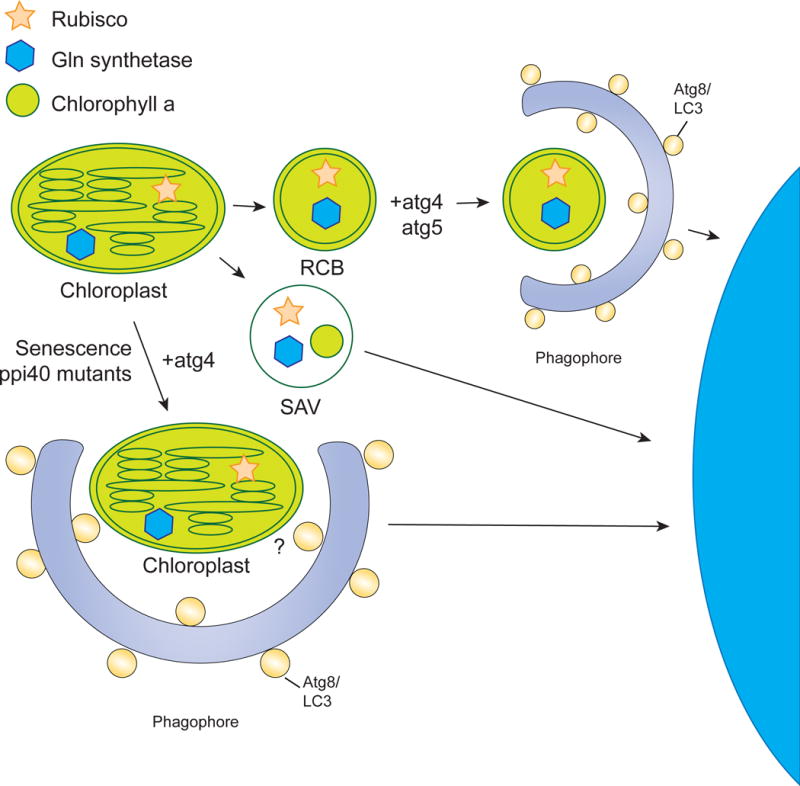
The chloroplast can be degraded in a piecemeal fashion or through whole organelle autophagy. Piecemeal degradation occurs via RCBs and SAVs. RCBs are derived from the chloroplast envelope and contain rubisco and Gln synthetase. The RCB is surrounded by the isolation membrane in the cytoplasm. This process has been shown to be dependent on atg4 and atg5. SAVs are small lytic vacuoles that also degrade parts of the chloroplast and have been shown to contain rubisco, Gln synthetase, and chlorophyll a. However, they have not been shown to use the autophagy machinery. A decrease in the total number of chloroplasts during senescence has been shown to involve the degradation of the entire chloroplast, a process that is blocked in atg4 mutants. However, the cargo receptor for this process has not been identified.
Much like the nucleus, the chloroplast can be degraded in a piecemeal fashion or through whole organelle autophagy. The piecemeal degradation of chloroplasts can occur via Rubisco-containing bodies (RCBs) and senescence associated vacuoles (SAVs). RCBs are double membrane bound bodies derived from the choloroplast envelope that contain Rubisco and Gln synthetase. After pinching off from the choloroplast, the RCB is further surrounded by other membrane structures such as the isolation membrane in the cytoplasm. The piecemeal degradation of choloroplasts by RCBs is important for protein recycling in both development and during abiotic stress responses, and seems to be common among plants. RCB-mediated autophagy has been shown to be dependent on atg4 and atg5 (Ishida et al., 2008; Wada et al., 2009). SAVs have also been implicated in the piecemeal autophagy of chloroplasts. These small, lytic vacuoles are found only in senescing tissues and, similar to RCBs, contain stromal proteins such as Rubisco and Gln synthetase. Unlike RCBs, however, they also contain chlorophyll a. Also unlike RCBs, there is no evidence that SAVs use any autophagic machinery.
A decrease in the total number of chloroplasts occurs in the late stages of senescence. Electron microscopy studies have indicated that this could be occurring through the degradation of the entire chloroplast by autophagy, a process termed chlorophagy (Minamikawa et al., 2001; Wittenbach et al., 1982). In atg4 mutants, this decrease in chloroplast number is impaired (Wada et al., 2009). In addition to senescence, mutations that impair chloroplasts can also lead to the degradation of whole chloroplasts, possibly through chlorophagy. For example, partially degraded chloroplasts were observed in the vacuole in Arabidopsis plastid protein import Tic40 (ppi40) mutants (Niwa et al., 2004). As these were observed in a starvation-independent manner, the authors suggested that plants can remove these defective chloroplasts by autophagy for organelle quality control.
Little is known about the mechanisms underlying chlorophagy and, thus, many important questions remain. Whether chlorophagy involves cargo receptor proteins, for example, is unknown. Plants contain putative autophagy cargo receptors, including a homolog of NBR1 (AtNBR1 in Arabidopsis) (Svenning et al., 2011), and AIM-containing proteins, such as tryptophan-rich sensory protein (TSPO)-related membrane protein (Hachez et al., 2014; Vanhee et al., 2011) and Arabidopsis Atg8-interacting proteins ATI1 and ATI2 (Honig et al., 2012). It remains unclear if any of these proteins could be involved in chlorophagy, and future studies should resolve if selective removal of chloroplasts involves mechanisms that are similar to those used in selective removal of organelles in animals.
Conclusions
Here we have highlighted our understanding of the selective autophagy of organelles. Though the stimuli inducing organelle-specific autophagy are diverse, the basic mechanisms by which these organelles are degraded are similar in many cases. Common mechanisms involve cargo selection proteins that recognize the target organelle, often through binding to ubiquitin that is associated with proteins on the surface of the organelle, and LC3/Atg8 either directly or through interacting partners. Although some of these pathways are becoming clear, some selective autophagy pathways lack the molecular mechanisms. Since inter-organelle interactions are at the interface of selective autophagy, such as ER as a source of autophagosome membrane for mitophagy, and are critical for diverse molecular functions (Prinz, 2014), it is interesting to consider how “innocent bystanders” are not recruited for degradation during selective clearance of organelles. It is possible that innocent bystanders are actually degraded by selective autophagy along with their counterparts, or could even be extruded after initial incorrect consumption. Elucidation of these details should yield important, clinically relevant information, such as how clearance of harmful, damaged organelles is necessary to avoid harmful human pathologies including cancer, neurodegeneration and inflammatory diseases.
References
- Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 2013;126:1905–1912. doi: 10.1242/jcs.091181. [DOI] [PubMed] [Google Scholar]
- Akinduro O, Sully K, Patel A, Robinson DJ, Chikh A, McPhail G, Braun KM, Philpott MP, Harwood CA, Byrne C, et al. Constitutive Autophagy and Nucleophagy during Epidermal Differentiation. J Invest Dermatol. 2016;136:1460–1470. doi: 10.1016/j.jid.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Anding AL, Baehrecke EH. Autophagy in Cell Life and Cell Death. Curr Top Dev Biol. 2015;114:67–91. doi: 10.1016/bs.ctdb.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol. 2013;15:1067–1078. doi: 10.1038/ncb2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013;126:939–952. doi: 10.1242/jcs.114819. [DOI] [PubMed] [Google Scholar]
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563–566. doi: 10.1038/nature14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efe JA, Botelho RJ, Emr SD. Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol Biol Cell. 2007;18:4232–4244. doi: 10.1091/mbc.E07-04-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré JC, Burkenroad A, Burnett SF, Subramani S. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 2013;14:441–449. doi: 10.1038/embor.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré JC, Krick R, Subramani S, Thumm M. Turnover of organelles by autophagy in yeast. Curr Opin Cell Biol. 2009;21:522–530. doi: 10.1016/j.ceb.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré JC, Manjithaya R, Mathewson RD, Subramani S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev Cell. 2008;14:365–376. doi: 10.1016/j.devcel.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:e00947. doi: 10.7554/eLife.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Hachez C, Veljanovski V, Reinhardt H, Guillaumot D, Vanhee C, Chaumont F, Batoko H. The Arabidopsis abiotic stress-induced TSPO-related protein reduces cellsurface expression of the aquaporin PIP2;7 through protein-protein interactions and autophagic degradation. Plant Cell. 2014;26:4974–4990. doi: 10.1105/tpc.114.134080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- Honig A, Avin-Wittenberg T, Ufaz S, Galili G. A new type of compartment, defined by plant-specific Atg8-interacting proteins, is induced upon exposure of Arabidopsis plants to carbon starvation. Plant Cell. 2012;24:288–303. doi: 10.1105/tpc.111.093112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YH, Chen LMW, Yang JY, Yang WY. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun. 2013;4:2111. doi: 10.1038/ncomms3111. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kominami E, Tanaka K, Komatsu M. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy. 2008;4:1063–1066. doi: 10.4161/auto.6826. [DOI] [PubMed] [Google Scholar]
- Ishida H, Yoshimoto K, Izumi M, Reisen D, Yano Y, Makino A, Ohsumi Y, Hanson MR, Mae T. Mobilization of rubisco and stroma-localized fluorescent proteins of chloroplasts to the vacuole by an ATG gene-dependent autophagic process. Plant Physiol. 2008;148:142–155. doi: 10.1104/pp.108.122770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Höke A, Dawson VL, Dawson TM, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. Embo J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tormediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katarzyna ZR, Suresh S. Autophagic degradation of peroxisomes in mammals. Biochem Soc Trans. 2016;44:431–440. doi: 10.1042/BST20150268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USa. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kondo-Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, Hashimoto A, Inagaki F, Ohsumi Y, Okamoto K. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J Biol Chem. 2012;287:10631–10638. doi: 10.1074/jbc.M111.299917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács AL, Réz G, Pálfia Z, Kovács J. Autophagy in the epithelial cells of murine seminal vesicle in vitro. Formation of large sheets of nascent isolation membranes, sequestration of the nucleus and inhibition by wortmannin and 3-ethyladenine. Cell Tissue Res. 2000;302:253–261. doi: 10.1007/s004410000275. [DOI] [PubMed] [Google Scholar]
- Kovács AL, Pálfia Z, Réz G, Vellai T, Kovács J. Sequestration revisited: integrating traditional electron microscopy, de novo assembly and new results. Autophagy. 2007;3:655–662. doi: 10.4161/auto.4590. [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim Biophys Acta. 2009;1793:1404–1412. doi: 10.1016/j.bbamcr.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Krick R, Muehe Y, Prick T, Bremer S, Schlotterhose P, Eskelinen EL, Millen J, Goldfarb DS, Thumm M. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–4505. doi: 10.1091/mbc.E08-04-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvam E, Goldfarb DS. Nucleus-vacuole junctions and piecemeal microautophagy of the nucleus in S. cerevisiae. Autophagy. 2007;3:85–92. doi: 10.4161/auto.3586. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- Lin W, Wittenbach VA. Subcellular localization of proteases in wheat and corn mesophyll protoplasts. Plant Physiol. 1981;67:969–972. doi: 10.1104/pp.67.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012a;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- Liu XH, Gao HM, Xu F, Lu JP, Devenish RJ, Lin FC. Autophagy vitalizes the pathogenicity of pathogenic fungi. Autophagy. 2012b;8:1415–1425. doi: 10.4161/auto.21274. [DOI] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. Embo J. 2013;32:2336–2347. doi: 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjithaya R, Nazarko TY, Farré JC, Subramani S. Molecular mechanism and physiological role of pexophagy. FEBS Lett. 2010;584:1367–1373. doi: 10.1016/j.febslet.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene. 1997;192:245–250. doi: 10.1016/s0378-1119(97)00084-x. [DOI] [PubMed] [Google Scholar]
- McLelland GL, Lee SA, McBride HM, Fon EA. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;114 doi: 10.1083/jcb.201603105. jcb.201603105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. Embo J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijaljica D, Prescott M, Devenish RJ. A late form of nucleophagy in Saccharomyces cerevisiae. PLoS ONE. 2012;7:e40013. doi: 10.1371/journal.pone.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millen JI, Krick R, Prick T, Thumm M, Goldfarb DS. Measuring piecemeal microautophagy of the nucleus in Saccharomyces cerevisiae. Autophagy. 2009;5:75–81. doi: 10.4161/auto.5.1.7181. [DOI] [PubMed] [Google Scholar]
- Minamikawa T, Toyooka K, Okamoto T, Hara-Nishimura I, Nishimura M. Degradation of ribulose-bisphosphate carboxylase by vacuolar enzymes of senescing French bean leaves: immunocytochemical and ultrastructural observations. Protoplasma. 2001;218:144–153. doi: 10.1007/BF01306604. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359–362. doi: 10.1038/nature14506. [DOI] [PubMed] [Google Scholar]
- Motley AM, Nuttall JM, Hettema EH. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. Embo J. 2012;31:2852–2868. doi: 10.1038/emboj.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010a;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010b;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa Y, Kato T, Tabata S, Seki M, Kobayashi M, Shinozaki K, Moriyasu Y. Disposal of chloroplasts with abnormal function into the vacuole in Arabidopsis thaliana cotyledon cells. Protoplasma. 2004;223:229–232. doi: 10.1007/s00709-004-0037-7. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, Inagaki F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13:1211–1218. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- Nordgren M, Francisco T, Lismont C, Hennebel L, Brees C, Wang B, Van Veldhoven PP, Azevedo JE, Fransen M. Export-deficient monoubiquitinated PEX5 triggers peroxisome removal in SV40 large T antigen-transformed mouse embryonic fibroblasts. Autophagy. 2015;11:1326–1340. doi: 10.1080/15548627.2015.1061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgren M, Wang B, Apanasets O, Fransen M. Peroxisome degradation in mammals: mechanisms of action, recent advances, and perspectives. Frontiers in Physiology. 2013;4 doi: 10.3389/fphys.2013.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara K, Sekito T, Niimi K, Ohsumi Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem. 2008;283:23972–23980. doi: 10.1074/jbc.M803180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Y, Munro S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol Biol Cell. 2010;21:3998–4008. doi: 10.1091/mbc.E10-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku M, Sakai Y. Pexophagy in yeasts. Biochim Biophys Acta. 2016;1863:992–998. doi: 10.1016/j.bbamcr.2015.09.023. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjϕrkϕy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Park YE, Hayashi YK, Bonne G, Arimura T, Noguchi S, Nonaka I, Nishino I. Autophagic degradation of nuclear components in mammalian cells. Autophagy. 2009;5:795–804. doi: 10.4161/auto.8901. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USa. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA. Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J Cell Biol. 2014;205:759–769. doi: 10.1083/jcb.201401126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde KR, Smith HL, Chau KY, Schapira AHV. PINK1 disables the antifission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol. 2016;213:163–171. doi: 10.1083/jcb.201509003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010a;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010b;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Rawi Al S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USa. 2016;113:4039–4044. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts P, Moshitch-Moshkovitz S, Kvam E, O’Toole E, Winey M, Goldfarb DS. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:129–141. doi: 10.1091/mbc.E02-08-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167–178. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol. 2014;204:869–879. doi: 10.1083/jcb.201312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent G, van Zutphen T, Shatseva T, Zhang L, Di Giovanni V, Bandsma R, Kim PK. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J Cell Biol. 2016;214:677–690. doi: 10.1083/jcb.201511034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- Schuck S, Gallagher CM, Walter P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci. 2014;127:4078–4088. doi: 10.1242/jcs.154716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USa. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. Embo J. 1999;18:5234–5241. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji JY, Kikuma T, Arioka M, Kitamoto K. Macroautophagy-mediated degradation of whole nuclei in the filamentous fungus Aspergillus oryzae. PLoS ONE. 2010;5:e15650. doi: 10.1371/journal.pone.0015650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015;22:419–432. doi: 10.1038/cdd.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani S. A mammalian pexophagy target. Nat Cell Biol. 2015;17:1371–1373. doi: 10.1038/ncb3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. Embo J. 2014;33:2142–2156. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. Embo J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenning S, Lamark T, Krause K, Johansen T. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy. 2011;7:993–1010. doi: 10.4161/auto.7.9.16389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takáts S, Nagy P, Varga Á, Pircs K, Kárpáti M, Varga K, Kovács AL, Hegedűs K, Juhász G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013;201:531–539. doi: 10.1083/jcb.201211160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka C, Tan LJ, Mochida K, Kirisako H, Koizumi M, Asai E, Sakoh-Nakatogawa M, Ohsumi Y, Nakatogawa H. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J Cell Biol. 2014;207:91–105. doi: 10.1083/jcb.201402128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, Kominami E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol Biol Cell. 1999;10:1367–1379. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till A, Lakhani R, Burnett SF, Subramani S. Pexophagy: the selective degradation of peroxisomes. Int J Cell Biol. 2012;2012:512721–18. doi: 10.1155/2012/512721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi DN, Walker CL. The peroxisome as a cell signaling organelle. Curr Opin Cell Biol. 2016;39:109–112. doi: 10.1016/j.ceb.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, Buss F. The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog. 2015;11:e1005174. doi: 10.1371/journal.ppat.1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Vanhee C, Zapotoczny G, Masquelier D, Ghislain M, Batoko H. The Arabidopsis multistress regulator TSPO is a heme binding membrane protein and a potential scavenger of porphyrins via an autophagy-dependent degradation mechanism. Plant Cell. 2011;23:785–805. doi: 10.1105/tpc.110.081570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USa. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada S, Ishida H, Izumi M, Yoshimoto K, Ohsumi Y, Mae T, Makino A. Autophagy plays a role in chloroplast degradation during senescence in individually darkened leaves. Plant Physiol. 2009;149:885–893. doi: 10.1104/pp.108.130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, et al. The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell. 2016;63:781–795. doi: 10.1016/j.molcel.2016.08.021. [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenbach VA, Lin W, Hebert RR. Vacuolar localization of proteases and degradation of chloroplasts in mesophyll protoplasts from senescing primary wheat leaves. Plant Physiol. 1982;69:98–102. doi: 10.1104/pp.69.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur ELF. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci USa. 2014;111:E4439–E4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita SI, Abe K, Tatemichi Y, Fujiki Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy. 2014;10:1549–1564. doi: 10.4161/auto.29329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ni HM, Guo F, Ding Y, Shi YH, Lahiri P, Fröhlich LF, Rülicke T, Smole C, Schmidt VC, et al. Sequestosome-1/p62 is Associated with Autophagic Removal of Excess Hepatic Endoplasmic Reticulum in Mice. J Biol Chem. 2016 doi: 10.1074/jbc.M116.739821. jbc.M116.739821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USa. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ney PA. NIX induces mitochondrial autophagy in reticulocytes. Autophagy. 2008;4:354–356. doi: 10.4161/auto.5552. [DOI] [PubMed] [Google Scholar]
- Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait-Mulder J, Lee JH, Paull TT, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17:1259–1269. doi: 10.1038/ncb3230. [DOI] [PMC free article] [PubMed] [Google Scholar]