

Abstract
Clostridium haemolyticum is the causal agent of bacillary hemoglobinuria in cattle, goat, sheep, and ruminants. In this study, we report the first recorded human-infecting C. haemolyticum strain collected from an 18-year-old woman diagnosed with acute lymphoblastic leukemia. After failure of traditional techniques, only next-generation sequencing (NGS) technology in combination with bioinformatics, phylogenetic, and pathogenomics analyses revealed that our King Faisal Specialist Hospital and Research Center (KFSHRC) bacterial isolate belongs to C. haemolyticum species. KFSHRC isolate is composed of 1 chromosome and 4 plasmids. The total genome size is estimated to be 2.7 Mbp with a low GC content of 28.02%. Comparative pathogenomics analysis showed that C. haemolyticum KFSHRC isolate is a potential virulent pathogenic bacterium as it possesses the virulence factors necessary to establish an infection, acquire essential nutrients, resist antimicrobial agents, and tolerate hostile conditions both in the human host and in its surrounding environment. These factors are included in the main chromosome in addition to novel recombination of the plasmids, and they could be the reason for the incidence of that human infection. This work demonstrated the importance of using NGS in medical microbiology for pathogen identification. It also demonstrates the importance of sequencing more microbial samples and sharing this information in public databases to facilitate the identification of pathogenic microbes with better accuracy.
Keywords: Clostridium haemolyticum, C. novyi sensu lato, next-generation sequencing techniques, bioinformatics, phylogeny, pathogenomics, acute lymphoblastic leukemia, human infection
Introduction
Clostridium haemolyticum is the causal organism of the fatal disease bacillary hemoglobinuria in cattle, goat, sheep, and ruminants.1–3 Clostridium haemolyticum is a widespread bacterial species that is commonly found in both soil and water sediments4 and is mainly characterized by the production of beta toxin. However, it can also produce alpha toxin when infected with the phage derived from its close relative Clostridium novyi type A.5 Beta toxin produced by C. haemolyticum is serologically identical to that of C. novyi type B.6 Clostridium haemolyticum is closely related to animals and human pathogens C. novyi and Clostridium botulinum. However, little information is available about the genetic relationships among these species on a genomic level.7,8
In the last decade, several Clostridial genomes have been sequenced, including the genomes of Clostridium difficile, Clostridium perfringens, C. novyi-NT, C. botulinum types A, B, E, and F and group III, and recently C. haemolyticum.9–15 These genomes range in size between 2.5 and 6 Mb with a low GC content. In addition to the bacterial chromosome, a number of plasmids are usually observed, usually 3 per genome. The extrachromosomal elements, such as plasmids and circular prophages, play pivotal role in the diseases caused by these bacteria in animals and humans.4,9,15 The high similarity of the observed sequenced chromosomes of C. botulinum (group III), C. novyi, and C. haemolyticum has led to the proposal of a collective “genospecies” named C. novyi sensu lato.9
The identification of the Clostridium species relies on 16S ribosomal RNA sequencing and DNA-DNA hybridizations.8,9 However, these techniques have limitations regarding (sub-) species characterization,16 and they cannot be extended to reveal the included pathogenicity factors. The use of next-generation sequencing (NGS) technology has provided a solution to overcome these limitations. Next-generation sequencing enables high throughput sequencing of whole genomes at affordable time and cost.
In this study, we report a human-infecting C. haemolyticum strain collected from an 18-year-old female diagnosed with acute lymphoblastic leukemia (ALL) and identified using NGS techniques. The identification and characterization of this strain would not have been possible without the use of NGS techniques, especially with this fastidious hard-to-culture bacteria. As we will explain in detail in the article, the genome of our C. haemolyticum strain shows a conserved core chromosome with the presence of plasmids, bacteriophages, and transposons. It also includes the pathogenic factor that could explain the reason of the incidence in the patient.
Materials and Methods
Subject and ethical statement
An 18-year-old female was diagnosed in June 2011 with ALL, and she started chemotherapy and had stem cell transplant in September 2011. In May 2013, she presented with fever and right hip pain. She was found to have right hip osteomyelitis and relapse of ALL. Several blood and tissue cultures were negative for bacteria, mycobacteria, and fungi. She continued to have persistent fever despite different courses of empirical broad-spectrum antimicrobial treatments. However, the empirical antimicrobial treatment failed, but she responded to 6 weeks of pipracillin/tazobactam and doxycycline. Two sets of blood cultures were taken for this patient. The first blood culture was taken on May 17, 2013, and the second on January 2014. None grew in any bacterial or fungal culture media despite the prolonged incubation and adding different media and nutrients. Both acid-fast and modified acid-fast stains were negative. Mycobacterium culture was also negative. The failure of growth can be attributed to the need for anaerobic growth condition for Clostridium spp. or the induction of lytic phases of the lysogenic bacteriophages.17
To further investigate the case, we submitted a request for study to the ethical review board of King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Kingdom of Saudi Arabia, and a written informed consent from the patient for participating in this study was obtained. The study was approved by the ethical review board. In addition, this study received approval from the Office of Research Affair at KFSHRC with reference number 2160098.
DNA extraction, quantification, and sequencing
DNA was extracted from blood culture broth (sample obtained in January 2014) using the manual extraction Gentra Puregene kit (Qiagen CAT# 158389; Autogen INC, Holliston, MA, USA) and quantified using the NanoDrop spectrophotometer. The quality of subjected DNA sample was determined by loading 150 mg of diluted DNA in 1% agarose E-gel (Invitrogen, Paisley, UK). We have conducted 2 sequencing runs using the Personal Genome Machine (PGM) sequencer from Life Technologies (Thermo Fisher, Carlsbad, CA, USA). Briefly, library was prepared using 50 ng of extracted DNA universal primers and AmpliSeq HiFi mix (Thermo Fisher). The 15-cycle amplification product was subjected to digestion using FuPa reagent (Thermo Fisher) and ligated with universal adapters. Purified barcoded libraries were quantified by real-time polymerase chain reaction (qPCR) and normalized to 100 pM. Then, they were subjected to emulsion polymerase chain reaction (ePCR) using the Ion PGM Template OT2 400 and the Ion OneTouch System. The ePCR template was enriched using the OneTouch ES (Thermo Fisher) to produce Ion Sphere particles ready for sequencing as single-ended read library. The library was then loaded to the Ion 318 Chip (Thermo Fisher) and sequenced using the PGM sequencer. The output of the sequencer is a set of single-ended reads with expected maximum length of 400 bp.
Draft genome accession number
Data from our draft genome of KFSHRC-CH1 isolate were deposited in National Center for Biotechnology Information (NCBI)-GenBank with an accession number LSZB01000000.
Bioinformatics analysis workflow
We have developed an analysis pipeline to identify the suggested pathogen and annotate it. The basic steps of this pipeline are summarized in Figure 1. First, the quality of the reads was assessed and reads with quality score less than 20 bp were trimmed out. Any read with less than 30 bp was discarded. The reads were then passed to the program MetaPhlAn18 for primary identifications of microbial families included in the samples based on unique and clade-specific marker genes. In parallel to running MetaPhlAn analysis, we ran the BLAST program to map each read to the nonredundant nucleotide database of NCBI. We observed high-rate reads mapped to the human genome, which is expected as a result of contamination with human DNA during the sample preparation (Table 1).
Figure 1.

Analysis pipeline.
Table 1.
Basic read information for each sequencing run.

To validate and eliminate reads from host DNA, we conducted direct mapping of the whole reads to the reference human genome (version hg19). For this purpose, we used the program TMAP (https://github.com/iontorrent/TMAP). TMAP is a version of BWA tuned for IonTorrent sequencing, where basic flow signals are taken into account. A read is considered a human if it is aligned to the human reference genome, with at least 80% sequence identity. For the nonhuman reads, we performed additional analysis steps: first, we conducted de novo assembly of the nonhuman reads using the program MIRA (version 4)19,20 and computed related assembly statistics like N50 at different contig lengths. Second, we mapped the reads back to the bacterial genomes thought to be the pathogen; these are the top-ranked bacteria based on MetaPhlAn, BLAST results, and related taxa analysis. Coverage and related mapping statistics are computed to evaluate the similarity to the target bacterial genome. The integration of the different tools and execution of the whole pipeline are achieved through python scripts developed in house. A version of this pipeline is currently being imported in the workflow system Tavaxy21 to be used by other researchers. To further investigate the deleted regions, we retrieved the genome annotation from the GenBank and investigated the missing genes. We used Mauve tool for multiple alignment of conserved genomic sequence comparison with Rearrangement22 and CoCoNUT for the genome comparison and analysis.23
Phylogenetic analysis and taxonomic assignment
The 16S rDNA sequences of our isolate were used to construct a phylogenetic relationship with other Clostridium species. We acquired partial 16S rDNA sequences of selected Clostridium species from GenBank. These sequences were first visualized using the BioEdit software. The program clustalW was used to provide primary alignment to help us curate the sequences and remove noncommon terminals. (In clustalW, default parameters were used; gap opening penalty = 10, gap extension penalty = 5, delay divergent sequences = 40%.)
In addition, we used other alignment methods, namely, MAFFT multiple sequence alignment program available online at http://www.ebi.ac.uk/Tools/msa/mafft/, PRRN (the best-first iterative refinement strategy with tree-dependent partitioning) multiple sequence alignment program available online at http://www.genome.jp/tools/prrn, and the program MUSCLE within the MEGA6 package. We compared the resulting alignments, and the final alignments were improved manually and prepared in FASTA, MEGA formats using format converter tool v2.2.5 available online at http://www.hiv.lanl.gov/content/sequence/FORMAT_CONVERSION/form.html.
The final alignment we used was the one computed using MUSCLE in the MEGA software.
To establish the relationships among taxa, a phylogenetic tree was constructed using the Maximum Likelihood (ML) method based on the Jukes-Cantor model that best fits the data according to Akaike Information Criterion (AIC) criterion.24 The MEGA6 program was used to conduct phylogenetic analysis.25
To further confirm the taxonomic assignment, we used the system QIIME26 for 16S rRNA-based taxonomy assignment. The workflow used in this system was based on aligning the input NGS reads to a built-in database of variable regions of rRNA sequences. The output of the system was compared to the closely related strain obtained by the phylogeny analysis. The results were consistent and confirmed that our input organism is closely related to C. haemolyticum.
Gene annotation and pathogenomics comparative analysis
Virulence gene sequences and functions, corresponding to different major bacterial virulence factors of selected pathogens, were collected from GenBank and validated in virulence factors of pathogenic bacteria database at http://www.mgc.ac.cn/VFs/.27 For this step, we used scripts developed in-house to retrieve the annotation from the text files and compared them with each other and with the pathogenicity database. The results were then arranged in a tabular format showing present/absent factors in each pathogen.
Results
Genome sequencing and sequence analysis
General genome features
Data from our draft genome of KFSHRC-CH1 isolate (accession number LSZB01000000) have shown high similarity of 86.03% to the genome of C. haemolyticum NCTC 9693. Our KFSHRC-CH1 isolate genome size is comparably small in relation to other clostridium genomes with a circular chromosome of 2.7 Mbp and a low GC content of 28.02% (Figures 2 and 3). The presence of 3 plasmids, namely, p2Ch9693, p3Ch9693_1, and p3Ch9693_2, was confirmed. Plasmid p1Ch9693 of C. haemolyticum and all of the plasmids of C. botulinum BKT015925 were missing in our genome. The remaining reads in our genome which did not map to C. haemolyticum or C. botulinum BKT015925 are mapped well to plasmid pCLG1 of the bacteria C. botulinum D str. 1873. The current bacteria could include some horizontal gene transfer from other (Clostridium) species. This result needs a confirmation by amplifying larger segments of the genome at hand that include segments from different species. But in our case we could not have a declaration to further aliquot of the submitted samples.
Figure 2.

The annotation tracks for our main chromosome, represented by circles from outer to inner, include (1) open reading frames (ORFs) on the forward strand (length ⩾100 codons), (2) ORFs on the reverse strand (length ⩾100 codons), (3) Clostridium botulinum genome BLAST alignment, (4) Clostridium haemolyticum genome BLAST alignment, (5) GC content, (6) GC skew (+/−), and (7) the concatenated contigs of our chromosome.
Figure 3.

The annotation tracks of our plasmidome (collection of our plasmids), represented by circles from outer to inner, include (1) open reading frames (ORFs) on the forward strand (length ⩾100 codons), (2) ORFs on the reverse strand (length ⩾100 codons), (3) Clostridium botulinum plasmid BLAST alignment, (4) Clostridium haemolyticum plasmid BLAST alignment, (5) GC content, 6) GC skew (+/−), and (7) our concatenated plasmid regions.
Pathogen identification and phylogenetic analysis
The primary analysis of MetaPhlAn showed that C. botulinum BKT015925 is the most dominant species in the sample (Figure 4). The appearance of other bacterial species in the MetaPhlAn diagram is explained by the similarity of other bacteria to C. botulinum. Furthermore, basic local alignment search tool nucleotide (Blastn) results from a long assembled contigs query of 749933 bp showed that our organism is similar to C. botulinum BKT015925 with an E-value of 0.0, percentage identity of 93%, query cover of 97%, and total score of 1.059e+06. However, during the preparation of this manuscript, the genome of C. haemolyticum NCTC 9693 has been released.15 This has dramatically changed the picture; the Blast results (Table 2) show that most of the sequencing reads were mapped toward C. haemolyticum bacterium, whereas the second organism with highest number of reads mapped toward was C. botulinum BKT015925. We noticed reads mapped to the C. botulinum plasmid NC_015426.1 and not to C. haemolyticum and the presence of some C. haemolyticum plasmids. These results suggest that our isolate which is a new variant of C. haemolyticum may have acquired some genetic element from C. botulinum through plasmid transfer or any means of horizontal gene transfer, although further genome assembly and analysis on the gene level would resolve this question.
Figure 4.

MetaPhlAn primary identification of the tested taxon.
Table 2.
Numbers of primary and secondary hits of Clostridium haemolyticum KFSHRC genome corresponding to reference genomes and plasmids of C. haemolyticum and Clostridium botulinum.

The table entries include the top 10 taxa and the number of reads mapped to them. The Column titled “Primary hit” counts how many reads yielded the respective organism as first hit. The Column titled “Secondary hit” counts how many reads yielded the respective organism as second hit.
Same scenario was observed when constructing the phylogenetic relationship between our isolate and other known Clostridial strains. Before the release of C. haemolyticum NCTC 9693, 16S rDNA-based ML phylogenetic tree (Figure 5) showed that our isolate is intermediate taxa in a clade that contains C. botulinum group c isolates and C. heamolyticum strains with a bootstrap value of 95%. However, after the addition of C. haemolyticum NCTC 9693 16Sr DNA sequence to the analysis, ML phylogenetic tree (Figure 6) showed that our isolate is located in a clade containing only C. haemolyticum isolates. Thus, phylogenetic analysis using 16S rDNA suggests that our KFSHRC-CH1 isolate belongs to species C. haemolyticum and is closely related to the strain NCTC 9693. Similarly, QIIME has classified our 16S rRNA as C. haemolyticum with a consensus identity of 99% to 100%. Figure 7 shows the percentage identity of each variable region of the rRNA and its assignment to C. haemolyticum.
Figure 5.

16S rDNA-based maximum likelihood phylogenetic tree without the presence of Clostridium haemolyticum NCTC 9693.
Figure 6.

16S rDNA-based Maximum likelihood phylogenetic tree in the presence of Clostridium haemolyticum NCTC 9693.
Figure 7.

Percentage identity of alignment of our rRNA to Clostridium haemolyticum as reported by QIIME.
The assembled isolate genome (Table 3) is composed of 801 contigs with a total length of ≈2.9 Mbp and N50 of 24 759 bp. The length of the largest contig is about 134 677 Kbp. To further confirm the results obtained from using the raw reads, we used the assembled contigs as queries against BLAST database. Table 4 shows the top 10 hits sorted by total (nonoverlapping) alignment length. The results in this table are consistent with the results in Table 2 for mapping our reads to C. botulinum and C. haemolyticum.
Table 3.
Metrics for the assembly of the nonhuman reads using MIRA Assembler.
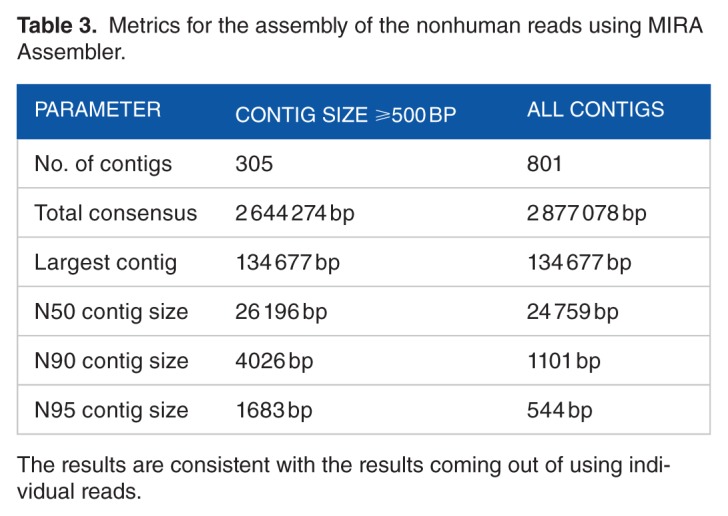
Table 4.
The top 10 hits sorted by total (nonoverlapping) alignment length.

For further confirmation, we mapped the bacterial (nonhuman) reads against the 2 alleged species C. botulinum and C. haemolyticum and their plasmids (Table 5; Figures 2 and 3). Results showed that 91% of the main chromosome of the C. haemolyticum genome is covered by our reads, and 3 out of 4 plasmids are well covered (97.38%–99.99%). Only plasmid p1Ch9693 was poorly covered with our reads (4.04%). As for C. botulinum, only 76% of the main bacterial chromosome is covered by our reads. All the 5 plasmids are not well covered (the mostly covered ones are plasmids 2 and 3, 27.8% and 30.27%, respectively). These mapping results further suggest that the major pathogen is more related to C. haemolyticum NCTC 9693. In addition, when analyzing the regions not mapped to both genomes, we found that the deleted regions include great match of about 82% to the plasmid pCGL1 of the bacteria C. botulinum D str. 1873 (Table 4). This suggests the inclusion of this plasmid in our genome by means of a horizontal gene transfer.
Table 5.
Mapping the Clostridium haemolyticum KFSHRC genome reads to Clostridium botulinum and C. haemolyticum (runs 1 and 2).

We also compared our draft genome to C. botulinum BKT015925 and to C. haemolyticum NCTC 9693 using the genome alignment programs Mauve and CoCoNUT (pairwise comparison option, computing local alignments with at least 85% identity, and minimum alignment length of 100 bp). The alignments that were common between the 2 aligners were considered. The results were as follows: 95% (2 499 026 bp) of our genome was aligned to C. haemolyticum NCTC 9693 genome (gap size = 108 630 bp), whereas 72% (2 317 723 bp) of it was aligned to C. botulinum BKT015925 (gap size = 889 869). These results confirm that our genome is more closely related to C. haemolyticum NCTC 9693 than to C. botulinum. Figure 8 shows plots representing alignment of contigs.
Figure 8.

(A) Alignment of Clostridium haemolyticum KFSHRC against Clostridium botulinum BKT015925 genome. (B) Alignment of C. haemolyticum KFSHRC against C. haemolyticum NCTC 9693 genome.
Genomic composition of KFSHRC-CH1 isolate
We compared the genes in C. botulinum BKT015925, C. haemolyticum, and those in plasmid pCGL1 from C. botulinum D str. 1873 with those in our isolate genome (Table 6). The full gene list is provided in Supplementary Material. The total number of annotated genes in our isolate is 1960. Of these, 1590 of them are common among C. botulinum BKT015925, C. haemolyticum, and pCGL1; 119 are common in C. haemolyticum; 127 and 124 are shared between our isolate and pCGL1 of C. botulinum, respectively. The common genes between our isolate and C. botulinum BKT015925 (1590+127 = 1717) are larger than those between ours and C. haemolyticum (1709 genes). However, the number of genes in C. botulinum BKT015925 that are not present in our isolate (956) is much higher than that in C. haemolyticum (119). This means our genomes loses big fraction of the genetic content of C. botulinum BKT015925 and C. haemolyticum.
Table 6.
N umber of genes present or absent in our genome and other inquiry genomes and plasmids.

Pathogenomics comparative analysis
Generally, our isolate showed more resemblance to both C. botulinum and C. haemolyticum. Our isolate and C. botulinum contained fibronectin-binding protein that was not detected in C. haemolyticum, whereas S-layer protein was only detected in C. botulinum and not in both C. haemolyticum and KFSHRC-CH1 isolate. KFSHRC-CH1 isolate and C. haemolyticum lacked the presence of Botulinum neurotoxin (BoNT) that was only present in C. botulinum.
Furthermore, our KFSHRC-CH1 isolate showed absolute similarity with C. haemolyticum when investigating plasmidborne toxin-producing genes. Results showed the absence of genes atx, cpb2, and tetX encoding for BoNT, Beta2 toxin, and tetanus toxin (TeTx), respectively (Tables 7 and 8).
Table 7.
Comparative pathogenomics of Clostridium spp. including our KFSHRC isolate (chromosomal genes).

Table 8.
Comparative pathogenomics (toxins) of Clostridium spp. including our KFSHRC isolate (plasmid genes).

Our analysis showed obvious divergence between our isolate and BKT015925 C. botulinum in many of the studied features. For example, we observed 6 types of bacterial toxins in our isolate, namely, neurotoxin type C1 (BoNT), alpha toxin (clostripain), C2 toxin, Epsilon toxin B Bacteriocin, Boclp (clostripain), and type D neurotoxin gene of phage d-18, whereas the 6 types of observed bacterial toxins in isolate BKT015925 were Tetanolysin O, bont C/D, alpha toxin (clostripain), C2 toxin, Epsilon toxin, and Bacteriocin. Moreover, we were able to detect the presence of 8 different clustered regularly interspaced short palindromic repeats (CRISPR) proteins, whereas only 3 proteins were observed with isolate BKT015925. In addition, we were able to detect genes related to sporulation, stress tolerance, and adherence virulence factors in isolate KFSHRC that were not mentioned in isolate BKT015925 (Table 9).
Table 9.
Comparison of KFSHRC and BKT015925 Clostridium botulinum isolates for the presence of putative genes coding for virulence proteins, antibiotic resistance, and CRISPR proteins.

Bacterial virulence factors
Genome analysis showed that our KFSHRC-CH1 isolate contains several chromosomal and plasmid-borne virulence factor genes that brand it to be virulent pathogenic bacterium. These virulence factors include mobility-related factors, toxinencoding genes, lipoproteins enzymes, phospholipase enzymes, drug resistance genes, hemolysin genes, stress tolerance factors, sporulation ability factors, and chemotaxis-related factors (Table 9). We observed the presence of 33 encoding flagella-associated genes. As expected, all flagella encoding genes were located on the bacterial chromosome (S1 Table). In addition, several toxin and hemolysin encoding genes were observed (S2 Table), and 41 drug, heavy metal, and antibiotic resistance genes were identified in our isolate (S3 Table). These include metallo-beta-lactamase superfamily proteins, multidrug resistance proteins, and, most interestingly, vancomycin b-type resistance protein (vanW). Moreover, we observed the presence of several stress tolerance-related proteins, such as carbon starvation protein carbon starvation protein (CstA), heat shock protein GrpE, chemical damaging agent resistance protein C, and several UV damage repair proteins (S4 Table). Furthermore, speculation ability factors were also detected (S5 Table). In addition, several genes and proteins involved in iron uptake and metabolism were detected in our isolate (S6 and S7 Tables).
CRISPR-associated and phage-related proteins
We observed the presence of several CRISPR-associated proteins such as Cas3, Cas5, CRISPR-associated helicase Cas3 domain protein, and Cas6, which provides acquired immunity against foreign DNA. In addition, we observed the presence of several phage proteins located both on the bacterial chromosome and on p1BKT015925 and p4BKT015925 (Table 9; S8 and S9 Tables).
Plasmidome
We observed evidence of the presence of DNA sequences belonging to the following plasmids—p2Ch9693,p3Ch9693_1, and p3Ch9693_2 of C. haemolyticum and plasmid pCLG1 of the bacteria C. botulinum D str. 1873 (Figure 3). The estimated approximate size of KFSHRC-CH1 isolate plasmidome is ≈214 Kbp with a low GC content of 29.85%. These plasmids contained genes for bacterial toxin production, such as C2 toxin component I and component II, putative epsilon toxin type B, clostripain, and alpha toxin. In addition, we observed thermolabile hemolysin gene in the plasmidome. We also observed metallo-betalactamase-resistant and carbamate kinase-resistant genes, in addition to several integrase catalytic subunits, 2F-recombinase, several putative IS transposases, and transposase mutator family proteins. Some phage-related encoding genes were also found.
Discussion
Clostridium botulinum (group III), C. novyi, and C. haemolyticum are notorious pathogens that cause botulism, gas gangrene/black disease, and bacillary hemoglobinuria, respectively. There is a very close genetic relationship among these species that has resulted in the term C. novyi sensu lato in reference to them.7,8 Generally, these species gain their pathogenic characters, such as the botulinum neurotoxin and the novyi alpha toxin, from their large plasmidome that consists of plasmids and circular prophages.9–15 Genomic comparisons between these species revealed 4 separate lineages, which did not rigorously draw a parallel with the species designations. Moreover, because of the frequent movement of plasmids and toxin genes across the lineage boundaries, the lineages and species are entwined.15 Skarin and Segerman15 explained the way in which the reorganization of the botulinum toxin and the novyi alpha toxin genes within the plasmidome can alter the pathogenic characteristics of any strain within C. novyi sensu lato. In this study, we identify a C. haemolyticum strain (C. novyi sensu lato) isolated blood sample of a morbid 18-year-old female diagnosed with ALL and characterize its genome using NGS techniques. Our isolate’s bacterial chromosome size has a small resemblance to other clostridium genomes with a circular chromosome of 2.7 Mbp and a low GC content of 28.02% but is comparable with C. botulinum C/D strain 08-BKT015925, 2.77 Mbp and 28%, and C. novyi A strain C. novyi-NT, 2.55 Mbp and 28%.10 As we stated before, this is the first report of C. haemolyticum (C. novyi sensu lato) isolated humans. Clostridium haemolyticum is a pathogen of cattle, goat, sheep, and ruminants.1–3 Other clostridium species such as C. botulinum C/D strain 08-BKT015925 was first isolated from poultry in Sweden at 2008, and C. novyi A strain C. novyi-NT was first isolated from gas gangrene in 1920. Previous phylogenetic studies in this group have shown that a strain of C. botulinum (group III), C. novyi, and C. haemolyticum species genetically more closely associated with strains from other species than the strains from its own species.7,8,15 Our results based on genomic analysis and 16S rRNA showed that our isolate belongs to C. haemolyticum and shows relative similarity to C. botulinum C/D strain 08-BKT015925 and close relation to C. novyi-NT. Thus, we can conclude that our KFSHRC-CH1 isolate belongs specifically to C. haemolyticum, a member of C. novyi sensu lato (Figure 8). Comparative pathogenomic analysis showed that our isolate exhibited more resemblance to C. haemolyticum and C. botulinum BKT015925 over any other studied C. botulinum strains (data not shown). These strains included C. botulinum ATCC 19397 (serotype A), C. botulinum ATCC 3502 (serotype A), C. botulinum Eklund 17B (serotype B), C. botulinum Hall (serotype A), C. botulinum Langeland (serotype F), C. botulinum Loch Maree (serotype A3), and C. botulinum Okra (serotype B1). Furthermore, our KFSHRC-CH1 isolate showed absolute similarity with C. haemolyticum for the investigated plasmidborne toxin-producing genes. These further affirm our previous conclusion about the identity of the studied isolate. Further analysis showed clear deviation between our C. haemolyticum KFSHRC-CH1 isolate and C. botulinum BKT015925 (group III) strain, the extensively studied strain. This divergence was clear in many genomic aspects, such as CRISPR proteins. Our results showed that KFSHRC-CH1 isolate contains Cas1, Cas2, Cas3, Cas4, Cas5, Cas6, Cst1, and CRISPR-associated protein, whereas only 3 proteins were observed with the isolate BKT015925. The presence of different CRISPR genes and proteins in KFSHRC and C. botulinum BKT015925 reflects divergent history of viral infection on both isolates. Further assertion of this conclusion is the observed characteristics such as lipoproteins, phospholipases, hemolysins, lipases, peptidases, proteases, phage proteins, toxins, and antibiotic resistance genes (Table 9). In addition, our pathogenomic analysis confirmed C. haemolyticum KFSHRC-CH1 isolate to be a potential virulent pathogenic bacterium that possesses virulence factors that are necessary to establish an infection, acquire essential nutrients, resist antimicrobial agent, and tolerate hostile conditions in a human host and its surrounding environment. For instance, KFSHRC-CH1 isolate possesses 6 types of bacterial toxins, namely, alpha-clostripain, C2 toxin, Epsilon toxin B bacteriocin, Boclp, and type D neurotoxin gene of phage d-18. For example, C2 toxin is an intracellular toxin that causes suppression of actin polymerization in addition to the obliteration of the actin cytoskeleton.28 Moreover, Epsilon toxin belongs to the aerolysin family that is also known as β-pore-forming toxins and increases vascular permeability that leads to edema and congestion in many organs such as lungs and kidneys. In addition, its neuronal damage can be lethal.29,30 Several encoding flagella-associated and adherence genes were present in our isolate; flagella plays an important role in the swimming motility of pathogens toward the infection site and in biofilm formation and other pathogenic adaptations.31–35 Hemolysin-encoding genes were observed in our isolate. For example, alpha-hemolysin aids in the lysis of red blood cells through the destruction of their cell membrane. This toxin kills cells by binding with their outer membrane that is followed by the oligomerization of the toxin monomer and water-filled channels that are accountable for osmotic phenomena, cell depolarization, and loss of important molecules, such as adenosine triphosphate, leading to their death.36 Our results showed that the KFSHRC-CH1 isolate includes a new recombination of plasmids that is associated with different hosts in mammals and birds. This new recombination facilitated human infection by our isolate. Our plasmidome and gene identification analysis indicated the presence of the following plasmids—p2Ch9693, p3Ch9693_1, and p3Ch9693_2 of C. haemolyticum and plasmid pCLG1 of the bacteria C. botulinum D str. 1873. Generally, plasmids pCLG1 and pCLG2 are found in the strain C. botulinum D (1873). These were first isolated from pork in Chad. However, pCLG1 plasmid shows high similarity to the plasmid pC2C203U28 isolated from the strain C. botulinum C-203U28 that contains the genes for the C2 toxin.4 It is worth mentioning that the source of isolation and location of C. botulinum C-203U28 are unknown. In addition, the 5 remaining plasmids are first isolated from poultry in Sweden from the strain C. botulinum C/D 08-BKT015925. In this article, we present a further proof for the assumption made by Skarin and Segerman15 that there is a deep genomic complexity inside the C. novyi sensu lato group that contains the intertwined species C. haemolyticum, C. botulinum (group III), and C. novyi. Moreover, this complexity is mainly contingent with phages and is associated with plasmids carrying toxin genes over lineage boundaries, leading to the creation of distinctly new pathogens with different pathogenic capabilities.
Conclusions
In this study, we present the first reported human infection with C. haemolyticum isolate, which we refer to as KFSHRC-CH1. Our isolate contains a new recombination of plasmids and circular prophages that lead to the creation of a distinctly different pathogen capable of infecting humans with deteriorated immune system. The identification of this isolate was performed using the NGS technique. Our pathogenomic analysis demonstrated that our isolate is a potential virulent pathogenic bacterium as it acquired all the factors needed to establish infection, acquire needed nutrients, resist antimicrobial agent, and tolerate adverse conditions in a human host and its surrounding environment.
Acknowledgments
All NGS experiments and analysis were performed as part of the Saudi Human Genome Program (SHGP) at KACST and KFSHRC. Moreover, we would like to acknowledge Dr. Nada Al-Tassan for her valuable comments, and editing work on this publication.
Footnotes
PEER REVIEW: Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 2141 words, excluding any confidential comments to the academic editor.
FUNDING: The author(s) received no financial support for the research, authorship, and/or publication of this article.
DECLARATION OF CONFLICTING INTERESTS: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
- – ATMS: Involved in study conception and design, data analysis and interpretation. Involved in drafting the manuscript or revising it critically for important intellectual content. Preparing the final approval of the version to be published.
- – MAH: Involved in study design. Involved in acquisition of data, or analysis and interpretation of data; preparation and involved in drafting the manuscript.
- – MS: Involved in acquisition of data, or analysis and interpretation of data.
- – SA: Involved in acquisition of sample and wet laboratory work.
- – MM: Involved in acquisition of sample and wet laboratory work.
- – MA: Involved wet laboratory work and DNA sequencing.
- – AH: Involved in study conception and design. Preparing the final approval of the version to be published.
- – HT: Involved in study conception and design. Over all study supervision. Involved in drafting the manuscript or revising it critically for important intellectual content. Preparing the final approval of the version to be published.
REFERENCES
- 1.Smith LD. The Pathogenic Anaerobic Bacteria. 2nd ed. Springfield, IL: Charles C. Thomas; 1975. pp. 271–280. [Google Scholar]
- 2.Stockdale CR, Moyes TE, Dyson R. Acute post-parturient haemoglobinuria in dairy cows and phosphorus status. Aust Vet J. 2005;83:362–366. doi: 10.1111/j.1751-0813.2005.tb15635.x. [DOI] [PubMed] [Google Scholar]
- 3.Shinozuka Y, Yamato O, Hossain MA, et al. Bacillary hemoglobinuria in Japanese black cattle in Hiroshima, Japan: a case study. J Vet Med Sci. 2011;73:255–258. doi: 10.1292/jvms.10-0231. [DOI] [PubMed] [Google Scholar]
- 4.Nishida S, Nakagawara G. Isolation of toxigenic strains of Clostridium novyi from soil. J Bacteriol. 1964;88:1636–1640. doi: 10.1128/jb.88.6.1636-1640.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schallehn G, Eklund MW. Conversion of Clostridium novyi type D (C. haemolyticum) to alpha toxin production by phages of C. novyi type A. FEMS Microbiol Lett. 1980;7:83–86. [Google Scholar]
- 6.Oakley CL, Warrack GH, Clarke PH. The toxins of Clostridium oedematiens (Cl. novyi) J Gen Microbiol. 1947;1:91–107. doi: 10.1099/00221287-1-1-91. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura S, Kimura I, Yamakawa K, Nishida S. Taxonomic relationships among Clostridium novyi types A and B, Clostridium haemolyticum and Clostridium botulinum type C. J Gen Microbiol. 1983;129:1473–1479. doi: 10.1099/00221287-129-5-1473. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki Y, Takikawa N, Kojima A, Norimatsu M, Suzuki S, Tamura Y. Phylogenetic positions of Clostridium novyi and Clostridium haemolyticum based on 16S rDNA sequences. Int J Syst Evol Microbiol. 2001;51:901–904. doi: 10.1099/00207713-51-3-901. [DOI] [PubMed] [Google Scholar]
- 9.Skarin H, Håfström T, Westerberg J, Segerman B. Clostridium botulinum group III: a group with dual identity shaped by plasmids, phages and mobile elements. BMC Genomics. 2011;12:185. doi: 10.1186/1471-2164-12-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu T, Ohtani K, Hirakawa H, et al. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci U S A. 2002;99:996–1001. doi: 10.1073/pnas.022493799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bettegowda C, Huang X, Lin J, et al. The genome and transcriptomes of the anti-tumor agent Clostridium novyi-NT. Nat Biotechnol. 2006;24:1573–1580. doi: 10.1038/nbt1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sebaihia M, Peck MW, Minton NP, et al. Genome sequence of a proteolytic (Group I) Clostridium botulinum strain hall A and comparative analysis of the clostridial genomes. Genome Res. 2007;17:1082–1092. doi: 10.1101/gr.6282807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith TJ, Hill KK, Foley BT, et al. Analysis of the neurotoxin complex genes in Clostridium botulinum A1–A4 and B1 strains: BoNT/A3,/Ba4 and/B1 clusters are located within plasmids. PLoS ONE. 2007;2:e1271. doi: 10.1371/journal.pone.0001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skarin H, Segerman B. Plasmidome interchange between Clostridium botulinum, Clostridium novyi and Clostridium haemolyticum converts strains of independent lineages into distinctly different pathogens. PLoS ONE. 2014;9:e107777. doi: 10.1371/journal.pone.0107777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol. 2007;45:2761–2764. doi: 10.1128/JCM.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brazier JS, Duerden BI, Hall V, et al. Isolation and identification of Clostridium spp. from infections associated with the injection of drugs: experiences of a microbiological investigation team. J Med Microbiol. 2002;51:985–989. doi: 10.1099/0022-1317-51-11-985. [DOI] [PubMed] [Google Scholar]
- 18.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chevreux S, Pfisterer B, Suhai T. Automatic assembly and editing of genomic sequences. In: Suhai S, editor. Genomics and Proteomics—Functional and Computational Aspects. 2nd ed. New York, NY: Kluwer Academic; Plenum Publishers; 2000. pp. 51–65. [Google Scholar]
- 20.Chevreux B. MIRA assembler. www.chevreux.org/projects_mira.html.
- 21.Abouelhoda M, Issa S, Ghanem M. Tavaxy: integrating Taverna and galaxy workflows with cloud computing support. BMC Bioinformatics. 2012;13:77. doi: 10.1186/1471-2105-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangement. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abouelhoda MI, Kurtz S, Ohlebusch E. CoCoNUT: an efficient system for the comparison and analysis of genomes. BMC Bioinformatics. 2008;9:476. doi: 10.1186/1471-2105-9-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 25.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Virulence Factors of Pathogenic Bacteria Data Base. Beijing, China: Institute of Pathogen Biology, CAMS&PUMC; 2003–2016. [Assessed February 23, 2016]. http://www.mgc.ac.cn/VFs/ [Google Scholar]
- 28.Aktories K, Barth H. Clostridium botulinum C2 toxin—new insights into the cellular up-take of the actin-ADP-ribosylating toxin. Int J Med Microbiol. 2004;293:557–564. doi: 10.1078/1438-4221-00305. [DOI] [PubMed] [Google Scholar]
- 29.Cole AR, Gibert M, Popoff M, Moss DS, Titball RW, Basak AK. Clostridium perfringens epsilon-toxin shows structural similarity to the pore-forming toxin aerolysin. Nat Struct Mol Biol. 2004;11:797–798. doi: 10.1038/nsmb804. [DOI] [PubMed] [Google Scholar]
- 30.Bokori-Brown M, Savva CG, Fernandes da Costa SP, Naylor CE, Basak AK, Titball RW. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011;278:4589–4601. doi: 10.1111/j.1742-4658.2011.08140.x. [DOI] [PubMed] [Google Scholar]
- 31.Feldman M, Bryan R, Rajan S, et al. Role of flagella in pathogenesis of pseudomonas aeruginosa pulmonary infection. Infect Immun. 1998;66:43–51. doi: 10.1128/iai.66.1.43-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 33.Ciacci-Woolwine F, McDermott PF, Mizel SB. Induction of cytokine synthesis by flagella from gram-negative bacteria may be dependent on the activation or differentiation state of human monocytes. Infect Immun. 1999;67:5176–5185. doi: 10.1128/iai.67.10.5176-5185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adamo R, Sokol S, Soong G, Gomez MI, Prince A. Pseudomonas aeruginosa flagella activate airway epithelial cells through asialoGM1 and toll-like receptor 2 as well as toll-like receptor 5. Am J Respir Cell Mol Biol. 2004;30:627–634. doi: 10.1165/rcmb.2003-0260OC. [DOI] [PubMed] [Google Scholar]
- 35.Dasgupta N, Wolfgang MC, Goodman AL, et al. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis. Pseudomonas aeruginosa Mol Microbiol. 2003;50:809–824. doi: 10.1046/j.1365-2958.2003.03740.x. [DOI] [PubMed] [Google Scholar]
- 36.McGillivray DJ, Heinrich F, Valincius G, et al. Structure of functional Staphylococcus aureus α-hemolysin channels in tethered bilayer lipid membranes. Biophys J. 2009;96:1547–1553. doi: 10.1016/j.bpj.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]