

Abstract
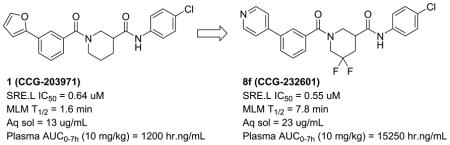
We recently reported the development of a novel inhibitor of Rho-mediated gene transcription (1, CCG-203971) that is efficacious in multiple animal models of acute fibrosis, including scleroderma, when given intraperitoneally. The modest in vivo potency and poor pharmacokinetics (PK) of this lead, however, make it unsuitable for long term efficacy studies. We therefore undertook a systematic medicinal chemistry effort to improve both the metabolic stability and the solubility of 1, resulting in the identification of two analogs achieving over 10-fold increases in plasma exposures in mice. We subsequently showed that one of these analogs (8f, CCG-232601) could inhibit the development of bleomycin-induced dermal fibrosis in mice when administered orally at 50 mg/kg, an effect that was comparable to what we had observed earlier with 1 at a 4-fold higher IP dose.
Keywords: Scleroderma, fibrosis, antifibrotic, myofibroblast, Rho, MRTF
Graphical Abstract
It has been estimated that fibrotic diseases are responsible for up to 40% of all deaths worldwide1. Although the progression and pathology of fibrosis are well described (relentless and excessive accumulation of collagen-rich extracellular matrix (ECM)), the precise etiology of the disease remains obscure. Systemic sclerosis (scleroderma, SSc) is a fibrotic disease of the connective tissues that involves both the vascular and immune systems. The hallmark of SSc, and in fact of all fibrotic disease, is the transition of normal fibroblasts into myofibroblasts, which are characterized by the expression of alpha smooth muscle actin (α-SMA) and the production of ECM2. SSc-associated fibrosis is currently treated with immunosuppressants, which are of limited efficacy, have untoward side effects, and do not address the underlying pathology3. Recent research suggests that targeting fibroblast activation is likely to be a more effective strategy4.
Evidence is growing that fibroblast activation to myofibroblasts results from gene transcription stimulated by a common Rho-mediated signaling pathway that originates from divergent extracellular pro-fibrotic stimuli5–6. Specifically, Rho mediates the conversion of G-actin to F-actin, which releases G-actin-bound myocardin-related transcription factor (MRTF), resulting in accumulation of MRTF in the nucleus, where it binds to serum response factor (SRF) on the serum response element (SRE) promoter. This Rho/actin/MRTF/SRF pathway may in fact serve as a regulator of the fibrotic process used in wound healing 7, but dysregulation and/or overstimulation can lead to fibrosis. Recently, MRTF-A expression in the nucleus of dermal fibroblasts from SSc patients was shown to be increased relative to normal8.
We have discovered and developed a novel class of inhibitors of the Rho/MRTF/SRF pathway9–11. A lead compound from this effort, CCG-203971 (1, Table 1), has exhibited promising anti-fibrotic activity both in vitro and in vivo in several animal models of disease, including intestinal fibrosis12, pulmonary fibrosis13 and dermal fibrosis14. Despite these encouraging preliminary results, 1 has modest potency and poor pharmacokinetic (PK) properties that would make it a poor candidate for further development. We report here our efforts to improve the solubility and metabolic stability of 1 to facilitate additional in vivo studies, and the successful identification of an analog exhibiting oral antifibrotic activity in a murine model of bleomycin-induced dermal fibrosis.
Table 1.
Piperidine Analogs
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | X | Y | SRE.L IC50 (μM)a | MLM T1/2 (min)b | Sol (μg/mL)c | ACTA2 (%ctrl)d | ClogPe | TPSAf |
| 1 | H | CH2 | CH2 | 0.64 | 1.6 | 13.4 | 37 | 4.25 | 62.6 |
| 8a | H | CF2 | CH2 | 0.60 | 4.9 | 2.3 | 9.3 | 4.27 | 62.6 |
| 11a | H | CH2 | O | 3.1 | 3.49 | 71.8 | |||
| 11b | Me | CH2 | CH2 | 0.55 | 0.84 | 4.80 | 62.6 | ||
| 11c | F | CH2 | CH2 | 0.80 | 2.8 | 4.39 | 62.6 | ||
| 12d | H | CH2 | NAc | 21 | 2.78 | 82.9 | |||
| 13d | H | CH2 | NCO2Me | 2.7 | 3.40 | 92.1 | |||
| 22a | H | CHNHAc | CH2 | 5.0 | 2.88 | 91.7 | |||
| 22b | H | CHNHCO2Me | CH2 | 5.0 | 3.50 | 100.9 | |||
| 23 | H | C=CH2 | CH2 | 0.098 | 1.4 | 9.5 | 4.34 | 62.6 | |
| 24 | H | CHNMe2 | CH2 | 5.8 | 3.3 | 3.88 | 65.8 | ||
| 25 | H | C=O | CH2 | 3.9 | 3.43 | 79.6 | |||
Inhibition of Gα12-stimulated SRE.Luciferase activity (mean of n ≥ 3) in HEK293T cells.
Half-life in mouse liver microsomes.
Thermodynamic solubility in water.
Inhibition of ACTA2 expression (percent control) in TGF-β-stimulated human dermal fibroblasts (10 μM, 24 hrs, mean of n=3).
Calculated log partition coefficient.
Topological polar surface area (Å2).
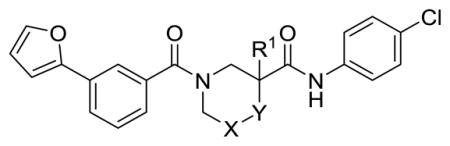
The half-life of 1 in mouse liver microsomes (MLM) is only 1.6 min (Table 1), indicating high susceptibility to oxidative metabolism. An analysis by SMARTCyp15–16 predicted that the primary sites for metabolism are the furan (a known structural alert17) and the 5-carbon of the piperidine-3-carboxylic acid. Metabolite id experiments on several analogs indicated oxidation of the piperidine ring and the left-hand aromatic ring as well as hydrolysis of the secondary carboxamide as major pathways (Supplemental). Our medicinal chemistry strategy thus had three major objectives: 1) stabilize the 3-furanylphenyl moiety to oxidation through substitution or replacement of one or both of the rings with heterocycles; 2) block oxidation of the piperidine carbons sterically or through fluorination; and 3) explore alternatives to the labile secondary amide. In most cases these changes would reduce ClogP, which we expected would also improve the modest aqueous solubility of 1.
Schemes 1 and 2 summarize the preparation of analogs that replace the 3-furanylphenyl of 1 with various heteroaromatics, and the inclusion of geminal difluoro on the piperidine ring. Piperidine carboxamide 211 was coupled with commercially available 3-bromo-heteroaryl carboxylic acids to provide amides 3 which could be reacted with 2-furanylboronic acid under Suzuki conditions to give final amides 4 (Scheme 1). If the requisite furan-2-yl-heteroaryl carboxylic acids were commercially available, they could be coupled directly with 2. Hydroxyacid 5 could be converted to anilide 6 in two steps (Scheme 2). The initial HATU coupling with 4-chloroaniline gave a mixture of lactone and hydroxyamides 6 that had to be further reacted under Weinreb conditions to drive to completion (Supplemental). Swern oxidation of the alcohol gave ketone 7 that was converted to the geminal difluoride with DAST. N-deprotection and amide coupling with diverse aromatic acids under standard conditions provided the difluoro analogs 8. Coupling of unsubstituted piperidine amide 2 with various aromatic acids gave the simple nipecotic amide derivatives 9.
Scheme 1.

Reagents and Conditions: (a) EDC, DIPEA, DMAP, Br-Het-CO2H, THF, 63%; (b) 2-furanylboronic acid, Pd(PPh3)4, Cs2CO3, Dioxane:H2O (4:1), 80 °C, 62% ; (c) EDC, DIPEA, DMAP, furan-2-yl-Het-CO2H, THF, 15–82%.
Scheme 2.

Reagents and Conditions: (a) 4-chloroaniline, HATU, Et3N, DMF; (b) 4-chloroaniline, Me3Al, CH2Cl2, 0 °C to RT, 74% over 2 steps; (c) (ClCO)2, DMSO, TEA, CH2Cl2, 69%; (d) DAST, CH2Cl2 −78 °C to 0 °C, 23%; (e) TFA, CH2Cl2, 84%; (f) Ar-CO2H, EDC, DIPEA, DMAP, THF, 46–98%.
The route to analogs incorporating heteroatoms into the piperidine ring or bearing substitution α to the carboxamide is presented in Scheme 3. Commercial acids 10a–d were converted to bis(amides) 11a–d through sequential EDC-mediated coupling with 4-chloroaniline, N-deprotection and further amide coupling with 3-furan-2-ylbenzoic acid. Hydrogenolysis of 11d followed by reaction with acetic anhydride or methyl chloroformate afforded the N-acyl analogs 12d and 13d. Replacements of the secondary carboxamide of 1 with reverse amide (16) and ether (19) were accomplished as described in Schemes 4 and 5, respectively.
Scheme 3.

Reagents and Conditions: (a) 4-chloroaniline, EDC, DIPEA, DMAP, THF, 55–84%; (b) HCl in dioxane or TFA in CH2Cl2, 54–100%; (c) 3-(furan-2-yl)benzoic acid, EDC, DIPEA, DMAP, THF, 49–94%; (d) 10% Pd/C, H2, MeOH, 40%; (d) Ac2O, DMAP, THF or ClCO2Me, Et3N, CH2Cl2, 55%.
Scheme 4.

Reagents and Conditions: (a) 4-chlorobenzoic acid, EDC, DIPEA, DMAP, THF, 83%; (b) TFA, CH2Cl2; (c) 3-(furan-2-yl)benzoic acid, EDC, DIPEA, DMAP, THF, 87% (2 steps).
Scheme 5.

Reagents and Conditions: (a) MsCl, Et3N, CH2Cl2, quant., (b) 4-chlorophenol, Cs2CO3, DMF, 75 °C, 65%; (c) TFA, CH2Cl2; (d) 3-(furan-2-yl)benzoic acid, EDC, DIPEA, DMAP, THF, 77% (2 steps).
Finally, analogs bearing substitution at the 5-carbon of the piperidine-3-carboxamide were prepared as summarized in Schemes 6 and 7. 5-hydroxy ester 20 was subjected to mesylation, displacement with azide, saponification, EDC-mediated amide coupling and N-deprotection to give 5-azido piperidine 21. Amidation of the secondary amine, Staudinger reduction and subsequent N-acylation provided amide 22a or carbamate 22b as mixtures of diastereomers. Ketone 7 could be methylenated under Wittig conditions, followed by N-deprotection and amidation, to give exo-methylene derivative 23. The carbonyl of 7 could also be reductively aminated, providing dimethylamino derivative 24 after N-deprotection/amidation. Finally, the carbonyl of 7 could be left in place during N-deprotection/acylation to give keto analog 25.
Scheme 6.

Reagents and Conditions: (a) CH3SO2Cl, DIPEA, CH2Cl2; (b) NaN3, DMF, 80 °C, 16h, 59% (2 steps); (c) 1:1 2N NaOH/MeOH, 95%; (d) 4-chloroaniline, EDC, DIPEA, DMAP, THF 75%; (e) TFA, CH2Cl2, 99%; (f) 3-(furan-2-yl)benzoic acid, EDC, DIPEA, DMAP, THF, 83%; (g) PPh3, THF/H2O (2:1), 70 °C, 6h, 72%; (h) CH3COCl, Et3N, CH2Cl2, quant. or CH3CO2Cl, Et3N, CH2Cl2, 84%.
Scheme 7.

Reagents and Conditions: (a) (CH3)2NH, NaBH(OAc)3, HOAc, 4h, 98%; (b) CH3P(Ph)3Br, KOtBu 30 min, 69% (c)TFA, CH2Cl2; (d) 3-(furan-2-yl)benzoic acid, EDC, DIPEA, DMAP, THF 10–83% (2 steps).
All new analogs were tested for their ability to block the Rho/MRTF/SRF pathway in HEK293T cells transfected with a luciferase reporter driven by the MRTF-selective SRE.L promoter, a modification of an assay that has previously been described by us (see Supplemental). Selected active analogs were then assayed for stability to incubation with MLM and for thermodynamic aqueous solubility. Data are summarized in Tables 1–4 along with key calculated physical properties: ClogP and topological polar surface area (TPSA).
Table 4.
a. Heteroaryl Carboxamide Analogs
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | HET | X | Y | R | SRE.L IC50 (μM) | MLM T1/2 (min) | Sol (μg/mL) | ACTA2 (%ctrl) | ClogP | TPSA |
| 1 | 2-furanyl | CH | CH | H | 0.64 | 1.6 | 13.4 | 37 | 4.25 | 62.55 |
| 8b | 1-Me-5-imidazoyl | CH | CH | F | 0.37 | 16 | 14.9 | 110 | 3.24 | 67.2 |
| 8c | 2-imidazolyl | CH | CH | F | 5.5 | 3.39 | 78.1 | |||
| 8d | 2-pyridyl | CH | CH | F | 1.0 | 9.5 | 3.9 | 37 | 4.38 | 62.3 |
| 8e | 3-pyridyl | CH | CH | F | 0.90 | < 3 | 4.00 | 62.3 | ||
| 8f | 4-pyridyl | CH | CH | F | 0.55 | 7.8 | 23.1 | 31 | 4.00 | 62.3 |
| 8g | 2-pyrimidinyl | CH | CH | F | 3.0 | 11.9 | 75.4 | 65 | 4.05 | 75.2 |
| 8h | 5-pyrimidinyl | CH | CH | F | 3.2 | 3.29 | 75.2 | |||
| 8i | 1-pyrazolyl | CH | CH | F | 0.68 | 4.8 | 23 | 140 | 3.65 | 67.2 |
| 8j | 4-pyrazolyl | CH | CH | F | 4.5 | 13 | 3.52 | 78.1 | ||
| 8k | 4-pyridyl | CF | CH | F | 0.53 | 6.9 | 9.8 | 14 | 4.14 | 62.3 |
| 8l | 4-pyridyl | N | CH | F | 1.2 | 7.7 | 62 | 190 | 2.78 | 75.2 |
| 8m | 2-furanyl | CH | N | F | 7.1 | 2.5 | 3.44 | 75.4 | ||
| 8n | 2-furanyl | N | CH | F | 3.5 | 3.0 | 3.06 | 75.4 | ||
| 9c | 1-imidazolyl | CH | CH | H | 5.2 | 2.46 | 67.2 | |||
| 9i | 1-pyrazolyl | CH | CH | H | 2.9 | 1.7 | 3.62 | 67.2 | ||
| 9j | 4-pyrazolyl | CH | CH | H | 3.0 | 5.6 | 73 | 73 | 3.49 | 78.1 |
| 9o | 3-pyrazolyl | CH | CH | H | 0.92 | 3.4 | 3.88 | 78.1 | ||
Assay descriptions provided in Table 1.
Substitution (F or Me) alpha to the secondary carboxamide was first explored to attenuate hydrolysis (11b and 11c). Although SRE.L potency was maintained, there was no significant improvement in stability to MLM, which contain hydrolyzing enzymes. Incorporation of heteroatoms into the piperidine ring (11a, 12d, 13d) diminished potency in every case. We next explored substitution on the piperidine 5-carbon, a site specifically predicted by SMARTCyp to be susceptible to oxidation. Geminal difluorination (8a) did not improve potency, but lengthened the half-time in MLM by a factor of three. Acyl or alkyl nitrogen substitution, tested as mixtures of diastereomers (22a–b, 24) induced marked losses in potency. A carbonyl at the 5-carbon (25) was similarly detrimental, but interestingly, the corresponding exo methylene (23) improved SRE.L potency nearly 10-fold (IC50 = 98 nM). Unfortunately, stability to MLM was not improved.
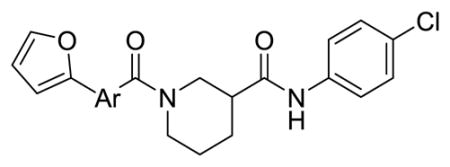
Table 2 summarizes our attempts to replace the benzamide phenyl ring of 1 with heterocycles, which was expected to improve metabolic stability by lowering overall ClogP. All 5-membered heterocycles (4d–g), along with pyrimidine 4c, were significantly less active than 1. Pyridine analogs 4a and 4b, however, maintained more activity, but did not possess measurably improved metabolic stability.
Table 2.
a. Heterocyclic Replacements for Benzamide
| |||||
|---|---|---|---|---|---|
| Cmpd | Ar | SRE.L IC50 (μM) | MLM T1/2 (min) | ClogP | TPSA |
| 1 |
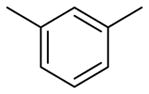
|
0.64 | 1.6 | 4.25 | 62.6 |
| 4a |
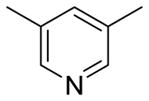
|
2.9 | < 3 | 3.03 | 75.4 |
| 4b |
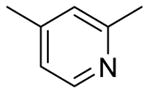
|
1.9 | < 3 | 3.41 | 75.4 |
| 4c |
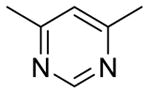
|
15.3 | 3.09 | 88.3 | |
| 4d |
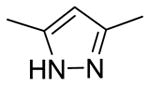
|
5.3 | 2.86 | 91.2 | |
| 4e |
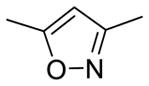
|
6.8 | 2.92 | 88.6 | |
| 4f |
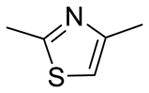
|
6.2 | 3.56 | 103.7 | |
| 4g |
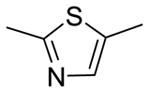
|
5.8 | 3.33 | 103.7 | |
Assay descriptions provided in Table 1.
Additional analogs designed to avoid secondary amide hydrolysis are presented in Table 3. N-methylation (26) reduced activity by 10-fold. The reverse amide 16 similarly lost too much potency. Interestingly, ether 19 retained all of the activity of amide 1, but was also similarly unstable to MLM, suggesting that the primary pathway for in vitro clearance is oxidative rather than hydrolytic.
Table 3.
a. Primary Carboxamide Replacements
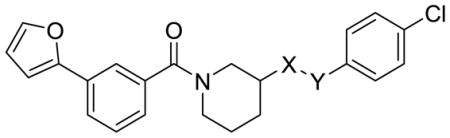
| ||||||
|---|---|---|---|---|---|---|
| Cmpd | X | Y | SRE.L IC50 (μM) | MLM T1/2 (min) | ClogP | TPSA |
| 1 | C=O | NH | 0.64 | 1.6 | 4.25 | 62.55 |
| 16 | NH | C=O | 4.9 | 3.92 | 62.6 | |
| 19 | CH2 | O | 0.81 | 1.8 | 4.73 | 42.7 |
| 26b | C=O | NMe | 8.6 | 4.11 | 53.8 | |
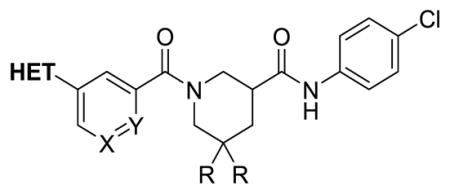
We then focused our attention on replacing the oxidatively labile phenyl furan (Table 4). Analogs 9c, 9i, 9j and 9o have pyrazole or imidazole installed as furan bioisosteres with lower lipophilicity and electron density. 3-pyrazole analog 9o maintained strong activity in the SRE.L assay, but was only marginally more stable in the MLM, suggesting that oxidation of the piperidine ring remained an issue. The most metabolically stable pyrazole 9j unfortunately had diminished SRE.L activity.
The remaining analogs in Table 4 (8b–n) represent our attempts to combine two or more of the most successful modifications from the earlier SAR (furan replacement, geminal difluorination of the piperidine, replacement of the benzamide phenyl with pyridine) into single analogs. Greatest retention of SRE.L potency was realized with pyridine or N-Me imidazole replacements for the furan in combination with difluorination of the piperidine (8b, 8d–f, 8k).
Several of these analogs also had improved half-lives in MLM, especially 8b and 8d. Most polar replacements for the furan (e.g. imidazole 8c, pyrimidines 8g and 8h, and 4-pyrazole 8j) resulted in unacceptably diminished potency in the SRE.L assay. The sole exception was 1-pyrazole 8i. Attempts to replace the benzamide phenyl of 8a with pyridine (8m, 8n) were similarly unsuccessful.
Selected analogs that were both active in the SRE.L assay and had improved stability to MLM were advanced to a primary fibroblast model of pro-fibrotic gene expression. Primary human dermal fibroblasts were stimulated with TGF-β, a pro-fibrotic cytokine that increases ACTA2 gene expression (α-SMA protein), a marker of the fibroblast to myofibroblast transition2, and treated with 10 μM of selected compounds to determine the reduction in ACTA2 mRNA as measured by qPCR. Results are included in Tables 1 and 4. Initial geminal difluoro analog 8a, despite equivalent potency in the primary SRE.L assay, displayed much greater inhibition of ACTA2 expression at 10 μM than 1 (9% control vs 37% control). Olefin 23 also exhibited stronger efficacy in the fibroblast assay than 1, consistent with its much greater SRE.L potency. Eight new analogs in Table 4 were tested, and their efficacy varied widely. There is no clear correlation with SRE.L potency, although no compound with an SRE.L IC50 > 1 μM inhibited ACTA2 expression by more than 50%. A mild correlation of ACTA2 expression inhibition with ClogP, however, is evident (Supplemental Figure S2), suggesting that a minimum lipophilicity is needed for good permeability into the fibroblasts. In contrast, ClogP overall correlated only very weakly with SRE.L activity and solubility, and not at all with MLM T1/2 (Supplemental Figures S3–S5).
Three new compounds possessing both greater efficacy than 1 in the fibroblasts and longer half-lives in MLM were progressed to short (7 hr) PK studies in mice to determine if they could achieve plasma exposure superior to that of 1 (Table 5). Analyses were initially performed following single intraperitoneal (IP) injection at a dose of 10 mg/kg. All displayed marked improvements in AUC over 1 (from 8-fold to over 20-fold). 8f and 8k had particularly high exposures, so were re-analyzed after single oral gavage at 20 mg/kg. The nearly equivalent dose-normalized exposures achieved by 8f following either IP or PO administration, as well as its superior solubility, encouraged us to progress this compound to our initial in vivo efficacy model of dermal fibrosis.
Table 5.
Single dose plasma exposure in mice
| Route | IP | PO | ||
|---|---|---|---|---|
| Cmpd | AUCa | AUCa | Concb (0.5 h) | Concb (7 h) |
| 1 (CCG-203971) | 1200 | |||
| 8a (CCG-222740) | 9200 | |||
| 8f (CCG-232601) | 15251 | 33719 | 14.4 | 2.4 |
| 8k (CCG-257081) | 26700 | 36385 | 13.2 | 1.3 |
Estimated area under the plasma concentration curve (0–7 hrs, hr.ng/mL) following a single dose by intraperitoneal injection (10 mg/kg) or oral gavage (20 mg/kg).
Plasma concentration at time indicated (μM).
We previously reported that 1 can attenuate the development of bleomycin-induced dermal fibrosis in mice with 14 days of IP dosing (100 mg/kg bid)14. We repeated this fibrosis prevention study with 8f, dosed PO at 50 mg/kg qd. Results are presented in Figure 1 and indicate efficacy at reducing the increases in both dermal thickness (−55%) and hydroxyproline content (−62%) induced by bleomycin treatment alone. Significantly, these results are comparable to what was achieved by 1 in our previous study but at one-fourth the total daily dose and by oral administration.
Figure 1.

8f inhibits bleomycin-induced skin fibrosis in mice. Three groups of mice (n=8) were treated for 14 days with one of the following daily protocols: Control: intracutaneous injections of PBS; Bleo: intracutaneous injections of bleomycin with concurrent oral gavage of vehicle; or Bleo + 8f: intracutaneous injections of bleomycin with concurrent oral gavage of 50 mg/kg 8f.
In conclusion, starting with a lead inhibitor 1 of the Rho/MRTF/SRF signaling pathway possessing poor solubility and metabolic stability, we undertook a systematic campaign to improve the physical and PK properties to facilitate testing in long-term animal models of fibrosis. Guided by both computational prediction and actual metabolite id studies, we modified areas of the molecule expected to be highly susceptible to metabolism in vivo, including the piperidine core and the heteroarylbenzamide moiety. Our major strategies centered on reducing the electron density of the aromatic rings, incorporating fluorine and reducing overall ClogP. We were successful in identifying several analogs with equipotent or better SRE.L activity and markedly improved plasma exposure levels upon IP or PO dosing in mice. In this work, analog 8f (CCG-232601) was shown to attenuate the development of bleomycin-induced dermal fibrosis in mice upon oral dosing at one-fourth the daily IP dose of 1 that had demonstrated comparable efficacy in an earlier experiment. Another new analog from this effort, 8a (CCG-222740), has recently been shown by Yu-Wai-Man et al to prevent scar tissue formation in a preclinical model of conjunctival fibrosis18. We believe these results further support that our Rho/MRTF/SRF inhibitors are a powerful new set of tools for studying the prevention and treatment of fibrosis in animal models, and represent potential clinical antifibrotics.
Supplementary Material
Acknowledgments
The research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award numbers R01AR066049 (SDL) and T32AR007080 (P-ST), and also by the National Cancer Institute of the National Institutes of Health under award number P30CA046592 (embedding and cutting of skin samples from the bleomycin study was performed by the UMCCC Histology/Immunohistochemistry Core. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Support was also provided by the Scleroderma Foundation, and the Michigan State University Fibrosis Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wynn TA. Fibrosis under arrest. Nat Med. 2010;16(5):523–5. doi: 10.1038/nm0510-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180(4):1340–55. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leask A. Emerging targets for the treatment of scleroderma. Expert Opin Emerg Drugs. 2012;17(2):173–9. doi: 10.1517/14728214.2012.678833. [DOI] [PubMed] [Google Scholar]
- 4.Sivakumar P, Ntolios P, Jenkins G, Laurent G. Into the matrix: targeting fibroblasts in pulmonary fibrosis. Curr Opin Pulm Med. 2012;18(5):462–9. doi: 10.1097/MCP.0b013e328356800f. [DOI] [PubMed] [Google Scholar]
- 5.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11(5):353–65. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsou PS, Haak AJ, Khanna D, Neubig RR. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: focus on Rho GTPase-regulated gene transcription. American journal of physiology. Cell physiology. 2014;307(1):C2–13. doi: 10.1152/ajpcell.00060.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandbo N, Lau A, Kach J, Ngam C, Yau D, Dulin NO. Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-beta. Am J Physiol Lung Cell Mol Physiol. 2011;301(5):L656–66. doi: 10.1152/ajplung.00166.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiwen X, Stratton R, Nikitorowicz-Buniak J, Ahmed-Abdi B, Ponticos M, Denton C, Abraham D, Takahashi A, Suki B, Layne MD, Lafyatis R, Smith BD. A Role of Myocardin Related Transcription Factor-A (MRTF-A) in Scleroderma Related Fibrosis. PLoS One. 2015;10(5):e0126015. doi: 10.1371/journal.pone.0126015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6(8):2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 10.Evelyn CR, Bell JL, Ryu JG, Wade SM, Kocab A, Harzdorf NL, Hollis Showalter HD, Neubig RR, Larsen SD. Design, synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG-1423. Bioorg Med Chem Lett. 2010;20:665–672. doi: 10.1016/j.bmcl.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell JL, Haak AJ, Wade SM, Kirchhoff PD, Neubig RR, Larsen SD. Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett. 2013;23(13):3826–32. doi: 10.1016/j.bmcl.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PD. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflammatory bowel diseases. 2014;20(1):154–65. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, Kim KK, Keshamouni VG, White ES, Zhou Y, Higgins PD, Larsen SD, Neubig RR, Horowitz JC. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol. 2015;185(4):969–86. doi: 10.1016/j.ajpath.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haak AJ, Tsou PS, Amin MA, Ruth JH, Campbell P, Fox DA, Khanna D, Larsen SD, Neubig RR. Targeting the myofibroblast genetic switch: inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. The Journal of pharmacology and experimental therapeutics. 2014;349(3):480–6. doi: 10.1124/jpet.114.213520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rydberg P, Gloriam DE, Zaretzki J, Breneman C, Olsen L. SMARTCyp: A 2D Method for Prediction of Cytochrome P450-Mediated Drug Metabolism. ACS Medicinal Chemistry Letters. 2010;1(3):96–100. doi: 10.1021/ml100016x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rydberg P, Olsen L. Ligand-Based Site of Metabolism Prediction for Cytochrome P450 2D6. ACS Medicinal Chemistry Letters. 2012;3(1):69–73. doi: 10.1021/ml200246f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Structural Alert/Reactive Metabolite Concept as Applied in Medicinal Chemistry to Mitigate the Risk of Idiosyncratic Drug Toxicity: A Perspective Based on the Critical Examination of Trends in the Top 200 Drugs Marketed in the United States. Chem Res Toxicol. 2011;24(9):1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 18.Yu-Wai-Man C, Spencer-Dene B, Lee RM, Hutchings K, Lisabeth EM, Treisman R, Bailly M, Larsen SD, Neubig RR, Khaw PT. Local delivery of novel MRTF/SRF inhibitor prevents scar tissue formation in a preclinical model of fibrosis. Scientific Reports. 2017 doi: 10.1038/s41598-017-00212-w. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.