

Abstract
Uveal melanoma is the most common malignancy of the eye, but little is known about its underlying genetic defects. Melanomas of uveal origin, unlike those of the skin, are rarely familial and have not been linked consistently to mutations in tumor suppressor genes. Here, we investigated the Rb pathway in uveal melanoma. Most tumors displayed strong immunostaining for Rb and p16, suggesting that they were not mutationally inactivated. However, Rb was frequently phosphorylated at serine-807 and serine-811, and cyclin D1 was expressed in many of the tumors. Mutation of these serine residues prevented cyclin D-dependent phosphorylation from inactivating Rb in cultured cells. We conclude that Rb is frequently inactivated in uveal melanoma by phosphorylation of residues in the COOH-terminal region that regulate its activity, and one mechanism for this phosphorylation is overexpression of cyclin D.
Introduction
Uveal melanoma is the most common cancer of the eye and leads to metastatic death in up to 53% of patients (1). Unlike cutaneous melanoma, little is known about the underlying genetic changes in uveal melanoma. Rb3 is the prototype tumor suppressor protein and is a major target for mutations in cancer (2). Rb is inactivated in most tumors, either by mutation of the Rb gene or by hyperphosphorylation of the protein as a result of mutations elsewhere in the Rb pathway (e.g., loss of p16, overexpression of cyclin D or cdk4; Ref. 2). Mutations in the Rb pathway, particularly those that affect p16 and cdk4, play an important role in cutaneous melanoma (3), but there is little evidence that these mutations are prevalent in uveal melanoma. Germ-line mutations in p16 are extremely rare in uveal melanoma patients, even among those with a family history of melanoma (4–6). Loss of heterozygosity at the chromosome 9p21 locus has been reported in up to 32% of uveal melanomas, but mutation of the p16 gene is rarely observed (7, 8). Likewise, germ-line or tumor mutations in cdk4 are rare in uveal melanoma (5, 9). The status of Rb itself has not been investigated adequately in this tumor. In the present study, we have used immunohistochemical analysis in 32 tumor specimens and transcriptional assays in cultured cells to examine the Rb pathway in uveal melanoma. By immunostaining, both Rb and p16 were expressed in the vast majority of tumors. Cyclin D was also expressed in most melanoma cells, and immunostaining with a phospho-Rb antibody revealed that two specific serine residues in the COOH-terminal region of Rb were frequently phosphorylated in these tumors. In transcriptional repression assays, these serine residues were required for cyclin D-mediated inactivation of Rb. Thus, our results suggest that the tumor suppressor activity of Rb is frequently inhibited in uveal melanoma by phosphorylation of specific residues in the COOH-terminal region of Rb, and that one mechanism for this phosphorylation is overexpression of cyclin D.
Materials and Methods
Immunohistochemistry
Immunohistochemistry was performed using the streptavidin-biotin method with the Vector ABC Elite kit (Vector Laboratories, Inc., Burlingame, CA). Specimens consisted of paraffin-embedded sections of 32 enucleated globes containing melanomas involving the choroid and ciliary body. Four-μm sections were obtained, deparaffinized, rehydrated with ethanol, and treated with 0.3% hydrogen peroxide and methanol to inhibit endogenous peroxidase activity. Heat-induced antigen retrieval was performed using microwave treatment in citrate buffer (Rb and p16 antibodies) or EDTA (cyclin D1 antibody) for 15 min. Primary antibodies were applied at 4°C overnight. Antibodies for Rb (C-15, 1:50 dilution; and IF-8, 1:40 dilution) and p16 (F-12; 1:75 dilution) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The antibody for phospho-Rb-serine 807/811 (1:25 dilution) was obtained from New England Biolabs, Inc. (Beverly, MA). The antibody for cyclin D1 (NCL-CYCLIN D1-GM, 1:40 dilution) was obtained from Novocastra Laboratories Ltd. (Newcastle Upon Tyne, United Kingdom). Positive controls included normal choroidal melanocytes (Rb and p16 antibodies), a mantle cell lymphoma (cyclin D1 antibody), and p16-null U20S osteosarcoma cells that constitutively hyperphosphorylate Rb (phospho-Rb antibody). Negative controls included Rb-null C33A cervical carcinoma cells (Rb antibody), U2OS cells (p16 antibody), and normal choroidal melanocytes (phospho-Rb and cyclin D1 antibodies). The secondary antibody alone was used as an additional negative control for all antibodies. The percentage of positive cells was estimated by counting at least 200 cells in at least eight ×40 fields for each specimen. In most cases, at least two sections from each tumor were analyzed for each antibody.
Transcription Assays and Plasmid Constructs
For CAT assays, 0.2 μg of the reporter plasmid pSVEC-G (Gal4 sites upstream of the SV40 enhancer and the E1b TATA box driving the CAT gene) along with 0.5 μg of the indicated expression vectors was transfected into Rb-null C33A cells in a total of 10 μg of DNA by the calcium phosphate method as described previously (10). A phosphorimager was used to quantify CAT activity. Expression plasmids included the following Gal4-tagged Rb proteins: G-A (domain A, amino acids 379–602), G-B (domain B, amino acids 620–792), and RbC (the COOH-terminal region, amino acids 767–928), as described previously (10). RbCΔ2 was created by subcloning an SspI/EcoRI (amino acids 767–928) fragment from PSM.2S (which contains serine-to-alanine substitutions at serine-807 and serine-811; Ref. 11) into the Gal4 DNA binding domain expression vector pM2. Rb was functionally reconstituted in these assays by coexpressing domains A and B and the COOH-terminal region on separate proteins, as described previously (10). Cyclin D was expressed as an RC.CMV vector.
Results
Immunohistochemistry
Using two separate antibodies that detect hypo- and hyperphosphorylated forms of Rb, 94% of tumors had strong nuclear staining (≥20% positive cells) with a mean of 64% positive cells/tumor (Table 1; Fig. 1A). Most normal choroidal melanocytes had positive nuclear staining. Immunostaining for p16 revealed strong nuclear expression (≥20% positive cells) in all cases, with a mean of 76% positive cells/tumor (Fig. 1B). Most normal choroidal melanocytes also had positive nuclear staining. For cyclin D1, all tumors contained cells with positive nuclear staining (mean, 18% positive cells; range, 1–60%; Fig. 1C). Strong staining (≥20% positive cells) was observed in 41% of tumors; 59% contained ≥5% positive cells. Normal choroidal melanocytes were negative, suggesting that detection of cyclin D1 in melanoma cells reflected abnormally high expression.
Table 1.
Summary of immunohistochemical analysis
| Histological classification | Positive tumorsa
|
Phospho-Rb (serine-807/811)b | ||
|---|---|---|---|---|
| Rb | p16 | Cyclin D1 | ||
| Spindle | 8/9 | 9/9 | 4/9 | 3.3% (0.2–7.0%) |
| Mixed | 11/12 | 12/12 | 7/12 | 1.5% (0.1–3.9%) |
| Epithelioid | 11/11 | 11/11 | 2/11 | 1.4% (0.1–5.1%) |
Tumors were scored positive if nuclear staining was present in ≥20% of cells.
Percentage of positive cells (and range) among tumors in each histological grade.
Fig. 1.
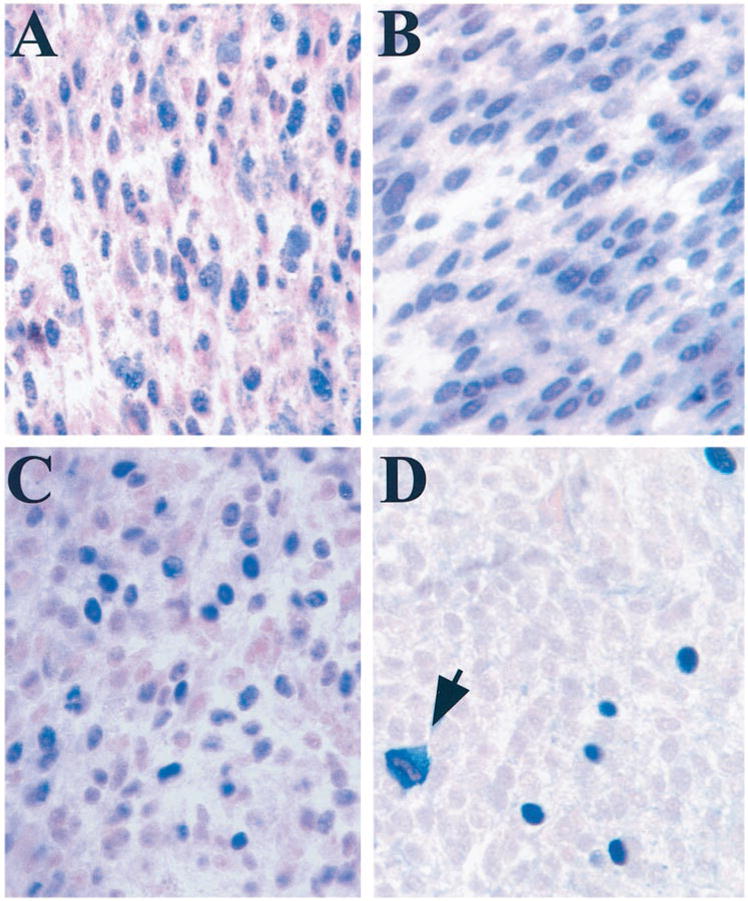
A, immunohistochemical staining of Rb in a representative uveal melanoma. Most melanoma cells had nuclear staining for Rb. ×40. B, immunohistochemical staining of p16 in a representative uveal melanoma. Most melanoma cells had nuclear staining for p16. ×40. C, immunohistochemical staining of cyclin D1 in a representative uveal melanoma. ×40. D, immunohistochemical staining for Rb phosphorylated on serine-807/811 in a representative uveal melanoma. The fraction of positive cells was similar to the fraction of cycling cells reported previously for uveal melanomas (12). Positively staining mitotic figures were observed frequently (arrow). ×40.
Using an antibody that specifically detects Rb that is phosphorylated at serine-807 and serine-811 (“phospho-Rb”), normal choroidal melanocytes were negative, but all tumors contained malignant cells with intense nuclear staining (Fig. 1D). The percentage of positive cells ranged from 0.1 to 5% of cells/tumor, consistent with the fraction of cycling cells in uveal melanomas stained for Ki-67 (12). Virtually all mitotic figures were positive and represented ~4% of all positive cells (Fig. 1D), further supporting the idea that phospho-Rb is expressed in cycling cells. There was a trend for increased cyclin D1 and phospho-Rb staining among melanomas of lower (spindle and mixed) histological grades (Table 1). Thus, although Rb does not appear to be mutated in most uveal melanomas, it is frequently phosphorylated on serine-807 and serine-811, and this phosphorylation may functionally inactivate Rb. Overexpression of cyclin D may be a common mechanism for maintaining Rb in a phosphorylated state in these tumors.
Transcription Assays
To determine the functional consequence of phosphorylating Rb on serine-807/811, we transfected Rb into an Rb-null cell line and measured active transcriptional repression, an activity that is required for Rb to arrest cells in G1 phase (13). Transfection of Rb repressed the activity of a CAT reporter by 85% (Fig. 2, Lane 2). Coexpression of cyclin D efficiently blocked this Rb repressor activity, presumably by activating endogenous kinases to phosphorylate Rb (Fig. 2, Lane 3). However, when serine-807 and serine-811 in Rb were converted to alanine, cyclin D was unable to block Rb repressor activity (Fig. 2, Lane 4). Phosphorylation of Rb was reduced in vitro when these serine residues were mutated (data not shown). Taken together, these results suggest that serine-807 and serine-811 are genuine targets for cyclin D-dependent phosphorylation, and that phosphorylation of these sites can inhibit Rb repressor activity.
Fig. 2.
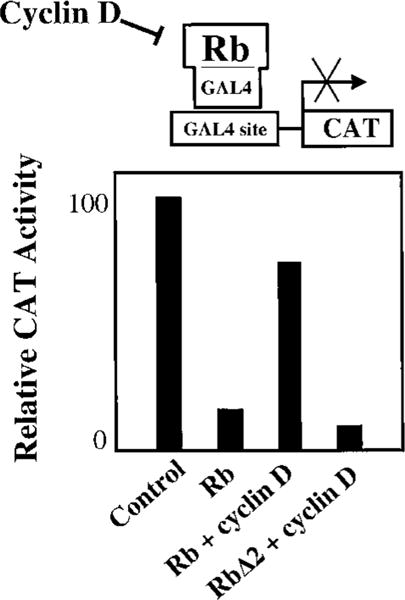
The phosphoacceptor sites serine-807 and serine-811 in the COOH-terminal region of Rb are required for cyclin D-mediated inhibition of Rb transcriptional repressor activity. To assay for active repression, Rb was fused to the DNA binding domain of Gal4 and coexpressed in Rb-null C33a cells, along with the pSVEC-G reporter containing Gal4 binding sites upstream of the SV40 enhancer. CAT activity from the reporter was measured with a phosphorimager. Rb, constructs containing wild-type Rb sequence (amino acids 379–928); RbΔ2, serine-807 and serine-811 have been converted to alanine to prevent phosphorylation of these sites. Cyclin D was coexpressed where indicated. Transfection of 0.5 μg of each expression vector (or vector control) and 0.2 μg of reporter was performed using the calcium phosphate method. Note that cyclin D blocks most of the transcriptional repression by Rb, but this effect is lost with mutation of serine-807/811.
Discussion
Rb inhibits proliferation by arresting cells in the G1 phase of the cell cycle (2). For cell division to occur, Rb is hyperphosphorylated and inactivated by cdks that interact with their cyclin partners to form active kinase complexes (2). cdks are in turn restrained by inhibitors such as p16, which blocks cdk4/6 and allows hypophosphorylated Rb to accumulate (14). The result of these interactions is a tightly regulated pathway that allows cell division only under appropriate physiological circumstances. In most cancers, this “Rb pathway” is disrupted such that Rb is inactivated, either by mutation of the Rb gene or by functional inactivation of Rb by hyperphosphorylation of the protein (2). In this study, we wished to determine the status of the Rb pathway in uveal melanoma.
We found that Rb is expressed strongly in most of the uveal melanomas, suggesting that the Rb gene is not commonly mutated in this cancer. However, we also found that serine-807 and serine-811 of Rb are often phosphorylated in these tumors, and this phosphorylation may block the tumor suppressor activity of Rb. Mutation of serine-807/811 prevented inhibition of Rb repressor activity by cyclin D-dependent phosphorylation. Furthermore, these sites have been shown to regulate Rb binding to the proto-oncogene c-abl (11), and this binding is important for tumor suppression by Rb (15). We showed previously that phosphorylation of two other sites in the COOH-terminal region (threonine-821/826) blocks active repression by Rb through induction of an intramolecular interaction that displaces histone deacetylases from the pocket (10). It is interesting that serine-807/811 can independently regulate active repression by Rb, possibly by inducing a similar intramolecular interaction. Taken together, our findings support the idea that Rb is functionally inactivated in uveal melanomas by phosphorylation of these (and potentially other) cdk phosphoacceptor sites.
One mechanism for inappropriately phosphorylating Rb is by mutation of p16 (2). However, we found no evidence for p16 inactivation in uveal melanoma. In one recent report, loss of heterozygosity at the p16 locus was observed in 24% of uveal melanomas, half of which had a homozygous deletion that included this locus (8). However, no mutations within the p16 gene were found, and no other evidence was presented that p16 was specifically targeted by these genetic rearrangements. Thus, most available evidence suggests that p16 is not a frequent target of inactivating mutations in uveal melanoma.
Another mechanism for hyperphosphorylating Rb is by overexpression of cyclin D (2). We found positive immunostaining for cyclin D in most tumors, whereas normal choroidal melanocytes were negative. Overexpression of cyclin D has been observed in a number of cancers as a result of amplification, translocation, or other rearrangement of the gene, and these mutations presumably contribute to tumorigenesis by activating endogenous cdk4/6 to phosphorylate Rb (2). In support of this possibility, we show that overexpression of cyclin D in cultured cells blocks active transcriptional repression by Rb, which is required for Rb to arrest cells in G1 (10, 13). Others have further shown that overexpression of cyclin D can overcome Rb-mediated tumor suppression in vivo (16). Therefore, the tumor suppressor function of Rb appears to be inhibited in uveal melanomas by phosphorylation of specific cdk phosphoacceptor sites, and this phosphorylation may be attributable to, at least in some cases, overexpression of cyclin D. Because some of the tumors were only weakly positive for cyclin D1, other proteins in the Rb pathway (e.g., cyclin D2, cyclin D3, cyclin E, or cdk4/6) may also be deregulated in some tumors.
Overexpression of cyclin D may also serve to deregulate the Rb pathway by another recently described mechanism (17). Cyclin D, when complexed with cdk4/6, can sequester p21 and p27 so that they are unavailable to inhibit cdk2 (Ref. 17; Fig. 3). Cyclin D also competes directly with p16 for binding to cdk4/6 (18, 19), as demonstrated in cultured uveal melanoma cells where p16 protein levels were normal, but p16-cdk4 complexes were not found as in normal choroidal melanocytes (20). Thus, cyclin D can activate cdk4/6 to phosphorylate Rb, and it can interfere directly and indirectly with several cdk inhibitors, resulting in the downstream activation of cdk2 and circumvention of the Rb checkpoint (Fig. 3). This study provides new insights into abnormalities of the Rb pathway in uveal melanoma, and it suggests that the molecular pathophysiology of this form of melanoma may be distinct from its cutaneous counterpart.
Fig. 3.
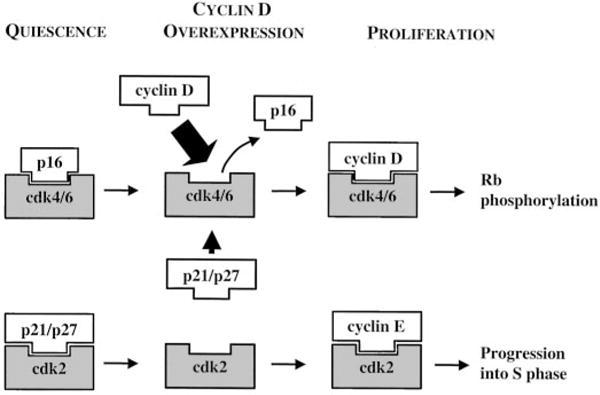
Overexpression of cyclin D may disrupt the Rb pathway at several points: (a) it competes with p16 for interaction with cdk4/6, which then becomes activated to phosphorylate Rb (top); (b) cyclin D-cdk4/6 complexes sequester the cdk inhibitors p21 and p27 so that they are unavailable to block cdk2 (bottom). Cyclin E-cdk2 can then act downstream of Rb to initiate cell cycle progression into S phase. See text for details.
Acknowledgments
We thank Belinda McMahan in the Immunomorphology Core Laboratory for performing immunohistochemistry and Dr. Morton Smith (University of Wisconsin) for assistance in obtaining tumor specimens.
Footnotes
These studies were supported by grants (to J. W. H.) from the NIH and Research to Prevent Blindness, Inc.
The abbreviations used are: Rb, retinoblastoma protein; cdk, cyclin-dependent kinase; CAT, chloramphenicol acetyltransferase.
References
- 1.Diener-West M, Hawkins BS, Markowitz JA, Schachat AP. A review of mortality from choroidal melanoma. II. A meta-analysis of 5-year mortality rates following enucleation, 1966 through 1988. Arch Ophthalmol. 1992;110:245–250. doi: 10.1001/archopht.1992.01080140101036. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ. Cancer cell cycles. Science (Washington DC) 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 3.Monzon J, Liu L, Brill H, Goldstein AM, Tucker MA, From L, McLaughlin J, Hogg D, Lassam NJ. CDKN2A mutations in multiple primary melanomas. N Engl J Med. 1998;338:879–887. doi: 10.1056/NEJM199803263381305. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Egan KM, Gragoudas ES, Kelsey KT. Constitutional alterations in p16 in patients with uveal melanoma. Melanoma Res. 1996;6:405–410. doi: 10.1097/00008390-199612000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Soufir N, Bressac-de Paillerets B, Desjardins L, Levy C, Bombled J, Gorin I, Schlienger P, Stoppa-Lyonnet D. Individuals with presumably hereditary uveal melanoma do not harbour germline mutations in the coding regions of either the P16INK4A, P14ARF or cdk4 genes. Br J Cancer. 2000;82:818–822. doi: 10.1054/bjoc.1999.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohta M, Nagai H, Shimizu M, Rasio D, Berd D, Mastrangelo M, Singh AD, Shields JA, Shields CL, Croce CM, Heubner K. Rarity of somatic and germline mutations of the cyclin-dependent kinase 4 inhibitor gene, CDK4I, in melanoma. Cancer Res. 1994;54:5269–5272. [PubMed] [Google Scholar]
- 7.Ohta M, Berd D, Shimizu M, Nagai H, Cotticelli MG, Mastrangelo M, Shields JA, Shields CL, Croce CM, Huebner K. Deletion mapping of chromosome region 9p21–p22 surrounding the CDKN2 locus in melanoma. Int J Cancer. 1996;65:762–767. doi: 10.1002/(SICI)1097-0215(19960315)65:6<762::AID-IJC9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 8.Merbs SL, Sidransky D. Analysis of p16 (CDKN2/MTS-1/INK4A) alterations in primary sporadic uveal melanoma. Invest Ophthalmol Vis Sci. 1999;40:779–783. [PubMed] [Google Scholar]
- 9.Tsao H, Benoit E, Sober AJ, Thiele C, Haluska FG. Novel mutations in the p16/CDKN2A binding region of the cyclin-dependent kinase-4 gene. Cancer Res. 1998;58:109–113. [PubMed] [Google Scholar]
- 10.Harbour JW, Luo RX, Dei Sante A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 11.Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 12.Bardenstein DS, Char DH, Kaleta-Michaels S, Kroll SM. Ki-67 and bromodeoxyuridine labeling of human choroidal melanoma cells. Curr Eye Res. 1991;10:479–484. doi: 10.3109/02713689109001755. [DOI] [PubMed] [Google Scholar]
- 13.Zhang HS, Postigo AA, Dean DC. Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFb, and contact inhibition. Cell. 1999;97:53–61. doi: 10.1016/s0092-8674(00)80714-x. [DOI] [PubMed] [Google Scholar]
- 14.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature (Lond) 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 15.Whitaker LL, Su H, Baskaran R, Knudsen ES, Wang JYJ. Growth suppression by an E2F-binding-defective retinoblastoma protein (RB): contribution from the RB C pocket. Mol Cell Biol. 1998;18:4032–4042. doi: 10.1128/mcb.18.7.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 18.Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature (Lond) 1998;395:237–243. doi: 10.1038/26155. [DOI] [PubMed] [Google Scholar]
- 19.Parry D, Mahony D, Wills K, Lees E. Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol. 1999;19:1775–1783. doi: 10.1128/mcb.19.3.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mouriaux F, Casagrande F, Pillaire MJ, Manenti S, Malecaze F, Darbon JM. Differential expression of G1 cyclins and cyclin-dependent kinase inhibitors in normal and transformed melanocytes. Invest Ophthalmol Vis Sci. 1998;39:876–884. [PubMed] [Google Scholar]