

Abstract
Aberrant expression of aurora kinase A is implicated in the genesis of various neoplasms, including acute myeloid leukemia. Alisertib, an aurora A kinase inhibitor, has demonstrated efficacy as monotherapy in trials of myeloid malignancy, and this efficacy appears enhanced in combination with conventional chemotherapies. In this phase I, dose-escalation study, newly diagnosed patients received conventional induction with cytarabine and idarubicin, after which alisertib was administered for 7 days. Dose escalation occurred via cohorts. Patients could then receive up to four cycles of consolidation, incorporating alisertib, and thereafter alisertib maintenance for up to 12 months. Twenty-two patients were enrolled. One dose limiting toxicity occurred at dose level 2 (prolonged thrombocytopenia), and the recommended phase 2 dose was established at 30mg twice daily. Common therapy-related toxicities included cytopenias and mucositis. Only three (14%) patients had persistent disease at mid-cycle, requiring “5+2” reinduction. The composite remission rate (complete remission and complete remission with incomplete neutrophil recovery) was 86% (nineteen of twenty-two patients; 90% CI 68–96%). Among those over age 65 and those with high-risk disease (secondary acute leukemia or cytogenetically high-risk disease), the composite remission rate was 88% and 100%, respectively. The median follow up was 13.5 months. Of those treated at the recommended phase 2 dose, the 12-month overall survival and progression-free survival were 62% (90% CI 33–81%) and 42% (90% CI 17–65%), respectively. Alisertib is well tolerated when combined with induction chemotherapy in acute myeloid leukemia, with a promising suggestion of efficacy. (clinicaltrials.gov Identifier:01779843).
Introduction
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy associated with poor outcomes. Current standard treatment includes remission induction chemotherapy, typically consisting of cytarabine combined with an anthracycline, an approach which has remained unchanged for more than 30 years.1–3 Over the last decade, a series of novel therapeutics have been combined with this chemotherapy backbone, with most yielding negative or inconsistent results.4–7 More recently, some improvement in outcomes have been noted with certain FLT3 tyrosine kinase inhibitors and antibody-drug conjugates8–10 in combination with induction chemotherapy; such benefits may be limited to certain subcategories of AML. For the majority of patients, new and effective approaches are still needed to enhance the current standard of care.
Aurora kinases are a family of serine-threonine kinases that regulate multiple phases of the mitotic signaling cascade.11 Aberrant upregulation of aurora A kinase (AAK) has been demonstrated in multiple malignancies, and its inhibition suppresses proliferation of neoplastic cells11,12 by triggering mitotic errors, aneuploidy, senescence, and apoptosis.13,14 This process appears especially applicable to cells during or shortly following exposure to cytotoxic chemotherapy.15–18
Alisertib is a potent, orally available inhibitor of AAK. Preclinical studies in AML cell lines, patient samples, and animal models have demonstrated potent cytotoxicity, diminished clonal survival, and promotion of apoptosis.15 Alisertib has been evaluated for safety and efficacy in multiple clinical studies,19–22 including those regarding hematological malignancy,23–26 and has been associated with clinical response. Alisertib has also been studied in combination with other neoplastic agents, where it was also demonstrated to be safe and well-tolerated.15–18
There has been growing interest in targeting AAK as a therapeutic approach in myeloid neoplasms, and preclinical studies have suggested that targeting AAK expression with alisertib promotes tumor sensitivity to cytarabine.15 A subsequent phase II trial of alisertib monotherapy demonstrated efficacy in a subset of patients with AML and high-grade myelodysplastic syndromes (MDS).27 Given the oncogenic role of aurora kinases, the activity of alisertib monotherapy in AML, and suggestion of synergy in combination with chemotherapies, we performed a phase I study to evaluate the safety and tolerability of alisertib combined with conventional induction chemotherapy for newly diagnosed AML.
Methods
This study (clinicaltrials.gov Identifier:01779843) was approved by the local institutional review board, and conducted in accordance with the declaration of Helsinki. Patients were eligible for enrollment if they were age 18 or older with previously untreated AML based on WHO criteria (≥ 20% bone marrow blasts).28 They were required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, cardiac ejection fraction of ≥50%, and intact organ function including aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase of <5 times the upper limit of normal, direct bilirubin of <2.0 mg/dL, and creatinine clearance of ≥40 mL/min. Standard cytogenetic and molecular testing was performed at diagnosis. Those with acute promyelocytic leukemia or with core-binding factors alterations, t(8;21) or inv(16)/t(16;16), were excluded.
Treatment
“7+3” induction chemotherapy included continuous cytarabine infusion at 100 mg/m2 on days 1 through 7, and idarubicin, dosed at 12 mg/m2 administered on days 1–3 by intravenous bolus. Starting on the day after the conclusion of cytarabine infusion, alisertib was administered orally according to dose level, twice daily (BID), for 7 days. A bone marrow biopsy was performed at mid-induction (between days 13 and 16). If bone marrow cellularity was ≥20% and myeloblast involvement was >5%, “5+2” reinduction, with continuous infusion of cytarabine at 100mg/m2 on days 1 to 5 and idarubicin 12 mg/m2 intravenously on days 1 and 2, was administered. If the bone marrow cellularity was <20% but with >5% myeloblasts, “5+2” reinduction was per clinician discretion. Patients receiving reinduction did not receive additional doses of alisertib during the remission induction period. A bone marrow evaluation was performed at the time of peripheral hematologic recovery (absolute neutrophil count (ANC) >1000/μL and platelet count >100,000/μL) or by day 40 (range 35–42) or day 60 (if “5+2” was administered) in the absence of optimal hematologic recovery. A marrow biopsy was also performed on clinical suspicion of resistant disease. Response criteria29 were categorized as complete remission (CR), complete remission with incomplete platelet recovery (CRp), complete remission with incomplete neutrophil recovery (CRi), or refractory disease.
Those achieving CR, CRi, or CRp, and eligible for allogeneic hematopoietic cell transplantation (HCT), could come off study treatment during follow up for that purpose; they were then followed for relapse and survival outcomes. Otherwise, responding patients were eligible for up to four cycles of consolidation therapy, at their clinician’s discretion. Consolidation therapy consisted of cytarabine intravenously dosed at 3 gm/m2 every twelve hours on days 1, 3, and 5 in patients < age 60, or 2 gm/m2 daily on days 1–5 in those ≥ age 60. Starting on day 6 of each consolidation cycle, they received BID alisertib according to dose level for 7 days. Once patients completed the cycles of consolidation and achieved count recovery, they were eligible for alisertib maintenance. This was administered BID according to the patient’s dose level on days 1–7 of 21-day cycles, and continued until 12 months after the start of induction, or until disease progression.
Patients were enrolled in one of three dose cohorts in a “3+3” dose-escalation design. The three alisertib dose levels were 10 mg, 20 mg, and 30 mg BID. Enrollment was stopped at dose levels until all three patients in a cohort were assessed for treatment-related dose-limiting toxicities (DLTs). The DLT period was from initiation up to day 40, or day 60 if “5+2” was administered. If no DLTs were experienced by the first three patients, three patients were treated at the next dose level. If one DLT was experienced, an additional three patients were enrolled at the same dose level. If fewer than 2 DLTs were experienced among the six, dose escalation was permitted. If 2 or more DLTs were experienced, the previous lower dose was deemed the maximum tolerated dose (MTD). Should DLTs not be encountered, the highest dose level (30 mg BID) within the protocol would be the recommended phase 2 dose (RP2D). Once the MTD or RP2D was identified, an additional six patients were to be treated at that dose level.
Toxicities were graded according to Common Terminology Criteria for Adverse Events (CTCAE version 4.0). Patients who experienced grade 3 or 4 non-hematologic toxicity related to the study drug, and which persisted for longer than 48 hours without resolution to ≤ grade 2, stopped alisertib. Grade 3 or 4 myelosuppression did not lead to alisertib cessation. DLTs were defined as any grade 4 or 5 non-hematologic toxicity, but excluding toxicities such as infection related to neutropenia, grade 4 fatigue or anorexia, and grade 4 nausea, vomiting, diarrhea, or electrolyte abnormalities which were reversible with the appropriate therapies. Grade 3 non-hematological toxicities that did not resolve to grade 2 by day 40 were also considered DLTs, unless they were attributed to persistent AML. Grade 4 neutropenia or thrombocytopenia at day 40 following induction, or day 60 if “5+2” reinduction was administered, were also considered DLTs, if the cytopenias were not thought to be related to the underlying leukemia.
Correlative Methods
Culture of primary human AML cells
Cryopreserved primary bone marrow samples, collected at baseline, were thawed and cultured for 36–48h on confluent irradiated (2,000 rads) mouse MS5 stromal cells in roswell park memorial institute (RPMI) medium supplemented with 20% fetal bovine serum (FBS), 1% penicillin/streptomycin, 50μM β-mercaptoethanol, and human cytokines including stem cell factor (SCF) (100ng/ml), interleukin (IL)-3 (10ng/ml), IL-6 (20ng/ml), thrombopoietin (TPO) (10ng/ml), and FLT3 ligand (FLT3L, 10ng/ml) (Peprotech). Once cells were proliferative, each sample was transferred to two new wells of irradiated stroma and left untreated, or treated with 50nM alisertib for 18 hrs.
Immunofluorescence Staining and Imaging
Patient leukemic cells were then spun at 1,000rpm for 3 minutes onto poly-lysine coated coverslips and fixed in ice-cold methanol. Coverslips were blocked in trisbuffered saline (TBS)/bovine serum albumin (BSA), stained for tubulin (dmlα: Sigma) and DNA (0.2 μg/mL 4′,6-diamidino-2-phenylindole (DAPI)), and mounted on coverslips using ProLong Antifade mounting medium (Molecular Probes). A Nikon Ti-E fluorescence microscope equipped with a 60x objective and a Zyla sCMOS camera was used to identify and capture images of mitotic cells in each sample. The identification and characterization of the mitotic spindle structure was performed on a minimum of 20 mitotic figures per condition.
Pharmacokinetics
Samples to determine the steady state minimum concentration of alisertib in plasma (Cminss) were obtained from each patient just prior to dosing on days 9, 11, and 14 during the first cycle. Peripheral blood (6mL) was drawn into plastic tubes containing freeze-dried K2EDTA, mixed by inversion, placed over ice until centrifuged (1,300 g, 10-min, 4°C), whereupon the plasma was removed and stored at −80°C until assayed. The concentration of alisertib in human plasma was determined by reversed-phase high performance liquid chromatography with tandem mass spectrometric detection as previously reported, with minor modifications.22 The analytical method was validated as recommended by current FDA Guidance for Industry: Bioanalytical Method Validation, May 2001, to document selectivity, carryover, accuracy, precision, absolute recovery, and matrix effects. Alisertib was determined with an interday accuracy of 100.8% and a precision of 1.3% at the 5.0 ng/mL lower limit of quantitation. Cminss was calculated for each patient as the average of the assayed concentration of alisertib in predose samples obtained on days 11 and 14. The geometric mean and standard deviation were calculated for Cminss values for patients evaluated at each dose level.
Statistical Analysis
The primary study objective was to determine the type, frequency, and severity of drug related toxicities and define the MTD/RP2D. The study utilized a 3+3 design, such that if the DLT rate exceeded 30% at a given level it was less likely that the alisertib dose would be escalated. The secondary objectives included assessment of the complete response rate (CR/CRi/CRp), and the rates of overall survival (OS) and progression-free survival (PFS) at one year.
OS was defined as the time from diagnosis to death and censored at the last known date alive. PFS was defined as the time from diagnosis to the first occurrence of progression or death and censored at the last known date alive and disease-free. For the two patients who died before the end of the DLT monitoring period, the date when they were deemed off-treatment was used for survival estimates. Survival was estimated for the trial and at the RP2D (cohort 3 and dose expansion) with the Kaplan-Meier method. The proportion of patients achieving a CR or CRi was estimated along with a 90% exact binomial confidence interval.
All grade 3 or higher toxicity was tabled regardless of attribution and dose cohort. The date for either count recovery or day 40 (D40), whichever was reported, were used to determine when toxicities occurred. Grade 3 or higher induction toxicities were tabled according to the dose cohort. Additionally, for patients proceeding to consolidation therapy, post-induction toxicities were tabled. Pharmacokinetic and correlative studies, as well as patient and disease characteristics, including the number of patients completing each stage of therapy (induction, consolidation, and maintenance) were presented descriptively.
Results
Patient Characteristics
Between May 2013 and January 2015, twenty-two patients were enrolled on study. Baseline patient characteristics are summarized in Table 1. The median age was 62.7 (range 41.7–75.1), and the majority were Caucasian and male. Eight patients (36%) were aged ≥ 65 years. Eight patients (36%) had secondary AML, six with preceding MDS, one with a preceding myeloproliferative neoplasm, and one with therapy-related AML. The majority of patients had intermediate-risk AML based upon cytogenetics (82%) or upon European LeukemiaNet (ELN) criteria (73%). The most frequent additional mutations identified were NRAS (27%), IDH2 R140 (23%), NPM1 (18%), IDH1 (14%), TP53 (14%), and FLT3-ITD (14%).
Table 1.
Patient and disease characteristics.
Treatment and Toxicity Profile
A total of twenty-two patients were enrolled: three patients (10mg BID), seven patients (20mg BID), six patients (30mg BID), and an additional six at the determined RP2D of 30mg BID. All patients experienced at least one expected grade 3/4 toxicity of anemia, leukopenia, thrombocytopenia, and febrile neutropenia. A single DLT, prolonged grade 4 thrombocytopenia (beyond 40 days), thought to possibly be related to alisertib, occurred at dose level two (20mg BID), prompting the enrollment of six patients at that dose level. No additional DLTs were observed. An expansion cohort of six patients was then added at 30mg BID. Two patient deaths occurred prior to the DLT assessment period. The first mortality, at dose level two, was caused by sepsis and thought to be unrelated to the study drug; this patient was replaced due to DLT assessment ineligibility. The second mortality was due to subarachnoid hemorrhage, again considered to be unrelated to the study drug; this patient was in the expansion cohort, and no replacement was performed.
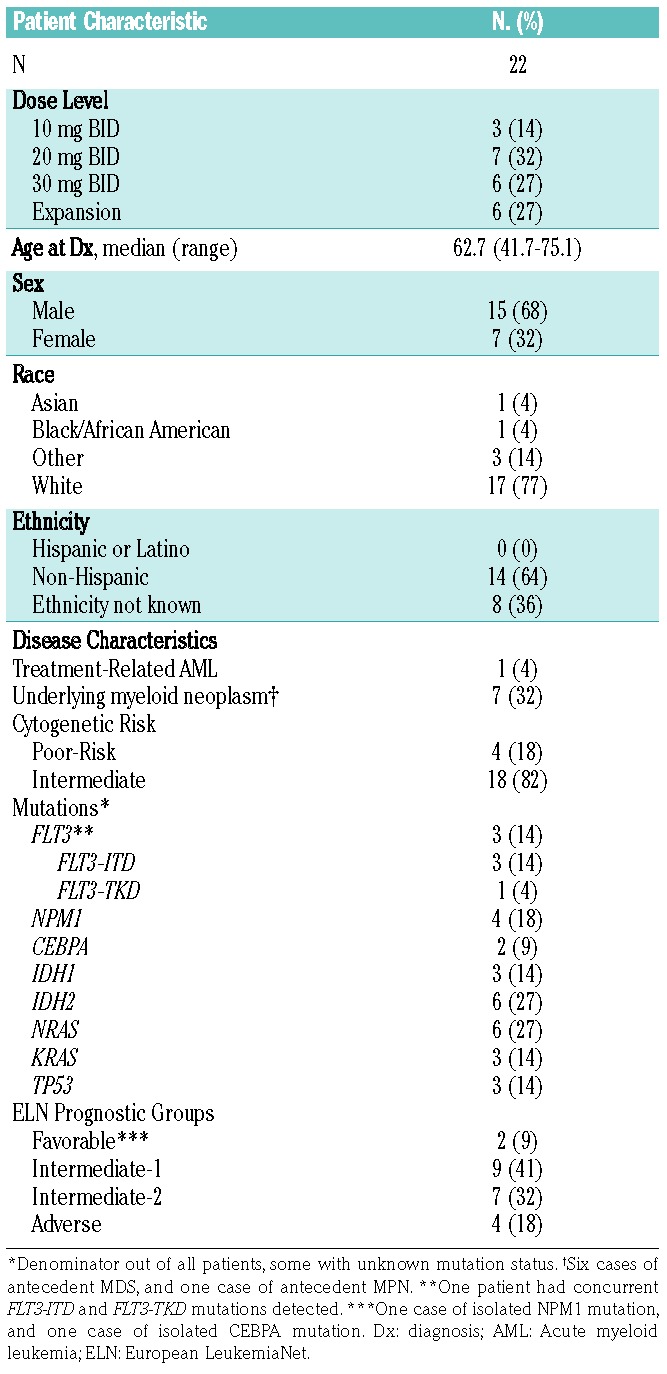
Alisertib was well tolerated. All grade 3 and higher non-hematologic toxicities, regardless of attribution, are summarized in Table 2. In addition to the DLT noted above and expected hematologic toxicities, other grade 4 toxicities included respiratory failure (one patient, 5%) and sepsis (two patients, 9%), neither of which were thought to be related to alisertib. The most frequent grade 3 toxicity was rash (four patients, 18%), which were also not felt to be related to alisertib. Non-hematologic toxicities possibly or probably attributable to alisertib are provided in Table 3. The large majority were deemed < grade 3, apart from two reversible episodes of grade 3 gastrointestinal toxicity. Mucositis was the most frequent toxicity with a possible relation to alisertib – this occurred in six patients (27%, all < grade 3). The median time to partial peripheral count recovery, as defined by an ANC of 500/mm3 and a platelet count of 50,000/mm3, was 33 days (range 26–48 days). The median time to full count recovery (defined as an ANC of 1000/mm3 and platelet count of 100,000/mm3) was 36 days (range 28–52 days).
Table 2.
Non-Hematologic Toxicities (grade 3 and higher) noted on study.
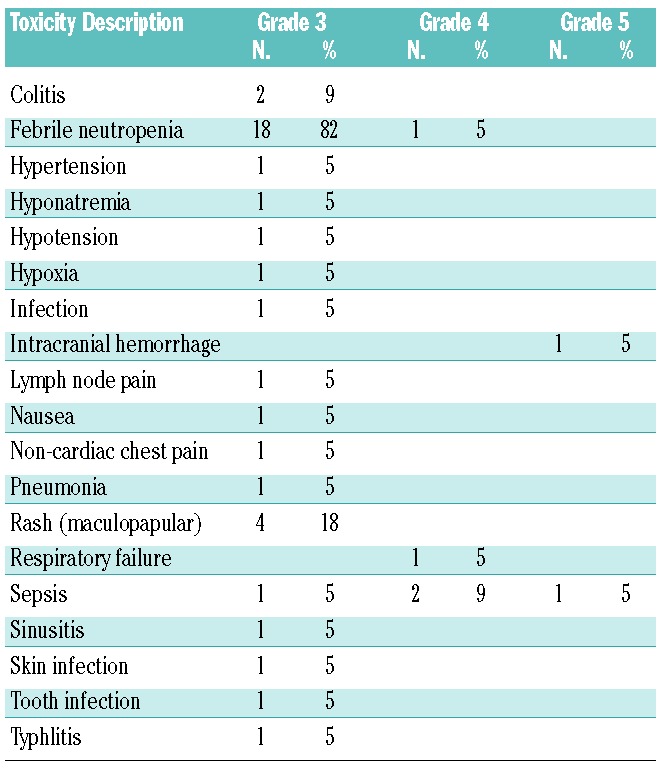
Table 3.
Non-hematologic toxicities on study by dose cohort, possibly or probably attributed to alisertib.

Efficacy
Treatment responses are summarized in Table 4, along with chromosomal and molecular features available at diagnosis for treated patients. Of the twenty-two patients treated, only three (14%) had persistent disease at mid-treatment, all of whom went on to receive “5+2” reinduction therapy per protocol. A remission was achieved in 86% (19/22; 90% CI 68–96%) of patients (64% CR, 23% CRi). Of the remaining three patients, two (9%) died during induction therapy and were inevaluable for marrow response, and one (5%) had refractory disease. The 12-month OS and PFS were 75% (90% CI 55–87%) and 54% (34–71%), respectively. Seven of the eight patients over age 65 years (87%) achieved a complete remission (six CR, one CRi, one death). Ten patients had high-risk AML, as defined as secondary/therapy-related AML and/or displaying poor-risk cytogenetic aberrations as per Medical Research Council (MRC) and ELN criteria; the overall remission rate for this group was 100% (80% CR, 20% CRi).
Table 4.
Course and outcome, by dose cohort, for patients on trial. Nineteen of twenty-two patients (86%) achieved a CR or CRi.

Ten patients (45%) received at least one cycle of cytarabine consolidation therapy on study; two patients (9%) received four cycles of consolidation therapy. Two patients were not able to proceed to consolidation, as per the discretion of their treating physician, due to ongoing infection. Four patients (18%) received alisertib as maintenance therapy following consolidation. At the time of manuscript preparation, six patients (27%) had experienced disease relapse and six patients (27%) had died (Figure 1). Among the four patients who received maintenance alisertib therapy, one experienced relapse and there were no instances of disease-related mortality. Ten patients (45%) went on to receive allogeneic HSCT after completing protocol-based therapy, four of whom relapsed with two ultimately dying from the disease in the protocol-determined follow-up period.
Figure 1.
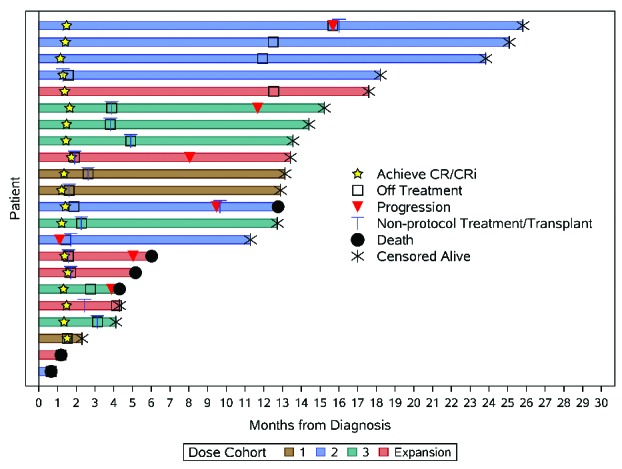
Summary plot of patient clinical course, sorted by follow up and treatment arm. CR: complete remission; CRi: remission with incomplete neutrophil recovery.
A total of twelve patients were treated at the RP2D (six from dose level three and six from the expansion cohort) of which eleven (92%; 90% CI 66–99%) achieved a CR/CRi. The median follow up for those remaining alive was 13.5 months. Figure 2 displays the OS and PFS for those treated at the RP2D. The 12-month OS for these patients was 62% (90% CI 33–81%) and the 12-month PFS was 42% (90% CI 17–65%).
Figure 2.
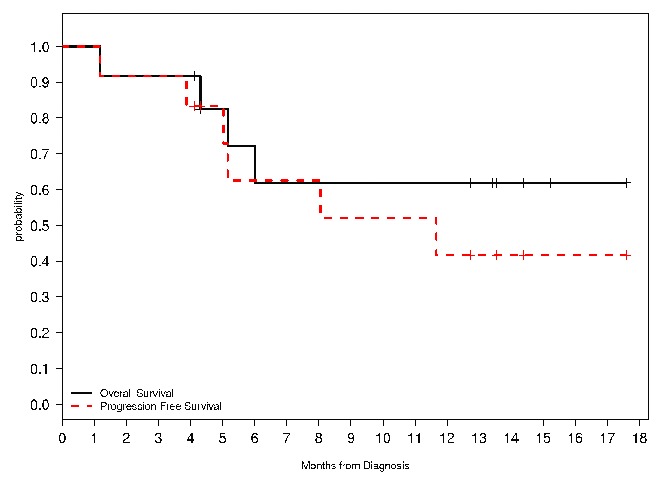
Overall (OS) and progression-free survival (PFS) estimates for those treated in cohort 3 and the expansion cohort. OS is noted in the solid black line, while PFS is the red dashed line.
Pharmacokinetics
The mean (±SD) Cminss of alisertib was 229±89 nM, 552±317 nM and 652±304 nM for patients treated with doses of 10 mg, 20 mg, and 30 mg BID, respectively.
Mitotic Spindle Studies
For a subset of patients, baseline samples of leukemic blasts were available for mitotic spindle study by immunofluorescence staining (Patients #4, #17, #20 and #22). Following 18 hours of treatment with 50nM of alisertib (a dose previously demonstrated to be optimal for studies in culture), all of these samples exhibited response (P<0.001). Treated samples demonstrated profound defects in mitotic spindle assembly, manifested as increases in mitotic cells exhibiting monopolar spindles and corresponding decreases in bipolar spindles (Figure 3), while untreated mitotic cells exhibited primarily bipolar or multipolar spindles.
Figure 3.
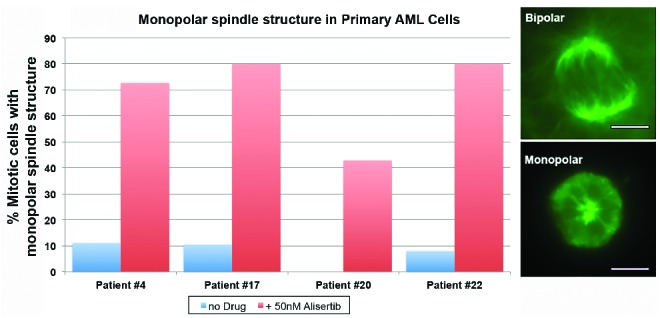
Spindle defects following alisertib treatment of baseline primary AML samples. Proliferative patient samples were left untreated, or treated with 50nM alisertib, and assessed for defects in mitotic spindle formation. The percentage of mitotic primary cells demonstrating defective monopolar spindle structures from untreated patient samples (blue bars) and those treated with alisertib (red bars) are demonstrated. All samples displayed an increase in monopolar spindle formation during mitosis following alisertib treatment. Representative images of bipolar and monopolar mitotic spindles in AML samples are shown in the panel on the right.
Discussion
The current therapeutic paradigm for AML is suboptimal for the majority of patients, and has remained unchanged for decades. Outcomes are particularly poor for certain higher risk subgroups, including older patients, those with secondary disease, and those with poor-risk cytogenetics. Effective novel approaches to the treatment of AML are necessary, and appear to be gradually emerging. Recent data has suggested that AAK inhibition with the targeted inhibitor alisertib may be an effective therapy across a range of cancers,19–24,26 including advanced myeloid malignancies.25 Others have found that the unique mitotic toxicity of alisertib may synergize with cytotoxic chemotherapy to enhance efficacy.15–18 Herein we report the results of a phase I study of alisertib combined with conventional induction chemotherapy for newly diagnosed AML.
The addition of alisertib to induction was well-tolerated without significant toxicity. Although limited by size in this phase I study, our data also suggests that the combination was promising, with higher response rates than those which have been historically reported.2,9,30,31 The composite rate of remission (CR and CRi) across all patients was 86%, and among those evaluable for response assessment, all but one achieved remission. More than one third of patients on study had high-risk AML, as established by karyotype or antecedent marrow process, and all of these patients achieved remission. All seven evaluable patients over age 65 also achieved remission. Mid-treatment bone marrow biopsies were performed according to the established approach for “7+3” induction, and only three of the twenty-two patients on study (14%) required reinduction for residual disease, a rate much lower than that previously reported with conventional induction.1,2,31 Among the high-risk patients studied, three patients had mutations impacting TP53, all with concurrent complex karyotypes. All achieved complete remission on study, and two went on to have stem cell transplantation. Nevertheless, none of the three were alive at 12 months, which is, unfortunately, consistent with the grim prognosis associated with the TP53 alteration.32–35
The pharmacokinetics of oral alisertib monotherapy has been characterized in phase I clinical trials.22,23 The drug has an apparent biological half-life of 19–23 hours, and steady-state pharmacokinetics is achieved within 7 days of repeated daily or twice daily dosing. The mean Cminss exhibited a relatively high degree of variability without a clear dose-dependent difference or even a reversal in magnitude between successive dose levels in some cases. Comparative data for the same dose levels evaluated in the present investigation have not previously been reported for the twice daily dosing schedule. Approximate values of the mean Cminss for the 50 mg BID were reported as 1,000 nM and 2,400 nM in the two single agent phase I studies. The Cminss of alisertib achieved with doses of 10, 20, and 30 mg BID in AML patients following the “7+3” remission induction regimen was in good general agreement with the expected drug levels based upon these prior early phase studies.
Aurora kinases are essential regulators of chromosomal alignment and separation during mitosis. Each of the enzymes, aurora A, B and C kinases, have key and coordinated functions in the mitotic process,36–38 including centrosome maturation, spindle assembly, chromosomal separation, and mitotic checkpoint regulation.36,39–42 AAK amplification has been detected across a range of malignancies, although its role in malignant transformation remains unclear.43–45 Overexpression of AAK may be insufficient, and some studies have suggested that additional concurrent activating mutations, in genes such as RAS, may be necessary to transform cells.46 A key contribution appears to be the role of AAK in chromosomal segregation, with overexpression leading to errors in this process. Aneuploidy, aberrant spindle formation, abortive mitoses, and other defects promote genetic instability and likely contribute to oncogenic transformation.38,43,47,48 In addition, an effect on tumor suppressor function may be important. AAK directly phosphorylates TP53, and amplification of the former may cause degradation of the latter.49–51 Nevertheless, given the broad impact on cell cycling and survival, the efficacy of AAK inhibitors in malignancy may be more generally related to anti-mitotic effects rather than to suppressing addiction to AAK activity.38
As a selective inhibitor of AAK, alisertib triggers the development of chromosomal defects and aneuploidy, with resulting cellular senescence and apoptosis in malignant cell lines.13,15 In multiple phase I and phase II clinical studies across solid tumor and hematologic malignancies,19–26 alisertib demonstrated a broad range of efficacy. In myeloid malignancies, a single-arm phase II study evaluated the efficacy of alisertib monotherapy in fifty-seven patients with advanced AML and high-grade MDS.25 Alisertib was administered at 50 mg twice daily for 7 consecutive days in 21-day cycles. The majority of patients had received prior therapies and had relapsed or refractory disease. Alisertib was well-tolerated, with the predominant grade 3/4 adverse events being febrile neutropenia, thrombocytopenia, anemia, and fatigue. Six responses were observed, all in AML patients, with five patients experiencing a partial remission, and one patient experiencing a prolonged CR lasting longer than a year. 49% of patients were also reported to have achieved stable disease.25
These findings suggested clinical activity with alisertib, now further supported by our data. Of twenty patients evaluable for response assessment, nineteen achieved remission. Only three of the twenty-two enrolled patients had persistent disease at mid-treatment with “7+3”. We also investigated available baseline samples from patients on study. In this small subgroup of four patients, spindle formation was clearly impacted by alisertib, with a marked increase in aberrant monopolar spindle formation. Intriguingly, all four of these patients went on to achieve remission on study, despite three of the four having high-risk karyotypic abnormalities and/or MDS-derived secondary AML. Additional study of primary samples from AML patients is necessary in order to establish trends, and these are currently ongoing.
The addition of alisertib was well-tolerated. Common toxicities on study were neutropenic fever, thrombocytopenia, and anemia (all expected with induction chemotherapy), and less common toxicities were rash and oral mucositis. Combining cytotoxic agents with induction chemotherapy raises concern for prolonged cytopenias; only one DLT, a case of prolonged thrombocytopenia, was noted on the study. Patients are frequently discharged following a degree of hematologic recovery rendering them clinically safe outside of the hospital, which at our institution typically includes an ANC of 500/mm3 and platelet transfusion independence. Encouragingly, among our patients, the median time to such a threshold was not greatly impacted, at 33 days. Nevertheless, we do acknowledge that there was a mild prolongation of time to peripheral count recovery, when compared to the approximate hospitalization duration expected for standard induction. However, this modest prolongation among our population did not contribute to morbidity or mortality in this study. The two deaths seen during induction, though related to cytopenias, occurred prior to day 30.
Our study was limited by its small size, being a phase I study with the key aim of determining tolerability and an MTD/RP2D. It was also limited by the relative heterogeneity of its population, with a mixture of high- and intermediate-risk AML cases. The mutational profile of patients in this small phase I study may also not fully reflect the larger population of AML patients. Additionally, a sizeable proportion of our patients went on to consolidative stem cell transplantation, and did not receive ongoing therapy on study. These trends are important since they can impact the interpretation of efficacy endpoints, a consideration which reinforces the need for further clinical study. Despite this, the overall outcomes for our cohort are favorable relative to historical outcomes, particularly given their age (median 63 years) and disease risk (36% with secondary AML and 18% with poor-risk cytogenetics). We demonstrated that alisertib combined with induction chemotherapy is safe and well-tolerated, and established an RP2D of 30mg BID in this setting. We also found that alisertib is safe among higher risk patients, with a promising suggestion of efficacy. All patients with high-risk AML (as defined by karyotype or antecedent marrow process), and all evaluable patients above age 65 achieved remission on study. Based on this promising data, we are currently conducting a phase II study of alisertib combined with “7+3” induction chemotherapy, specifically for newly diagnosed patients with higher risk AML (clinicaltrials.gov Identifier:02560025).
Supplementary Material
Acknowledgments
Takeda Pharmaceuticals kindly provided funding and a supply of alisertib for the conduct of this study.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/4/719
References
- 1.Rai KR, Holland JF, Glidewell OJ, et al. Treatment of acute myelocytic leukemia: a study by cancer and leukemia group B. Blood. 1981;58(6):1203–1212. [PubMed] [Google Scholar]
- 2.Yates J, Glidewell O, Wiernik P, et al. Cytosine arabinoside with daunorubicin or adriamycin for therapy of acute myelocytic leukemia: a CALGB study. Blood. 1982;60(2):454–462. [PubMed] [Google Scholar]
- 3.Yates JW, Wallace HJ, Jr, Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother Rep. 1973;57(4):485–488. [PubMed] [Google Scholar]
- 4.Garcia-Manero G, Tambaro FP, Bekele NB, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J Clin Oncol. 2012;30(18):2204–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24): 4854–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serve H, Krug U, Wagner R, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31(25):3110–3118. [DOI] [PubMed] [Google Scholar]
- 8.Röllig C, Müller-Tidow C, Hüttmann A, et al. Sorafenib versus placebo in addition to standard therapy in younger patients with newly diagnosed acute myeloid leukemia: results from 267 patients treated in the randomized placebo-controlled SAL-Soraml trial. Blood (ASH Annual Meeting Abstracts). 2014;124 (Abstract 6). [Google Scholar]
- 9.Stone RM, Mandrekar S, Sanford BL, et al. The multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (C) induction (ind), high-dose C consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18–60 with FLT3 mutations (muts): An international prospective randomized (rand) P-controlled double-blind trial (CALGB 10603/RATIFY [Alliance]). Blood (ASH Annual Meeting Abstracts). 2015;126 (Abstract 6). [Google Scholar]
- 10.Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508–1516. [DOI] [PubMed] [Google Scholar]
- 11.Malumbres M, Perez de Castro I. Aurora kinase A inhibitors: promising agents in anti-tumoral therapy. Expert Opin Ther Targets. 2014;18(12):1377–1393. [DOI] [PubMed] [Google Scholar]
- 12.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4(12):927–936. [DOI] [PubMed] [Google Scholar]
- 13.Hoar K, Chakravarty A, Rabino C, et al. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27(12):4513–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honda R, Korner R, Nigg EA. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003;14(8):3325–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly KR, Nawrocki ST, Espitia CM, et al. Targeting Aurora A kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a FOXO-dependent mechanism. Int J Cancer. 2012;131(11):2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sehdev V, Katsha A, Ecsedy J, Zaika A, Belkhiri A, El-Rifai W. The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer. 2013;119(4):904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sehdev V, Peng D, Soutto M, et al. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Mol Cancer Ther. 2012;11(3):763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huck JJ, Zhang M, Mettetal J, et al. Translational exposure-efficacy modeling to optimize the dose and schedule of taxanes combined with the investigational Aurora A kinase inhibitor MLN8237 (alisertib). Mol Cancer Ther. 2014;13(9):2170–2183. [DOI] [PubMed] [Google Scholar]
- 19.Cervantes A, Elez E, Roda D, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(17):4764–4774. [DOI] [PubMed] [Google Scholar]
- 20.Dees EC, Cohen RB, von Mehren M, et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res. 2012;18(17):4775–4784. [DOI] [PubMed] [Google Scholar]
- 21.Melichar B, Adenis A, Lockhart AC, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. 2015;16(4):395–405. [DOI] [PubMed] [Google Scholar]
- 22.Matulonis UA, Sharma S, Ghamande S, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol. 2012;127(1):63–69. [DOI] [PubMed] [Google Scholar]
- 23.Barr PM, Li H, Spier C, et al. Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J Clin Oncol. 2015;33(21):2399–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldberg SL, Fenaux P, Craig MD, et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk Res Rep. 2014;3(2):58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly KR, Shea TC, Goy A, et al. Phase I study of MLN8237–investigational Aurora A kinase inhibitor–in relapsed/refractory multiple myeloma, non-Hodgkin lymphoma and chronic lymphocytic leukemia. Invest New Drugs. 2014;32(3):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldberg SL, Fenaux P, Craig MD, et al. Phase 2 study of MLN8237, an investigational aurora A Kinase (AAK) inhibitor in patients with acute myelogenous leukemia (AML) or myelodysplastic syndromes (MDS). Blood (ASH Annual Meeting Abstracts). 2010;116 (Abstract 3273). [Google Scholar]
- 28.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 29.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21(24):4642–4649. [DOI] [PubMed] [Google Scholar]
- 30.Attar EC, Johnson JL, Amrein PC, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. 2013;31(7):923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361(13):1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Middeke JM, Herold S, Rucker-Braun E, et al. TP53 mutation in patients with high-risk acute myeloid leukaemia treated with allogeneic haematopoietic stem cell transplantation. Br J Haematol. 2016;172(6):914–922. [DOI] [PubMed] [Google Scholar]
- 34.Wattel E, Preudhomme C, Hecquet B, et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84(9):3148–3157. [PubMed] [Google Scholar]
- 35.Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci. 2007;120:2987–2996. [DOI] [PubMed] [Google Scholar]
- 37.Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786(1):60–72. [DOI] [PubMed] [Google Scholar]
- 38.Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015;34(5):537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hegarat N, Smith E, Nayak G, Takeda S, Eyers PA, Hochegger H. Aurora A and Aurora B jointly coordinate chromosome segregation and anaphase microtubule dynamics. J Cell Biol. 2011;195(7):1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marumoto T, Hirota T, Morisaki T, et al. Roles of aurora-A kinase in mitotic entry and G2 checkpoint in mammalian cells. Genes Cells. 2002;7(11):1173–1182. [DOI] [PubMed] [Google Scholar]
- 41.Marumoto T, Honda S, Hara T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278(51):51786–51795. [DOI] [PubMed] [Google Scholar]
- 42.Mori D, Yano Y, Toyo-oka K, et al. NDEL1 phosphorylation by Aurora-A kinase is essential for centrosomal maturation, separation, and TACC3 recruitment. Mol Cell Biol. 2007;27(1):352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3(1):51–62. [DOI] [PubMed] [Google Scholar]
- 44.Gu J, Gong Y, Huang M, Lu C, Spitz MR, Wu X. Polymorphisms of STK15 (Aurora-A) gene and lung cancer risk in Caucasians. Carcinogenesis. 2007;28(2):350–355. [DOI] [PubMed] [Google Scholar]
- 45.Sakakura C, Hagiwara A, Yasuoka R, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer. 2001;84(6):824–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatsuka M, Sato S, Kitajima S, et al. Overexpression of Aurora-A potentiates HRAS-mediated oncogenic transformation and is implicated in oral carcinogenesis. Oncogene. 2005;24(6):1122–1127. [DOI] [PubMed] [Google Scholar]
- 47.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2(1):21–32. [DOI] [PubMed] [Google Scholar]
- 48.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20(2):189–193. [DOI] [PubMed] [Google Scholar]
- 49.Katayama H, Sasai K, Kawai H, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36(1):55–62. [DOI] [PubMed] [Google Scholar]
- 50.Mao JH, Wu D, Perez-Losada J, et al. Crosstalk between Aurora-A and p53: frequent deletion or downregulation of Aurora-A in tumors from p53 null mice. Cancer Cell. 2007;11(2):161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nair JS, Ho AL, Schwartz GK. The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell cycle. 2012;11(4):807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.