

Abstract
Over the past decade, developments in high resolution mass spectrometry have enabled the high throughput analysis of intact proteins from complex proteomes, leading to the identification of thousands of proteoforms. Several previous reports on top-down proteomics (TDP) relied on hybrid ion trap–Fourier transform mass spectrometers combined with data-dependent acquisition strategies. To further reduce TDP to practice, we use a quadrupole-Orbitrap instrument coupled with software for proteoform-dependent data acquisition to identify and characterize nearly 2000 proteoforms at a 1% false discovery rate from human fibroblasts. By combining a 3 m/z isolation window with short transients to improve specificity and signal-to-noise for proteoforms >30 kDa, we demonstrate improving proteome coverage by capturing 439 proteoforms in the 30–60 kDa range. Three different data acquisition strategies were compared and resulted in the identification of many proteoforms not observed in replicate data-dependent experiments. Notably, the data set is reported with updated metrics and tools including a new viewer and assignment of permanent proteoform record identifiers for inclusion of highly characterized proteoforms (i.e., those with C-scores >40) in a repository curated by the Consortium for Top-Down Proteomics.
Keywords: top-down proteomics, gas-phase fractionation, Orbitrap, quadrupole, data-dependent acquisition, false-discovery rate, mass spectrometry, proteoform, AUTOPILOT, medium/high
Graphical Abstract
INTRODUCTION
In recent years, technological developments in high resolution mass spectrometry (MS) have substantially contributed to the growing adoption and impact of top-down proteomics (TDP).1 The conceptual strength of TDP derives from its ability to identify intact proteoforms,2 which are whole proteins with unique primary structures characterized by specific sets of genetic or enzymatic modifications. By interrogating proteoforms directly, TDP does not suffer the protein inference problem inherent to its counterpart,3 bottom-up proteomics (BUP), which relies on the identification of short peptides produced by enzymatic or chemical proteolysis.
Although BUP can identify thousands of protein families in a single liquid chromatography–mass spectrometry (LC–MS) experiment,4 TDP is closing the identification gap when supported by the use of sophisticated instrumentation such as hybrid mass spectrometers based on the latest developments in Fourier transform mass spectrometry (FTMS).5 In a 2013 research article focused on human H1299 cells, Catherman et al. reported the identification of more than 5000 proteoforms, which corresponded to over 1200 unique accession numbers (at a 1% estimated false-discovery rate, FDR). This result represents the current record for the coverage of the human proteome by TDP.6
In addition to instrumental advances, Durbin et al. recently implemented a novel instrument control software named AUTOPILOT.7 AUTOPILOT was specifically designed for TDP by incorporating online spectral deconvolution, limited database searching in real time, and creation of a project-wide exclusion list, all to reduce inefficiencies with commonly applied data-dependent acquisition (DDA) approaches originally developed for BUP. When applied to the analysis of the human proteome, AUTOPILOT reproduced the level of proteome coverage described by Catherman but with a fourfold reduction in the number of LC–MS runs required.8
Here, we describe the top-down study of the proteome of human fibroblasts (IMR90) using a benchtop quadrupole-Orbitrap mass spectrometer, the Q Exactive HF, equipped with a segmented quadrupole and an ultrahigh field Orbitrap mass analyzer.9 In this study, we utilize a 3 m/z isolation window for precursors instead of the ~15 m/z windows for prior TDP studies using a ion trap-FTMS instrument6,10,11 with space charge-limited isolation of precursors. Notably, many biologically relevant PTMs such as acetylation (+42 Da) and phosphorylation (+80 Da) have mass shifts large enough to allow single proteoform isolation using 3 m/z isolation windows for precursor charge states up to ~30+. The benefits of applying narrow quadrupole isolation for precise top-down proteoform characterization have been previously described in the literature but have so far been limited to targeted studies based on direct infusion of purified proteins.12,13 Alternatively, quadrupole-time-of-flight14 or quadrupole-Orbitrap15,16 mass spectrometers have been used for LC–MS experiments on complex mixtures of whole proteins but without quantifying the benefits of restricting precursor isolation width.
Seeking to increase the quality and coverage of proteins and proteoforms in TDP, we mimicked a data acquisition strategy for BUP wherein standard MS1 survey scans are replaced with consecutive selected ion monitoring (SIM) scans of fixed width and m/z. This approach, called a “SIM march” here, implements gas phase fractionation (GPF),17–19 originally developed on hybrid mass spectrometers to improve signal-to-noise ratio (SNR) and dynamic range for peptides by dividing MS1 signals into narrow scan windows. With SIM marching, we applied GPF to TDP using the AUTOPILOT framework to control decision making on-the-fly.7 Differently from the older “quad-march” concept developed for quadrupole–ion cyclotron resonance (ICR) instruments using ~60 m/z-wide scans,20 AUTOPILOT manages deconvolution and decision making after each single SIM scan. After MS1 spectral interpretation, AUTOPILOT either launches the next SIM scan or, if mass values corresponding to unknown or poorly characterized proteoforms are found, proceeds with tandem MS (MS2) by quickly varying activation energy to improve fragmentation quality.7
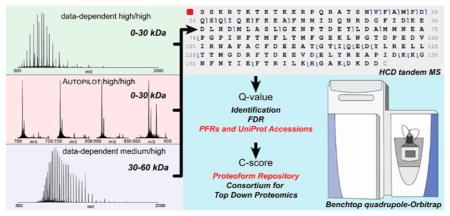
In this study, we also address a traditional issue of TDP, its limited performance for LC–MS2 of complex mixtures of proteoforms >30 kDa.11,21 Here, we investigate proteins up to ~60 kDa by detecting them in the Orbitrap mass analyzer but with dramatically reduced transient lengths to improve spectral SNR without significant losses in mass accuracy. Importantly, the technological improvements converge to produce a high-quality set of resultant proteoforms reported in a public repository using new quality metrics and reporting norms. Particularly, data analysis was carried out through a new portal, which assigns a characterization-score, or C-score,22 to every identified proteoform. The aim is to use these metrics to define a group of proteoforms characterized with high-confidence and give each a unique proteoform record (PFR, analogous to UniProt accession numbers for gene-specific identification of proteins). Creation of a highly annotated proteoform repository has begun and is hosted on the web-based infrastructure of the Consortium for Top-Down Proteomics (http://repository.topdownproteomics.org/).
EXPERIMENTAL SECTION
Cell Culture and Sample Preparation
Primary IMR90 human fibroblasts were cultured adherently in Dulbecco’s modified Eagle’s medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (Thermo Scientific, Rockford, IL). Confluent cells were trypsinized and subjected to two cycles of centrifugation (270 × g for 5 min) followed by resuspension with phosphate buffered saline (Thermo Scientific). Pellets were flash-frozen and stored at −80 °C. For protein extraction, pellets were resuspended in a cell lysis buffer composed of 4% SDS (w/v), 10 mM Tris-HCl (pH 7.8), 1 mM dithiothreitol, and 10 mM sodium butyrate, in the presence of a protease inhibitors cocktail (Thermo Scientific). Resuspended cells were boiled in lysis buffer under shaking for 10 min. Lysed cells were acetone-precipitated at −20 °C for 1 h, and protein pellets were resolubilized using 1% SDS (w/v). Proteins were quantified using the bicinchoninic acid assay (Pierce BCA Protein Assay Kit, Thermo Scientific).
Proteins were off-line separated by their molecular weight (MW) using a GELFREE 8100 Fractionation System (Expedeon, Harston, Cambridgeshire, UK). Both 10% and 8% GELFREE 8100 cartridges were employed for the separation of low (<30 kDa) and high (30–60 kDa) MW proteins, respectively. Two 10% lanes (for summing up 6 LC injections total/fraction) and one 8% lane (for 3 LC injections) were used, loading about 350 μg of protein on each, for a total of ~1 mg of protein. Ten microliters of each fraction collected from the gel-eluted liquid-fraction entrapment electrophoresis (GELFrEE) system23 was loaded onto an SDS-PAGE gel and visualized by silver staining according to a previously described protocol.24 Fractions 1–5 and 2–9 were used from 10% and 8% GELFrEE, respectively, to investigate the low and high MW portions of the cells’ proteome (Supplementary Figure S-1). SDS and other contaminants were removed by MeOH/CHCl3/H2O precipitation as previously described.25 After drying in a fume hood, protein pellets were resuspended in 25 μL of Solution A (see next paragraph for the composition). GELFrEE fractions collected from the 10% cartridge were pooled according to their elution order (e.g., fraction 1 of the first lane with fraction 1 of the second lane) to ensure enough material for at least six replicate injections of the precisely same protein mixture.
Liquid Chromatography–Mass Spectrometry
Resuspended protein fractions were further separated by nanocapillary high performance liquid chromatography (LC) online coupled to a nanoelectrospray ionization source. Reversed phase LC was carried out using a Dionex Ultimate 3000 chromatographic system (Thermo Scientific, Sunnyvale, CA) by applying a gradient of Solution B from 5–15% in 2 min, then from 15–50% in 50 min followed by a wash at 95% B for 5 min and a final re-equilibration phase at 5% B for 15 min. Solution A was composed of 4.8% acetonitrile in water in the presence of 0.2% formic acid, whereas Solution B consisted of 4.8% water in acetonitrile and 0.2% formic acid. All mobile phase components were LC–MS purity grade (Thermo Scientific). Two different column setups, both including a trap and an analytical column, were applied to the analysis of low or high molecular weight proteins. For the low MW fractions, both trap column (20 mm length, 150 μm i.d.) and analytical column (220 mm length, 75 μm i.d.) were in-house packed using PLRP-S stationary phase (5 μm particle size, Agilent, Santa Clara, CA). The trap was washed for 10 min with Solution A at 3 μL/min before the analytical gradient was applied using a 300 nL/min flow rate. For the high MW fractions, commercial monolithic columns were used (ProSwift RP-4H, 50 cm length, 100 μm i.d., Thermo Fisher Scientific) and combined with PepSwift traps (2 cm length, 200 μm i.d.). The trap column was washed for 3 min at 10 μL/min while the analytical gradient was subsequently carried out at 1 μL/min. Both setups used a column heater to maintain a constant temperature of 45 and 35 °C for low and high MW fractions, respectively. The outlet of the columns was coupled to a 15 μm i.d. electrospray emitter (New Objective, Woburn, MA), packed with ~0.5 mm of PLRP-S resin to prevent bubble formation, through a high voltage union to which a 1.9–2.1 kV potential was applied for generating nanoelectrospray.
All mass spectrometry measurements were carried out on an Orbitrap Q Exactive HF mass spectrometer (Thermo Scientific, San Jose, CA) operating in “protein mode”, with reduced pressure in the HCD cell and forced extended trapping of ions in the HCD cell (applying a 4–5 V axial potential). The instrument control software was either Xcalibur (Thermo Scientific) or a new version of AUTOPILOT, which worked as previously described7 but adapted to the Q Exactive (any differences from prior research are described in the Results section). Source region parameters included a temperature of 320 °C for the heated transfer capillary, 50% RF amplitude for the S-lens, and an offset of 15 V between the last element of the S-lens and the inject flatapole to promote desolvation and the removal of labile adducts. Acquisition parameters varied depending on scan type. Survey MS scans used a resolving power (r.p.) of 120 000 (at 200 m/z) for low MW fractions, which corresponded to a transient length of 256 ms (applying eFT). Conversely, high MW fractions were analyzed with a “short transient” of 8 ms, which corresponded to an r.p. of ~3750 (at 200 m/z) selected through the Developers Kit (Thermo Scientific). Importantly, the first isotope beat falls within the first 5 ms of the time domain transient. Longer durations of the transients such as 16 ms would only add noise to the transient, as the second beat falls far longer in time for large proteins.26 Broadband MS1 scans were acquired within a 500–2000 m/z window, 4 or 25 microscans (for high and low r.p. spectra, respectively), with a target AGC of 1e6 and a maximum injection time of 50 ms. Selected ion monitoring (SIM) scans, with width of 25 or 50 m/z, were collected averaging eight microscans using an AGC target of 5e4 and a maximum injection time of 400 ms. Most important parameters for survey scans are summarized in Table 1. Tandem MS (MS2) was performed via higher-energy collision dissociation (HCD)27 by using different energies depending on the experiment type and the analyzed fraction: normalized collision energy (NCE, equal to 25% or to 22% for low and high MW proteins, respectively), under data-dependent acquisition mode controlled by Xcalibur, or alternatively varied scan-by-scan by AUTOPILOT. Precursor selection was obtained through the resolving quadrupole with an isolation window of 3 m/z. All MS2 scans were collected with an r.p. of 60 000 (at 200 m/z) and setting the minimum m/z value to 400, an AGC target of 1e6, and a maximum injection time of 800 ms. Hereafter, we will refer to the experiments for low and high MW fractions as “high/high” or “medium/high” (or “hi/hi” and “med/hi” for short) by using a nomenclature that describes the r.p. settings applied to precursor and tandem MS scans, respectively. Historically, we apply instead the term “low/high” to indicate experiments performed in hybrid instruments (e.g., LTQ-Orbitrap) where MS1 scans are recorded in the ion trap. All experiments used the dynamic exclusion option; for DDA runs, an exclusion list of m/z values was set (with 60 s duration and 3 m/z width), whereas in AUTOPILOT, which performs accurate spectral deconvolution on-the-fly, a mass-based exclusion was applied for selected masses over a time interval of 120 s.
Table 1.
Main Instrument Parameters Used for Survey MS1 Scans during the Three Types of Data Acquisition Modes Used in This Studya
| data acq. logic | tech. rep.b | resolving power (at 200 m/z) | scan type | scan range | AGC target | microscans | max injection time |
|---|---|---|---|---|---|---|---|
| Top-2 hi/hid | all 3 | 120 000 | MS1 | 500–2000 m/z | 1.00 × 1006 | 4 | 50 ms |
| AUTOPILOT hi/hid | #1 | 120 000 | MS1 and random SIM | 500–2000 m/z MS1, 25 m/z SIMc | 1e6 MS1, 5e4 SIM | 4 MS1, 8 SIM | 50 ms |
| #2 | 120 000 | SIM march | 4 × 50 m/z, 700–900 m/z | 5.00 × 1004 | 8 | 400 ms | |
| #3 | 120 000 | SIM march | 8 × 25 m/z, 700–900 m/z | 5.00 × 1004 | 8 | ||
| Top-2 med/hie | all 3 | 3750 | MS1 | 500–2000 m/z | 1.00 × 1006 | 25 | 50 ms |
Note the study design and use of technical replicates.
Technical replicates, where the same sample was subjected three times to LC–MS.
SIM, selected ion monitoring.
hi/hi refers to use of high-resolution data for both MS1 and MS2 scans.
med/hi (short hand for “medium/high”) refers to use of nonisotopically resolved data at a “medium” level of resolution for MS1 and high-resolution data for fragment ions recorded in a MS2 scan.
Experimental Design and Data Analysis
The final output of searching in top-down proteomics is a set of identified proteins and proteoforms. Each identified protein maps to a unique accession number in the UniProt Knowledgebase and is assigned several statistical metrics including a q-value obtained from a procedure to determine a global false discovery rate (FDR, described below). Each proteoform is assigned a C-score as a metric of the quality of its characterization.22
Each experiment, defined as a single GELFrEE fraction analyzed using a specific data acquisition mode, was run in triplicate; the same acquisition parameters were used for all technical replicates when the mass spectrometer was controlled by the commercial software; conversely, variable settings were applied among the three replicates when AUTOPILOT was in control of the instrument (vide infra). RAW files were analyzed postacquisition with a newly implemented platform for searching, reporting, and FDR estimation described in detail by Shams et al.28 Briefly, a cRAWler algorithm was first used to associate deconvoluted MS2 spectra with the corresponding deconvoluted precursor masses. A dedicated cRAWler version was used for AUTOPILOT-generated RAW files to account for precursors determined by SIM rather than traditional MS1 scans. Deconvolution was performed using either Xtract (Thermo Scientific) or the kDECON algorithm,29 for high or medium resolution spectra of intact proteoforms, respectively. Grouped scans (i.e., precursor + MS2 fragments) were subsequently searched against a search space of ~107 candidate proteoforms created from a UniProt formatted text file of Homo sapiens (version: July 2014) allowing up to maximum of 11 post-translational modifications, PTMs, or sequence variations per proteoform. In the case of coisolated species, separate searches were run for each individual precursor mass. Three database searches were run using a Galaxy-based environment sequentially,28 including: (i) an Absolute mass search with precursor tolerance of 2.2 Da; (ii) a Biomarker search with 10 ppm precursor tolerance (equivalent to a “no-enzyme” search); and (iii) an Absolute mass search with 200 Da precursor tolerance applying the Delta Mode option to account for unexpected PTMs. All fragment ions were searched with 10 ppm tolerance.
Each forward hit consists of a proteoform with associated p-score,30 E-value,31 and C-score.22 An instantaneous q-value is also associated with each identified protein and proteoform, as a result of searching a decoy database (created starting from scrambled protein sequences) and using a FDR estimation.11,28 Distinct q-values are obtained separately for each of the three searches, with the FDR determined by running a single experiment against a scrambled set of candidate proteoforms; subsequently, the best p-score retrieved from the forward search is used to fit a gamma distribution to determine local FDR for forward hits from each search type. A global FDR estimation is finally generated according to the procedure described by Higdon et al.32 by using only the hit with the best q-value obtained from the three searches. Somewhat akin to assembling PSMs into protein groups in BUP, proteoforms are grouped into isoforms and their common UniProt accession numbers.28 Both lists are generated after dedicated FDR calculations. In compliance with the most recent HUPO guidelines,33 the herein reported results were filtered using a 1% FDR cutoff at both the proteoform and the accession number level (the isoform level is not reported). Lists of identified proteins and proteoforms are included in Supplementary Tables S-1, S-2, and S-3 and in new Top Down Reports that can be visualized with the TDViewer software, freely downloadable at http://topdownviewer.northwestern.edu. Given that FDR values are determined at protein and proteoform levels, we “anchor” proteoforms to only those that map to identified proteins for simplicity of reporting and viewing of results. In this manner, each proteoform in the file is assured to map to an accession that is also in the set of proteins identified. Graphical fragmentation maps were generated using either the TDViewer or ProSight Lite, available at http://prosightlite.northwestern.edu.34 Gene Ontology analysis was performed using the DAVID bioinformatics resources35 or STRING protein–protein interaction network database36 by uploading onto these web-based interfaces a list of UniProt accession numbers identified at 1% FDR as input. All RAW data files, the UniProt formatted text file used for generating the proteoform database, and the three .tdReport files associated with this study are available at http://massive.ucsd.edu/ with the identifier MSV000079913.
RESULTS
Data Acquisition Strategies
The distinct phases and aspects of top-down MS experiments performed here are recapitulated in Figure 1. In comparison to “top-2” data-dependent acquisition, which employed broadband MS1 to populate a list of precursors for MS2 fragmentation (Figure 1A), we also implemented a “SIM march” strategy consisting of a series of consecutive SIM scans (either 4 of 50 m/z or 8 of 25 m/z each) covering a total m/z window from 700–900 m/z. In the SIM march, the center of each SIM window is fixed (Figure 1B). The motivation for SIM scans is to improve the spectral dynamic range in limited portions of the m/z space where ions of denatured proteins of mass <30 kDa naturally fall due to their similar charge density. To avoid ion coalescence,37–39 which impairs the correct assignment of charge states and masses to proteoform precursors, the AGC target was reduced to 5e4; to counterbalance the reduction in the number of analyzed ions, the number of microscans was increased from 4 to 8 for SIM scans.
Figure 1.

Data acquisition strategies for top-down analysis of human proteins below 60 kDa. (A) Traditional data-dependent high/high experiments as well as medium/high experiments start with a broadband MS1 scan for the determination of precursors to be fragmented in a data-dependent top-2 fashion. Similarly, the standard version of AUTOPILOT (AP), employed as first technical replicate in the high/high study, uses by default a MS1-MS2 scheme. (B) The second and third technical replicates of the AUTOPILOT experiment are designed as a SIM march, that is, as a series of SIM scans to investigate an overall 200 m/z window between 700 and 900 m/z. Precursors are selected from online deconvolution of SIM scans. (C) Selected precursors, both from Xcalibur data-dependent or AUTOPILOT-driven acquisition, are quadrupole isolated with a narrow isolation window of 3 m/z units. (D) Selected proteoforms are subject to HCD activation with dedicated parameters for high or low MW proteins. (E) An off-line database search associates each proteoform with a C-score and determines its identification confidence through an FDR calculation based on q-values. Well characterized proteoforms are indicated by a unique PFR identifier.
Independently from the type of scan which determined their selection, precursor ions were then isolated using the resolving quadrupole with a 3 m/z isolation window (Figure 1C), activated, and fragmented via HCD (Figure 1D). Notably, in previous studies based on hybrid ion trap-Orbitrap mass spectrometers, we applied a 15 m/z isolation window to prevent potential space-charge effects. Finally, collected RAW files were analyzed as described in the Experimental Section to assign q-values for protein identification and C-scores for proteoforms asserted to be present in the sample (Figure 1E). Medium/high experiments, dedicated to the analysis of proteins with intact masses between 30 and 60 kDa, did not differ in their main design from high/high top-2 experiments. Notably, though, MS1 spectra were recorded with only 8 ms long transients in the Orbitrap mass analyzer to improve SNR for proteins >30 kDa. The three operation modes differ in a variety of acquisition parameters (summarized in Table 1), making a direct comparison of their duty cycles complicated. Especially under the control of AUTOPILOT, the acquisition logic is based on the number of new precursors found rather than the simpler “top-N” scheme. In typical DDA mode operation, ~0.5–0.7 s are spent for the survey scan, while up to ~3.8 s are spent for each MS2 event (depending on the abundance of the isolated proteoform). Recording of SIM scans with eight microscans required on average 1.8 s to acquire. In the central portion of the chromatographic gradient (corresponding to ~15–40 min of a LC–MS run), the instrument cycle time for DDA runs, measured as the time difference between two consecutive MS1 scans in each RAW file, has been calculated to be 1.94 and 2.18 s for high/high and medium/high experiments, respectively.
High-Throughput Proteoform and Protein Identification
The combination of results from three technical replicates for each of 18 GELFrEE fractions gave a total of 54 RAW data files collected, with the precise details of sample creation illustrated in Supplementary Figure S-1. Thirty RAW files were generated by high/high experiments, divided equally between those using data-dependent top-2 and AUTOPILOT-directed logic. The remaining 24 RAW files contain data collected from eight fractions from a 8% GELFrEE run and used the medium/high data acquisition logic described above.
Proteins
A summary of the search results is shown in Figure 2, and complete reports of identified proteins and proteoforms are provided in the three Top Down Report files (uploaded onto MassIVE with the RAW files) and Supplementary Tables S-1, S-2, and S-3. A total of 393 unique accession numbers were identified at 1% FDR (Figure 2A). A similar number of identifications was obtained from the technical triplicates of each data-dependent top-2 and AUTOPILOT (235 versus 204, respectively). A large degree of overlap is present between the two accession number lists (about 70%). Conversely, only 29 of the total 147 unique accession numbers resulting from medium/high experiments are shared with any high/high identifications.
Figure 2.

Summary of unique proteoforms and accession numbers identified at 1% FDR from 45 total RAW files. (A) Venn diagram for the total 393 unique accession numbers identified at a 1% protein-level FDR from 54 LC–MS runs. Note, ~80% of the proteins identified by medium/high experiments were not found in either of the two high/high modes of data acquisition. (B) Venn diagram of proteoforms identified at 1% FDR. Approximately 50% of identified proteoforms were shared between top-2 and AUTOPILOT high/high experiments, and low overlap was observed for the <30 kDa and 30–60 kDa portions of the fibroblast proteome interrogated here.
Proteoforms
Interestingly, when considering the Venn diagram for the 1872 unique proteoforms identified, the high degree of overlap observed at the protein level was not observed at the proteoform level (Figure 2B). By comparing data sets derived from top-2 versus AUTOPILOT high/high experiments, only about 34% of identified proteoforms were shared. Interestingly, medium/high experiments resulted in the identification of 171 proteoforms in the <30 kDa range (vide infra), but only 7% of the total 439 proteoforms derived from these experiments were identified in both medium/high and high/high LC–MS runs.
Effectiveness of SIM March in High/High Experiments
AUTOPILOT was used in two different modes (“MS1-MS2” and “SIM march”) for the three technical replicates. The overall numerical results in terms of identified proteoforms and proteins are similar to those obtained in the 0–30 kDa range by the data-dependent top-2 experiments. An in-depth look at the identification rate for new proteoforms identified in each single technical replicate for GELFrEE fractions 1–3 (those that accounted for the largest portion of total proteoforms) indicates a divergent trend between traditional data-dependent and AUTOPILOT-driven data acquisition, Figure 3. Specifically, the stochastic nature of data-dependent precursor selection leads to concentration of identifications in the first technical replicate, with the two consecutive runs being incapable of matching the identification rate of the first with both adding similar numbers of new proteoform to the total count (Figure 3A). On the contrary, the SIM march based on four SIM scans with width of 50 m/z each outperforms the standard AUTOPILOT runs for GELFrEE fractions 1 and 2, and trails in fraction 3 only slightly (Figure 3B). The implementation of the SIM march using eight SIM scans of 25 m/z width shows the worst proteoform collection rate in all cases. Overall, this suggests that the SIM march is competitive with and can outperform experiments based on the traditional MS1–MS2 scheme but likely only up to the point where the instrument duty cycle (expressed as total time needed to scan the full 700–900 m/z window) is not expanded excessively due to the use of too many narrow SIM scans. Note that the comparison with data-dependent top-2 experiments is made more realistic by the fact that the feature for the online generation of a global exclusion list in all AUTOPILOT experiments was disabled, and the online search by AUTOPILOT during each single LC–MS run more closely resembling the “dynamic exclusion” algorithm implemented in Xcalibur.
Figure 3.

Efficiency of identification of new proteoforms from a single GELFrEE fraction using three technical replicates under Xcalibur data-dependent or AUTOPILOT data acquisition. The number of new proteoforms identified in each technical replicate for GELFrEE fractions 1, 2, and 3 is normalized over the total number of new proteoforms identified in the single GELFrEE fraction of interest. (A) The data-dependent top-2 method shows that for the three fractions considered, the first technical replicate provides the highest number of new proteoforms, and the capability of the data-dependent method of finding new confident proteoforms decreases with the number of technical replicates. (B) The AUTOPILOT experiments show that the SIM march with 50 m/z windows (2nd technical replicate) outperforms the standard AUTOPILOT acquisition based on the MS1–MS2 scheme (1st technical replicate) in two fractions out of three. Conversely, the SIM march composed by eight SIM events (3rd technical replicate) produces the lowest number of new identified proteoforms.
Extending the Mass Range with Medium/High Experiments
Figure 4, panel A shows an example of MS1 data for an identified ~41 kDa proteoform. Despite the resolving power being ~4000, the mass accuracy was observed in this case to be ~2.5 ppm. In general, the difference between theoretical and experimental average mass is <1 Da, which corresponds to ~ 20–35 ppm for proteoforms in the 30–60 kDa range. However, as protein size increases from 5 to 30 kDa, their isotopic distributions converge toward a Gaussian-like shape (Supplementary Figure S-2). As this convergence occurs, peaks composed of unresolved isotopic distributions are read out with increasing accuracy as the average mass of a protein increases. For proteins larger than ~30 kDa measured at “medium” resolution, isotopic distributions become Gaussian enough to yield low ppm mass accuracy without any adjustment for fundamental peak shape (Supplementary Figure S-2). Therefore, for proteoforms >30 kDa, accurate mass determination is possible for these experiments due to the high AGC target value used in a Orbitrap mass analyzer compared with other analyzers capable of producing low resolution spectra (i.e., a linear ion trap), and to the shape of the unresolved peaks of large proteins, which are symmetrical with respect to a central axis so that the peak apex corresponds well to the position of the average mass (Figure 4B and Supplementary Figure S-2). Extension of this medium/high mode of data acquisition resulted in the identification of 147 proteins and 439 proteoforms, and for both categories only a minimal overlap with the results of high/high experiments was observed (Figure 2). The mass distributions shown in Figure 4, panel C clearly indicate an increase in ability to identify proteins and proteoforms in the >30 kDa range relative to prior reports.6
Figure 4.

Example of ~41 kDa protein identified from a medium/high experiment (8% GELFrEE fraction 4). (A) The broadband MS1 spectrum obtained using a short transient in the Orbitrap mass analyzer shows high spectral signal-to-noise ratio for a number of charge states from 32 to 55+. (B) The graphical fragment map shows that HCD fragmentation primarily sequenced the C-terminal region to lead to a high C-score of 255 for the proteoform PFR20440, whose experimental mass matches the theoretical one within 2.5 ppm. (C) Histogram of mass distribution for proteoforms identified at 1% FDR through medium/high, top-2 experiments; the distribution is centered around 35–40 kDa.
Using the C-score To Define High Quality Proteoforms
The data set presented here is composed of proteoforms designated by a PFR identifier and an associated C-score. Briefly, the C-score metric applies a Bayesian framework to score proteoforms.22 Following the definition given in the 2014 article, the characterization score can be employed to differentiate proteoforms into three groups, as shown in Figure 5. For all three different experiments reported here, the predominant group is that of uncharacterized proteoforms, which corresponds to about 50% of the total count. Similar relative ratios are then obtained for the partially characterized and fully characterized pools. Only the third group of proteoforms with C-scores >40 was uploaded into the repository, which archives proteoforms at tiered quality levels (http://repository.topdownproteomics.org/StarLevels).
Figure 5.

C-score distributions for the three experimental setups. Identified proteoforms are binned according to their associated C-scores. Panels A–C show C-score distributions for data-dependent high–high, AUTOPILOT high/high, and data-dependent medium/high results, respectively. Proteoforms with a C-score lower than 3 are considered statistically identified but not well characterized. Proteoforms with a C-score between 3 and 40 are defined as partially characterized, as the set of fragment ions used for their identification might be consistent also with the presence of one or more highly similar proteoform(s). Finally, proteoforms with a C-scores >40 are considered well characterized, and their respective PFRs are included in a top-down proteoform repository.
DISCUSSION
The presented study is focused on the implementation and evaluation of new avenues for performing top-down LC–MS of complex protein mixtures and on the comparison of new approaches with standard ones. Concerning the methods for instrument control and data acquisition, the three most relevant innovations are represented by narrow precursor isolation window on the LC time scale, the adoption of short time-domain transients for larger proteins, and the development of an AUTOPILOT-driven version of GPF dedicated to top-down (called SIM marching here). Figure 3 suggests that the series of 50 m/z-wide SIM scans is capable of detecting and identifying lower abundance proteoforms than standard top-2 data acquisition logic (DDA). Manual data analysis confirms that during a SIM march, the precursors selected for fragmentation frequently are low-intensity peaks unlikely to even be detected in a broadband MS1 (Supplementary Figure S-3). The identification of low-abundant proteoforms via SIM marching can also partially explain the limited overlap between the proteoform lists for traditional DDA and AUTOPILOT-driven experiments. The data of Figure 3 also imply that SIM marching <30 kDa performs at its best using SIM windows that are not excessively small (i.e., 50 m/z-wide outperformed 25 m/z-wide by identifying about two-fold more proteoforms). The observation that reducing the width of m/z window below 50 m/z units in GPF does not generally improve the global identification rate was also reported for bottom-up experiments.40 Once controlled by a fully operational version of AUTOPILOT, which would include online identified proteoforms into a project-wide exclusion list based on multiple LC–MS experiments, we speculate that the SIM march can be used, potentially in combination with sample prefractionation, to sharply increase coverage of the human proteome achievable by TDP.
Data acquisition based on short transients allowed the interrogation of proteins in the 30–60 kDa mass range with determination of precursor average masses with high accuracy. Importantly, HCD fragmentation has to be adapted by reducing NCE for higher mass proteins. When using short time-domain transients (a result of setting the instrument to acquire at ~4000 resolving power), individual peaks for protein charge state do not have an assigned charge and are therefore treated by default like singly charged ions; these are by default subjected to higher axial potentials in the HCD cell than multiply charged species. Without reducing the set NCE, this would ultimately induce over fragmentation of large proteins in medium/high DDA experiments. It is possible that experimental steps prior to MS analysis (e.g., GELFrEE fraction cleanup, protein resolubilization, LC performance) might have contributed to the limited number of identified proteoforms >45 kDa. Even with these limitations, this acquisition mode resulted in the identification of 369 proteoforms not otherwise detected during traditional data-dependent experiments. Further refinements of the medium/high approach will consider the role played by the AGC target value, which if increased beyond 1e6 might improve average signal and mass accuracy metrics without inducing detrimental effects due to high space charge.
The biological relevance of extending proteoform-resolved analysis to higher MW portions of the proteome can be considered using a gene ontology analysis on the medium/high proteoform set (Figure 6 and Supplementary Figure S-4). This confirms that the short transient method gave us access to proteins involved in metabolic pathways such as glycolysis/gluconeogenesis otherwise uncovered by traditional top-down analyses due to the high average molecular weight of the enzymes involved, Figure 6, panel B. Similar to high/high experiments, the medium/high analysis was able to capture molecular details such as uncommon PTMs like N-terminal trimethylation on ribosomal protein L23a (PFR16022, UniProt P62750, Supplementary Table S-3), which requires the action of a specific alpha-N-methyltransferase.41 Another example is a N-terminal myristoylation on NADH-cytochrome b5 reductase 3 (PFR20695, UniProt P00387–1, Supplementary Table S-3), which is generally identified by shotgun BUP only upon specific sample enrichment.42 Finally, in the medium/high data set the most commonly observed PTM is N-terminal acetylation, probably also due to the characteristics of HCD fragmentation, which generally provides high sequence coverage on the termini of proteoforms >30 kDa.
Figure 6.

Results of Gene Ontology analysis using DAVID Bioinformatics Resources. (A) First three functional protein groups ranked according to their p-values. Functional groups are based on the list of UniProt accession numbers identified at 1% FDR in medium/high experiments. Note that the UniProt accession numbers of the first two functional groups are largely overlapping. (B) Mass distribution of the 41 proteoforms referring to the eight UniProt accession numbers identified for the glycolysis pathway. (C) Summary of the identified proteoforms of the glycolysis-involved enzyme L-lactate dehydrogenase (P00338).
Despite its intrinsically exploratory nature, the complete data set of this study accounts for the identification of almost 400 unique accession numbers and 2000 proteoforms. The 1:5 ratio between identified gene products and proteoforms extends the ~1:3 or 1:4 ratio previously obtained on human cell lines studied by TDP.6,11 Importantly, this large scale study is the first where RAW files have been processed using a new data analysis platform crafted in a manner analogous to BUP, with multiple, hierarchically ordered aggregation levels associated with FDR calculations. As in bottom-up, two separate FDR estimations are performed at the peptide and at the protein level; here, the proteoform, isoform and accession lists are all subject to distinct FDR calculations, now captured and reported via the .tdReport files accompanying publication. Note that 80 proteoforms passed the 1% FDR threshold at the proteoform-level but did not map to any UniProt Accession reported at the 1% FDR at the protein level. Therefore, these 80 (average C-score of 52.6) were removed from the total count but are represented in the data of Supplementary Figure S-5. Note that the proteins and proteoforms reported here are the result of a new FDR estimation28 and application of quality metrics in a more structured manner than in previous studies. Indeed, the total number of accession numbers associated with the proteoforms passing the cutoff imposed by the FDR calculation at the first aggregation level would be 468, out of which 75 (or 16%) were subsequently filtered out by the aggregation across the proteoform, isoform, and protein levels during global FDR calculations levels.28
Integrated within this new pipeline, the C-score has been utilized to define the subgroup of proteoforms to be used for the creation of a highly confident proteoform repository. Considering proteoforms as the actual molecular effectors in cellular and molecular processes, initiatives such as the Human Proteome Project (HPP) and its articulations like the cell-based version of the HPP43 necessarily will have to rely on the high-quality characterization of proteoforms including localization of PTMs and presence of genetic modifications such as alternative splicing or single nucleotide polymorphisms. The distribution of C-score values for the present data set is such that only approximately one-fourth of the 1872 identified proteoforms will be included in the repository. Future large-scale studies based on different fragmentation techniques such as electron transfer dissociation (ETD)44 or ultraviolet photodissociation45 will have to clarify whether the limited number of fully characterized proteoforms reported for the present study is directly linked with the use of HCD fragmentation. The lack of correlation between C-score and q-values observed for this TDP study (Supplementary Figure S-6) might be partially ascribed to the fact that HCD, although efficient in generating abundant product ions useful for protein identification, typically cannot match electron-driven ion fragmentation techniques for generating a greater number of backbone fragmentation events. The high sequence coverage that results for ETD can translate into highly characterized proteoforms and therefore improved C-scores, particularly for cases harboring multiple modifications or polymorphisms.
CONCLUSIONS
Overall, we have shown that advances in instrument control software and data collection strategies, coupled with improved data analysis, can allow the effective use of a benchtop high resolution mass spectrometer for the top-down analysis of highly complex proteoform mixtures such as those presented by the human proteome. The use of efficient, benchtop instrumentation alongside improved software and more structured handling/reporting of proteoforms will advance top-down proteomics.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Grant No. P41 GM108569 for the National Resource for Translational and Developmental Proteomics (NRTDP), and Award No. GM067193 (N.L.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. L.F. would like to acknowledge the Swiss National Science Foundation for support of an Early Postdoc. Mobility fellowship. We would also like to thank all the members of the Kelleher Research Group for their help with this work.
Footnotes
ORCID
Neil L. Kelleher: 0000-0002-8815-3372
Notes
The authors declare the following competing financial interest(s): The authors declare a conflict and several are involved in software commercialization. Thermo Fisher Scientific is an Industrial Collaborator of the NRTDP.
All RAW data files, the UniProt formatted text file used for generating the proteoform database, and the three .tdReport files associated with this study are available at http://massive.ucsd.edu/ with the identifier MSV000079913.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00698.
Analytical SDS-PAGE gels run to visualize fractions from a GELFrEE separation of whole cell extracts of human IMR90 fibroblasts; mass accuracy of the “medium”-resolution approach to MS1 as a function of protein molecular weight; example of identifying a low-abundance proteoform via SIM marching; graphical output of STRING Gene Ontology analysis based on accession numbers of larger proteins identified by experiments run with “medium/high” data acquisition logic; distribution of all 1952 proteoforms identified at a 1% proteoform-level FDR; correlation between q-values and C-scores for the proteoforms identified by AUTOPILOT high/high experiments at 1% proteoform-level FDR (PDF)
List of identifications including entries and proteoforms for data-dependent high/high experiments (XLSX)
List of identifications including entries and proteoforms for AUTOPILOT high/high experiments (XLSX)
List of identifications including entries and proteoforms for data-dependent medium/high experiments (XLSX)
References
- 1.Toby TK, Fornelli L, Kelleher NL. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu Rev Anal Chem. 2016;9:499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith LM, Kelleher NL, Linial M, Goodlett D, Langridge-Smith P, Ah Goo Y, Safford G, Bonilla L, Kruppa G, Zubarev R, et al. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. The one hour yeast proteome. Mol Cell Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahlf DR, Compton PD, Tran JC, Early BP, Thomas PM, Kelleher NL. Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J Proteome Res. 2012;11:4308–4314. doi: 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catherman AD, Durbin KR, Ahlf DR, Early BP, Fellers RT, Tran JC, Thomas PM, Kelleher NL. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol Cell Proteomics. 2013;12:3465–3473. doi: 10.1074/mcp.M113.030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durbin KR, Fellers RT, Ntai I, Kelleher NL, Compton PD. Autopilot: an online data acquisition control system for the enhanced high-throughput characterization of intact proteins. Anal Chem. 2014;86:1485–1492. doi: 10.1021/ac402904h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M, Kelleher NL. Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J Proteome Res. 2016;15:976–982. doi: 10.1021/acs.jproteome.5b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A, Mann M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol Cell Proteomics. 2014;13:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD, Heffron F, Adkins JN, Pasa-Tolic L. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc Natl Acad Sci U S A. 2013;110:10153–10158. doi: 10.1073/pnas.1221210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li MX, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–U141. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Y, Fornelli L, Compton PD, Sharma S, Canterbury J, Mullen C, Zabrouskov V, Fellers RT, Thomas PM, Licht JD, Senko MW, Kelleher NL. Unabridged Analysis of Human Histone H3 by Differential Top-Down Mass Spectrometry Reveals Hypermethylated Proteoforms from MMSET/NSD2 Overexpression. Mol Cell Proteomics. 2016;15:776–790. doi: 10.1074/mcp.M115.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coelho Graca D, Hartmer R, Jabs W, Beris P, Clerici L, Stoermer C, Samii K, Hochstrasser D, Tsybin YO, Scherl A, Lescuyer P. Identification of hemoglobin variants by top-down mass spectrometry using selected diagnostic product ions. Anal Bioanal Chem. 2015;407:2837–2845. doi: 10.1007/s00216-015-8525-5. [DOI] [PubMed] [Google Scholar]
- 14.Chen B, Peng Y, Valeja SG, Xiu L, Alpert AJ, Ge Y. Online Hydrophobic Interaction Chromatography-Mass spectrometry for Top-down Proteomics. Anal Chem. 2016;88:1885–1891. doi: 10.1021/acs.analchem.5b04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valeja SG, Xiu L, Gregorich ZR, Guner H, Jin S, Ge Y. Three dimensional liquid chromatography coupling ion exchange chromatography/hydrophobic interaction chromatography/reverse phase chromatography for effective protein separation in top-down proteomics. Anal Chem. 2015;87:5363–5371. doi: 10.1021/acs.analchem.5b00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiu L, Valeja SG, Alpert AJ, Jin S, Ge Y. Effective protein separation by coupling hydrophobic interaction and reverse phase chromatography for top-down proteomics. Anal Chem. 2014;86:7899–7906. doi: 10.1021/ac501836k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spahr CS, Davis MT, McGinley MD, Robinson JH, Bures EJ, Beierle J, Mort J, Courchesne PL, Chen K, Wahl RC, Yu W, Luethy R, Patterson SD. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry I.Profiling an unfractionated tryptic digest. Proteomics. 2001;1:93–107. doi: 10.1002/1615-9861(200101)1:1<93::AID-PROT93>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 18.Scherl A, Shaffer SA, Taylor GK, Kulasekara HD, Miller SI, Goodlett DR. Genome-specific gas-phase fractionation strategy for improved shotgun proteomic profiling of proteotypic peptides. Anal Chem. 2008;80:1182–1191. doi: 10.1021/ac701680f. [DOI] [PubMed] [Google Scholar]
- 19.Breci L, Hattrup E, Keeler M, Letarte J, Johnson R, Haynes PA. Comprehensive proteornics in yeast using chromatographic fractionation, gas phase fractionation, protein gel electrophoresis, and isoelectric focusing. Proteomics. 2005;5:2018–2028. doi: 10.1002/pmic.200401103. [DOI] [PubMed] [Google Scholar]
- 20.Patrie SM, Ferguson JT, Robinson DE, Whipple D, Rother M, Metcalf WW, Kelleher NL. Top down mass spectrometry of < 60-kDa proteins from Methanosarcina acetivorans using quadrupole FRMS with automated octopole collisionally activated dissociation. Mol Cell Proteomics. 2005;5:14–25. doi: 10.1074/mcp.M500219-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Compton PD, Zamdborg L, Thomas PM, Kelleher NL. On the scalability and requirements of whole protein mass spectrometry. Anal Chem. 2011;83:6868–6874. doi: 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LeDuc RD, Fellers RT, Early BP, Greer JB, Thomas PM, Kelleher NL. The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. J Proteome Res. 2014;13:3231–3240. doi: 10.1021/pr401277r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran JC, Doucette AA. Gel-eluted liquid fraction entrapment electrophoresis: an electrophoretic method for broad molecular weight range proteome separation. Anal Chem. 2008;80:1568–1573. doi: 10.1021/ac702197w. [DOI] [PubMed] [Google Scholar]
- 24.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 25.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 26.Hofstadler SA, Bruce JE, Rockwood AL, Anderson GA, Winger BE, Smith RD. Isotopic Beat Patterns in Fourier-Transform Ion-Cyclotron Resonance Mass-Spectrometry - Implications for High-Resolution Mass Measurements of Large Biopolymers. Int J Mass Spectrom Ion Processes. 1994;132:109–127. [Google Scholar]
- 27.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 28.Sham DP, Early BP, Fellers RT, Greer JB, Thomas PT, Fornelli L, LeDuc RD, Shwab DJ, Kelleher NL. Accurate Estimation of False Discovery Rates for Protein and Proteoform Identification in Top Down Proteomics. submitted. [Google Scholar]
- 29.Durbin KR, Tran JC, Zamdborg L, Sweet SM, Catherman AD, Lee JE, Li M, Kellie JF, Kelleher NL. Intact mass detection, interpretation, and visualization to automate Top-Down proteomics on a large scale. Proteomics. 2010;10:3589–3597. doi: 10.1002/pmic.201000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat Biotechnol. 2001;19:952–957. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- 31.Fenyo D, Beavis RC. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal Chem. 2003;75:768–774. doi: 10.1021/ac0258709. [DOI] [PubMed] [Google Scholar]
- 32.Higdon R, Haynes W, Kolker E. Meta-analysis for protein identification: a case study on yeast data. OMICS. 2010;14:309–314. doi: 10.1089/omi.2010.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Omenn GS, Lane L, Lundberg EK, Beavis RC, Nesvizhskii AI, Deutsch EW. Metrics for the Human Proteome Project 2015: Progress on the Human Proteome and Guidelines for High-Confidence Protein Identification. J Proteome Res. 2015;14:3452–3460. doi: 10.1021/acs.jproteome.5b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fellers RT, Greer JB, Early BP, Yu X, LeDuc RD, Kelleher NL, Thomas PM. ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics. 2015;15:1235–1238. doi: 10.1002/pmic.201570050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 36.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorshkov MV, Fornelli L, Tsybin YO. Observation of ion coalescence in Orbitrap Fourier transform mass spectrometry. Rapid Commun Mass Spectrom. 2012;26:1711–1717. doi: 10.1002/rcm.6289. [DOI] [PubMed] [Google Scholar]
- 38.Tarasova IA, Surin AK, Fornelli L, Pridatchenko ML, Suvorina MY, Gorshkov MV. Ion coalescence in Fourier transform mass spectrometry: should we worry about this in shotgun proteomics? Eur Mass Spectrom. 2015;21:459–470. doi: 10.1255/ejms.1356. [DOI] [PubMed] [Google Scholar]
- 39.Werner T, Sweetman G, Savitski MF, Mathieson T, Bantscheff M, Savitski MM. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal Chem. 2014;86:3594–3601. doi: 10.1021/ac500140s. [DOI] [PubMed] [Google Scholar]
- 40.Chapman JD, Goodlett DR, Masselon CD. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom Rev. 2014;33:452–470. doi: 10.1002/mas.21400. [DOI] [PubMed] [Google Scholar]
- 41.Schaner Tooley CE, Petkowski JJ, Muratore-Schroeder TL, Balsbaugh JL, Shabanowitz J, Sabat M, Minor W, Hunt DF, Macara IG. NRMT is an alpha-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature. 2010;466:1125–1128. doi: 10.1038/nature09343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thinon E, Serwa RA, Broncel M, Brannigan JA, Brassat U, Wright MH, Heal WP, Wilkinson AJ, Mann DJ, Tate EW. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat Commun. 2014;5:4919. doi: 10.1038/ncomms5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelleher NL. A cell-based approach to the human proteome project. J Am Soc Mass Spectrom. 2012;23:1617–1624. doi: 10.1007/s13361-012-0469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhurov KO, Fornelli L, Wodrich MD, Laskay UA, Tsybin YO. Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem Soc Rev. 2013;42:5014–5030. doi: 10.1039/c3cs35477f. [DOI] [PubMed] [Google Scholar]
- 45.Shaw JB, Li WZ, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J Am Chem Soc. 2013;135:12646–12651. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.