

Abstract
Peripheral viral infections increase seizure propensity and intensity in susceptible individuals. We have modeled this comorbidity by demonstrating that the acute phase response (APR) instigated by an intraperitoneal (i.p.) injection of a viral mimetic, polyinosinic-polycytidylic acid (PIC), induces protracted hypersusceptibility to kainic-acid (KA)-induced seizures. We have further demonstrated that PIC challenge robustly increases the level of tonic extracellular glutamate and neuronal excitability in the hippocampus. The present study was undertaken to determine a relationship between tonic glutamate and seizure susceptibility following PIC challenge. Briefly, glutamate-sensing microelectrodes were permanently implanted into the CA1 of eight-week old female C57BL/6 mice. Following a three day recovery, APR was induced by i.p. injection of 12 mg/kg of PIC, while saline-injected mice served as controls. Tonic glutamate was measured at 1, 2, 3 and 4 days after PIC challenge. PIC challenge induced an approximately 4-fold increase in tonic glutamate levels measured after 24 h. The levels gradually declined to the baseline values within four days. 24 h after PIC challenge, the mice featured an approximately 3-fold increase in cumulative seizure scores and 2-fold increase in the duration of status epilepticus induced by subcutaneous (s.c.) injection of 12 mg/kg of KA. Seizure scores positively correlated with pre-seizure tonic glutamate. Moreover, seizures resulted in a profound (76%) elevation of extracellular glutamate in the CA1 of PIC-challenged but not saline-injected mice. Our results implicate the increase of extracellular glutamate as a mediator of seizure hypersusceptibility induced by peripheral viral challenge.
Keywords: Polyinosinic-polycytidylic acid, Extracellular glutamate, Seizures, Acute phase response, Hyperexcitability, Microelectrode arrays
Graphical Abstract
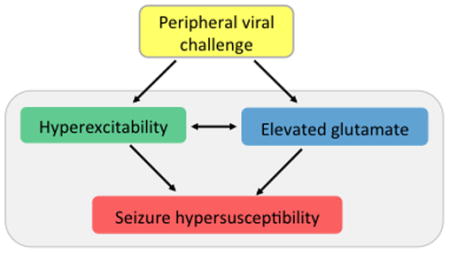
Intraperitoneal injection of a viral mimetic, PIC, results in a robust elevation of extracellular (tonic) glutamate in the hippocampus. Here, we showed that tonic glutamate levels positively correlate with the susceptibility to kainic acid (KA)-induced seizures. The elevation of tonic glutamate by PIC challenge likely results from the hyperexcitability of hippocampal neurons. In addition, tonic glutamate level might be increased directly by inflammatory milieu in the hippocampal parenchyma. Tonic glutamate enhances and/or sustains neuronal excitability likely through extrasynaptic receptors. We posit that both, neuronal hyperexcitability and elevated tonic glutamate contribute to the exacerbation of seizure susceptibility.
Introduction
Inflammatory mediators generated in the periphery are efficiently conveyed via circulation to the brain and modulate its function. A stellar example is the induction of sickness behavior by systemic inflammation/infection (Dantzer 2009). Although sickness behavior is an adaptive response that promotes survival and recovery, the underlying cellular and molecular alterations may be disadvantageous. For example, systemic infection burden increases seizure propensity in susceptible individuals (Tellez-Zenteno et al. 2005). Moreover, peripheral infections trigger exacerbations in multiple sclerosis (Buljevac et al. 2002), Alzheimer disease (Holmes 2013) and Parkinson disease (Ferrari & Tarelli 2011). Consequently, the elucidation of underlying mechanisms is of a paramount clinical importance.
We have developed a preclinical model to study the comorbid effect of peripheral viral infections on seizure propensity (Kirschman et al. 2011, Michalovicz & Konat 2014). This model entails intraperitoneal injection of mice with a viral mimetic, polyinosinic-polycytidylic acid (PIC) to induce the acute phase response (APR), which is an early reaction of the host to infections characterized by a fulminant elevation of blood borne inflammatory mediators. The PIC challenge results in a robust and protracted increase in the intensity and duration of kainic acid (KA)-induced seizures (Kirschman et al. 2011, Michalovicz & Konat 2014). Recently, we have shown (Hunsberger et al. 2016) that PIC challenge profoundly increases excitability of neuronal networks in the hippocampus, the ictal site for KA-induced seizures (Ben-Ari & Cossart 2000). Moreover, using the enzyme-based microelectrode array (MEA) technique we have shown that PIC challenge robustly upsurges extracellular glutamate levels in the hippocampus (Hunsberger et al. 2016). Although this finding indicated a plausible role of tonic glutamate in the development of seizure hypersusceptible phenotype, this notion could not have been tested since the measurements were performed in anesthetized animals.
The present study was undertaken to test the hypothesis that the susceptibility to KA-induced seizures is a function of extracellular glutamate levels in the hippocampi of conscious mice. Specifically, we posit that extracellular glutamate levels are predictive of the extent of status epilepticus. Temporal changes in extracellular glutamate levels induced by PIC challenge were monitored in the hippocampi of free-moving mice using permanently implanted MEAs. The levels of glutamate were correlated with the extent and duration of KA-induced seizures.
Materials & Methods
Animals
Eight-week-old female C57BL/6 mice obtained from Jackson labs (Bar Harbor, ME) were group housed with free access to food and water in a temperature- and humidity-controlled colony room under a 12:12 h light-dark cycle. Mice were weighed daily for the duration of the study. The Auburn University Animal Care and Use Committees has approved all experimental procedures.
Extracellular (tonic) glutamate measurement
Self-referencing, enzyme-based microelectrode arrays (MEA) were assembled and prepared for in vivo glutamate recordings as previously described (Burmeister et al. 2000, Burmeister et al. 2002). Briefly, the electrodes obtained from Quanteon (Nicholasville, KY) were coated with glutamate oxidase and calibrated as previously demonstrated in Hunsberger et al. (2016). To modify the MEA for recording in freely-moving awake animals, the MEA paddle was shortened and attached to a miniature omnetics connector (Omnetics Connector Corporation; Minneapolis, MN) to create a pedestal. This miniature omnetics connector is smaller and lighter than those used previously for in vivo recordings of freely moving animals (Hascup et al. 2008). This smaller model allows the animal to move more easily in space. The four sites on the MEA paddle were connected to the gold-plated pin on the connector by copper wires. To secure the paddle to the connector and to prevent the penetration of moisture, waterproof epoxy resin was applied, and the copper wires were tucked around the connector (Rutherford et al. 2007, Stephens et al. 2014). The completed pedestal was allowed to dry for at least 24 hours. An Ag/Cl reference electrode was also prepared and soldered to the gold-plated pin.
At commencement of the experiment (Day 1), mice were anesthetized with isoflurane (1–4% inhalation; continuous) and placed into a stereotaxic device (Stoelting, Wood Dale IL, USA). The MEA pedestal was implanted into either the right or left hippocampal cornu ammonis 1 (CA1) using the following coordinates: AP: −2.3 mm, ML: +/−1.7 mm, DV: 1.4 mm (Paxinos & Franklin 2012). The assembly was anchored with stainless steel screws, and after inserting the reference electrode, secured with four layers of acrylic resin (Lang Dental, Wheeling IL, USA). Immediately after surgery, subcutaneous (s.c.) injections of 1 mg/kg of bupivicaine were given to alleviate pain, and mice were placed on a heating pad until full recovery from anesthesia. To assuage inflammation, 2 mg/kg of Ketoprofen was subcutaneously (s.c.) injected on Day 1–3.
On Day 5, mice were placed in an observation chamber [17.5 in (L) x 17.5 in (W) x 14.5 in (H)], and the MEA pedestal was connected to the FAST-16 mkII system (Quanteon, Nicholasville, KY). After reaching a stable baseline (approximately 30 minutes), tonic glutamate levels, sampled approximately every 5 min, were measured for 1 h or longer (i.e., during seizures; see below).
Induction of APR and seizures
On Day 4, APR was induced by intraperitoneal (i.p.) injection of 12 mg/kg of ultrapure PIC (Invivogen, San Diego, CA) in saline. Mice injected with 100 μL of saline served as vehicle controls.
Seizures were induced on Days 5, 6, 7, and 8. Briefly, after obtaining pre-seizure glutamate measurements, mice were subcutaneously (s.c.) injected with 12 mg/kg of KA to induce status epilepticus (SE) under continuous glutamate recording. Mice injected with saline (50 μL) served as vehicle controls. Seizure severity was graded by blinded observers in 5-minute increments using the 6-step scale (Morrison et al. 1996). Seizures lasted approximately 100–200 minutes, and glutamate measurements were recorded throughout this period. Twenty minutes after seizures cessation, post-seizure glutamate was measured. Chronically implanted MEAs have been shown to reliably record glutamate in conscious, freely moving rodents with minimal damage to surrounding tissue for up to one week post-implantation (Rutherford et al. 2007, Hascup et al. 2008).
Study design and statistical analyses
Because of the long durations required for measuring extracellular glutamate concentrations on Days 5–8, mice were examined in a staggered manner. Mice were randomly selected from their home cage and assigned to a group on Day 1 (implantation of MEA) based on a table of simple randomization created a priori. During glutamate recordings (Days 5–8), the experimenter was blinded to treatment. Sample sizes were calculated using effect sizes from our previous study (Hunsberger et al. 2016), specifically CA1 differences in tonic glutamate in anesthetized PIC-challenged mice versus saline-injected controls. Using G*Power (ANOVA: Fixed effects, one-way with the following parameters: effect size=2.27; α=.05, power=0.8, number of groups=2), we obtained a recommendation for a total sample size of 6 (or 3 per group), which we increased to 5 to 7 animals per group.
Tonic glutamate levels were sampled every 5 min prior to KA injection (pre-seizure), after KA injection (seizure) or after seizure cessation (post-seizure), and expressed as average values for each period. All results were evaluated by repeated measures of ANOVAs using JMP (SAS, Cary, NC 27513). Within-subject measures included Day (Days 5–8) and phase (pre-seizure, during seizure, and post-seizure). A Grubb’s test was used to identify outliers. Significant omnibus tests were followed by t-test post hoc comparisons. Spearman correlations were run to determine the relation between tonic glutamate and seizure scores. Results are presented as mean ± SEM, and differences between groups are considered statistically significant at p ≤ 0.05.
Results
As shown in Fig. 1, PIC-challenge transiently decreased bodyweight 24 h after injection (Day 5) [Day*Group effect, F(7,182)=3.08, p=.004]. Thereafter, the bodyweight returned to control (SAL) levels. These results are concordant with the temporary body weight loss observed by the Cunningham group (Cunningham et al. 2007).
Figure 1. Temporal changes in body weight following MEA implantation and PIC challenge.

Mice were implanted with MEA pedestals on Day 1, and challenged with 12 mg/kg of PIC on Day 4. Saline-injected mice served as respective controls. Symbols represent means ± SEM from 13 to 15 animals. Asterisks denote significant differences from saline controls (**p≤ .01).
Our previous work demonstrates that PIC-challenge significantly increases tonic glutamate levels 24 hours after PIC injection in anesthetized mice (Hunsberger et al. 2016). To determine whether this increase is protracted, we monitored tonic glutamate up to 96 h after PIC-challenge. Fig. 2 shows that tonic glutamate level was transiently elevated at 24 h after PIC-challenge (Day 5) by approximately 3.7-fold as compared to the control (SAL) group [Group effect, F(1,9)=6.00, p=.04]. It tended to be still elevated thereafter, but the values were not significantly divergent from control values.
Figure 2. Changes in tonic glutamate levels induced by PIC challenge.
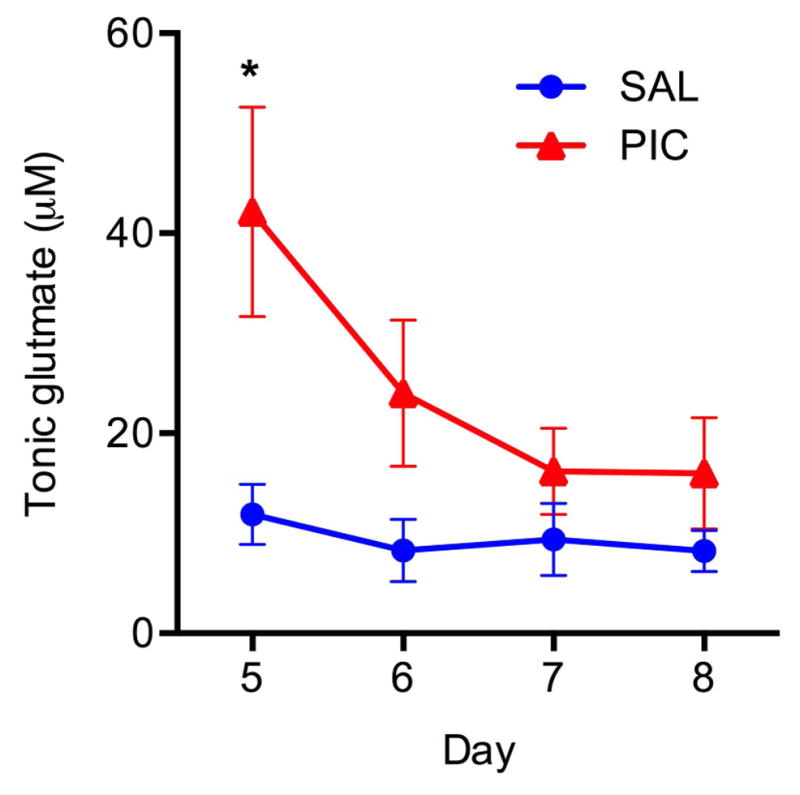
Mice implanted with MEA pedestals were challenged with PIC on Day 4 post-surgery (PIC), and tonic glutamate levels in the CA1 region was measured on Days 5, 6, 7 and 8. Saline-injected mice (SAL) served as controls. Symbols represent means ± SEM from 5 to 7 animals per group. Asterisks denote significant differences from saline controls (*p≤ .05).
As previously demonstrated in naïve mice (Kirschman et al. 2011, Michalovicz & Konat 2014), PIC-challenge resulted in a transient increase in the susceptibility to KA-induced seizures in the mice implanted with MEA (Fig. 3) as seen from the average seizure score (Day*Group effect, F(3,33)=15.98, p=.0001; Fig. 3A), seizure duration (Day*Group effect, F(3,33)= 14.17, p=.0001; Fig. 3B), and cumulative seizure score (Day*Group effect, F(3,33)=9.74, p=.0001; Fig. 3C). On Day 5, these values were significantly increased in PIC-challenged mice by approximately 1.5-, 2.0- and 2.9-fold (p’s<.001), respectively when compared to saline-injected controls. The values dropped to 1.3-fold (p<.001), 1.8-fold (p<.01) and 2.3-fold (p<.01) on Day 6, and returned to control levels on Day 7 (p’s>.05). However, the average seizure score fell further to approximately 75% of control (p<.01) on Day 8.
Figure 3. Enhancing effect of PIC-challenge on KA-induced seizures.

Mice implanted with MEA pedestals were challenged with PIC on Day 4 post-surgery, and status epilepticus (SE) was induced by subcutaneous injection of 12 mg/kg of KA on Days 5, 6, 7 and 8 (PIC group). Mice injected with saline in lieu of PIC served as controls (SAL). Seizures were expressed as average seizure score (a), seizure duration (b) and cumulative seizure score (c) Also, the effect of SE on the body weight was monitored (d). Symbols represent means ± SEM from 5 to 7 animals per group. Asterisks denote significant differences from saline controls (**p≤ .01, ***p≤ .001).
Moreover, repeated daily injections of KA did not significantly change the body weight of the animals (Fig. 3D), indicating no overt adverse effects of the SE. This is consistent with the relatively high resistance of C57BL/6 mice to KA-induced neurotoxicity (McKhann et al. 2003).
To determine if pre-seizure tonic glutamate levels would predict seizure severity, we analyzed data from Day 5 by the Spearman test. This analysis demonstrated that average seizure score (R2=.65, p=.001; Fig. 4A), cumulative seizure score (R2=.73, p=.0002; Fig. 4B) and seizure duration (R2=.64, p=.0012; Fig. 4C) significantly correlated with the pre-seizure tonic glutamate levels in the CA1 (Fig. 4).
Figure 4. The relationship between tonic glutamate level and seizure intensity.
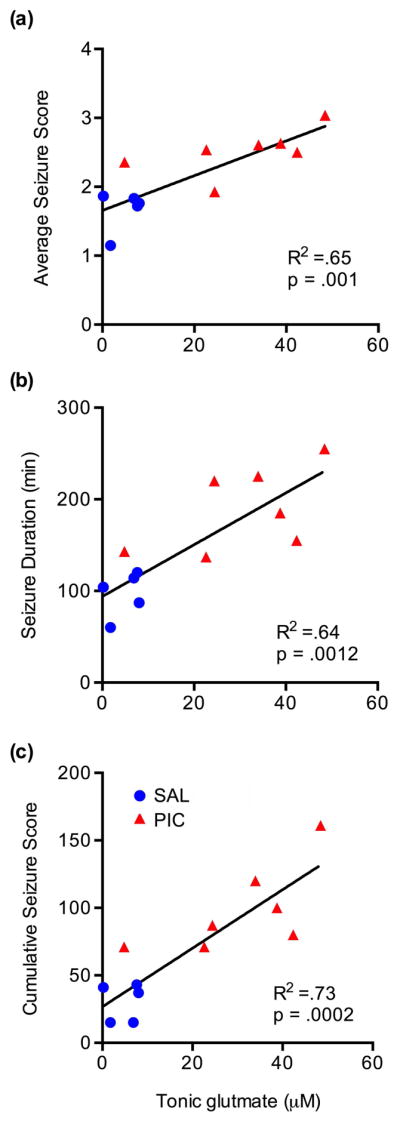
Tonic glutamate was determined in animals described in Fig. 3 before injection of KA. These values were plotted against the average seizure scores (a), seizure duration (b) and cumulative seizure scores (c). The correlation was analyzed by the Spearman’s rank correlation.
Next, we sought to determine the effect of seizures on extracellular glutamate levels. On Day 5 (24 hours after PIC injection), glutamate levels were examined in PIC-challenged (PIC) and saline-injected mice (SAL) across three phases, i.e., pre-seizure, seizure, and post-seizure (Fig. 5). Extracellular glutamate levels were significantly higher in PIC-challenged mice as compared to saline-injected controls (Group effect, F(1,9)=37.71, p=.0002), with a 7.7-fold (p<.001), 6-fold (p<.001) and 4.3-fold (p<.01) across the 3 phases, respectively. PIC-challenged mice exhibited a 76% increase in tonic levels during seizures, whereas the saline-injected mice did not [Phase*Group effect, F(2,18)=5.7, p=.01]. There were no differences between pre- and post-seizure glutamate levels in either PIC or SAL mice.
Figure 5. Seizure-induced changes in extracellular glutamate level.
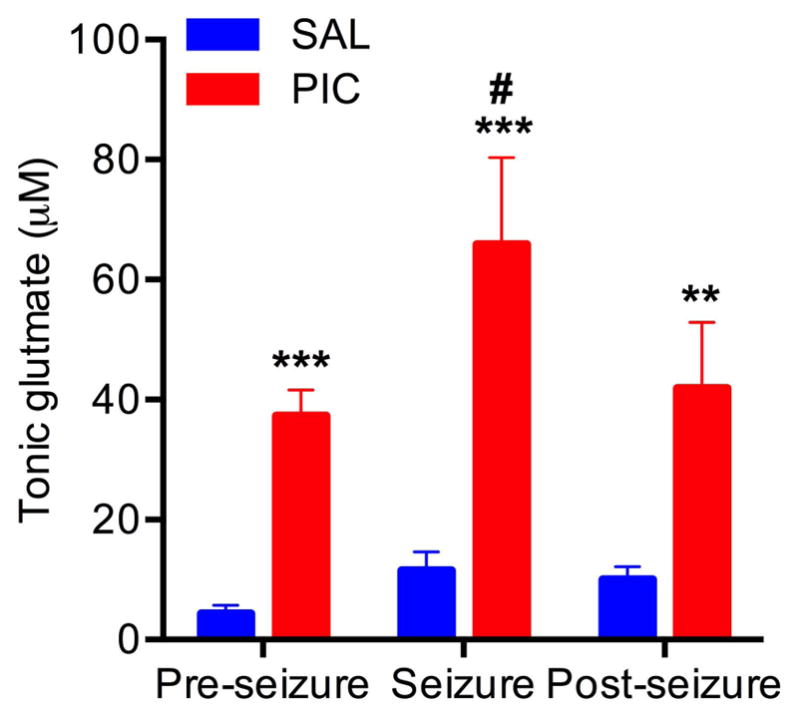
Mice implanted with MEA pedestals were challenged with PIC on Day 4 post-surgery (PIC group). Mice injected with saline in lieu of PIC served as respective controls (SAL). Seizures were induced by subcutaneous injection of 12 mg/kg of KA on Day 5. Tonic glutamate levels were measured prior, during, and after SE (for details see M&M). Bars represent means ± SEM from 5 to 7 animals per group. Asterisks denote significant differences from saline controls (**p≤ .01, ***p≤ .001). #denotes significant differences between pre-seizure and seizure (p≤ .05).
Discussion
In the present study, we adopted the paradigm of chronic implantation of MEA (Hascup et al. 2008) to monitor extracellular glutamate in the hippocampus of awake, freely behaving mice. In line with a minimal damage to adjacent brain tissue instigated by the MEA implantation (Rutherford et al. 2007), no overt morbidity was detectable in mice 2–8 days post-op. Moreover, there was no change in tonic glutamate level at 5–8 days post op (Fig. 2, SAL) further buttressing functional integrity of the brain tissue, as well as the patency of chronically implanted MEAs in awake animals (Rutherford et al. 2007, Hascup et al. 2008).
Tonic glutamate level in CA1 of awake control mice (Fig. 2, SAL) was approximately 3-fold higher than the level observed in mice anesthetized with isoflurane (Hunsberger et al. 2016). This is congruent with previous studies in rats that revealed approximately 3-fold higher levels in the striatum of freely moving vs. anesthetized animals; while the respective increase for the cortex was approximately 30-fold (Rutherford et al. 2007). These authors attributed this difference to the anesthesia, as the administration of urethane resulted in approximately 60% drop in the tonic glutamate level. Also, the administration of pentobarbital was shown to cause 42% decrease in rat cortical glutamate level (Dash et al. 2009). Together, these studies support the notion that anesthesia profoundly dampens glutamatergic transmission in the brain.
Congruent with our previous study (Hunsberger et al. 2016), also MEA-implanted mice responded to peripheral PIC challenge by increasing tonic glutamate in CA1 (Fig. 2). However, this increase was only approximately 3-fold, in contrast to approximately 10-fold increase in anesthetized mice (Hunsberger et al. 2016). The cause of this divergence is likely to involve complex neuromodulatory mechanism that might be dependent on the tonic levels of extracellular glutamate.
We have previously demonstrated that i.p. injection of PIC renders the brain hypersusceptibile to KA-induced seizures (Kirschman et al. 2011, Michalovicz & Konat 2014). We have further shown that this hypersusceptibility is concomitant with a robust increase in extracellular glutamate levels in the hippocampus, and with hyperexcitability of hippocampal neuronal circuitry (Hunsberger et al. 2016). The present study performed in awake animals has revealed that seizure intensity strongly correlate with pre-seizure tonic glutamate levels (Fig. 4). A similar correlation was previously observed in a divergent experimental system using intrahippocampal injection of 4-aminopyridine (4-AP) in freely behaving rats implanted with MEA (Stephens et al. 2014). Together, these results strongly implicate elevated extracellular glutamate as a causal factor in the development of seizure hypersusceptibility.
KA-induced status epilepticus increased extracellular glutamate in the CA1 of PIC-challenged mice (Fig. 5). Similar increase in glutamate level was observed in rabbit hippocampus following perfusion with KA (Lehmann et al. 1983). The elevation of extracellular glutamate has also been observed during 4-AP-induced seizures (Morales-Villagran et al. 2008). The underlying mechanism(s) likely entails enhanced excitatory activity that increases glutamate release. Moreover, a direct contribution of KA might also be envisaged as it has been shown to inhibit glutamate uptake (Fykse et al. 1992). Within 20 min after seizure cessation, extracellular glutamate dwindled to the pre-seizure levels in both PIC-challenged and saline controls, indicating a relatively rapid restoration of glutamate homeostasis.
In addition to tonic glutamate, seizure activity positively correlates with paroxysmal, low magnitude fluctuations in extracellular glutamate, referred to as glutamate transients (Stephens et al. 2014). Although we also detected a trend to increase the amplitude of glutamate transients in MEA-implanted mice during KA-induced status epilepticus, no significant correlation between seizures activity and glutamate transients was evident (results not shown). This difference might be related to the ictogenic factors and their route of administration, i.e., 4-AP injected into the hippocampus (Stephens et al. 2014) vs. KA injected subcutaneously (present study). Also, species-specificity might be considered as a contributing factor.
It is of interest whether the correlation between seizure severity and extracellular glutamate levels applies to other types of seizure, or is specific to the ictogenic activity of KA. This issue will be addressed in future studies.
In conclusion, the major finding of our study is that the hypersusceptibility to KA-induced seizures instigated by peripheral PIC challenge likely results from the elevation of extracellular glutamate in the hippocampus.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (MNR & GWK; U54GM104942), NIA (MNR; R15AG045812), Alzheimer’s Association (MNR; NIRG-12-242187), WVU Faculty Research Senate Grant (GWK) and WVU PSCOR Grant (GWK).
Abbreviations
- 4-AP
4-aminopyridine
- APR
acute phase response
- CA1
cornu ammonis 1
- i.p
intraperitoneal
- KA
kainic acid
- MEA
microelectrode arrays
- PPF
paired pulse facilitation
- PIC
polyinosinic-polycytidylic acid
- s.c
subcutaneous
- SAL
saline
Footnotes
Conflict of interest disclosure. The authors have no conflict of interest to declare.
References
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Buljevac D, Flach HZ, Hop WC, Hijdra D, Laman JD, Savelkoul HF, van Der Meche FG, van Doorn PA, Hintzen RQ. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 2002;125:952–960. doi: 10.1093/brain/awf098. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Moxon K, Gerhardt GA. Ceramic-based multisite microelectrodes for electrochemical recordings. Anal Chem. 2000;72:187–192. doi: 10.1021/ac9907991. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J Neurosci Methods. 2002;119:163–171. doi: 10.1016/s0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Teeling J, Felton L, Perry VH. The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C) Brain, behavior, and immunity. 2007;21:490–502. doi: 10.1016/j.bbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am. 2009;29:247–264. doi: 10.1016/j.iac.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash MB, Douglas CL, Vyazovskiy VV, Cirelli C, Tononi G. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J Neurosci. 2009;29:620–629. doi: 10.1523/JNEUROSCI.5486-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari CC, Tarelli R. Parkinson’s disease and systemic inflammation. Parkinsons Dis. 2011;2011:436813. doi: 10.4061/2011/436813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fykse EM, Iversen EG, Fonnum F. Inhibition of L-glutamate uptake into synaptic vesicles. Neuroscience letters. 1992;135:125–128. doi: 10.1016/0304-3940(92)90151-v. [DOI] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Pomerleau F, Huettl P, Gerhardt GA. Second-by-second measures of L-glutamate in the prefrontal cortex and striatum of freely moving mice. The Journal of pharmacology and experimental therapeutics. 2008;324:725–731. doi: 10.1124/jpet.107.131698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C. Review: systemic inflammation and Alzheimer’s disease. Neuropathol Appl Neurobiol. 2013;39:51–68. doi: 10.1111/j.1365-2990.2012.01307.x. [DOI] [PubMed] [Google Scholar]
- Hunsberger HC, Wang D, Petrisko TJ, Alhowail A, Setti SE, Suppiramaniam V, Konat GW, Reed MN. Peripherally restricted viral challenge elevates extracellular glutamate and enhances synaptic transmission in the hippocampus. J Neurochem. 2016 doi: 10.1111/jnc.13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschman LT, Borysiewicz E, Fil D, Konat GW. Peripheral immune challenge with dsRNA enhances kainic acid-induced status epilepticus. Metab Brain Dis. 2011;26:91–93. doi: 10.1007/s11011-011-9236-z. [DOI] [PubMed] [Google Scholar]
- Lehmann A, Isacsson H, Hamberger A. Effects of in vivo administration of kainic acid on the extracellular amino acid pool in the rabbit hippocampus. J Neurochem. 1983;40:1314–1320. doi: 10.1111/j.1471-4159.1983.tb13572.x. [DOI] [PubMed] [Google Scholar]
- McKhann GM, 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–561. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- Michalovicz LT, Konat GW. Peripherally restricted acute phase response to a viral mimic alters hippocampal gene expression. Metab Brain Dis. 2014;29:75–86. doi: 10.1007/s11011-013-9471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Villagran A, Medina-Ceja L, Lopez-Perez SJ. Simultaneous glutamate and EEG activity measurements during seizures in rat hippocampal region with the use of an electrochemical biosensor. Journal of neuroscience methods. 2008;168:48–53. doi: 10.1016/j.jneumeth.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. Mouse Brain in Stereotaxic Coordinates. Academic Press; 2012. [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J Neurochem. 2007;102:712–722. doi: 10.1111/j.1471-4159.2007.04596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens ML, Williamson A, Deel ME, Bensalem-Owen M, Davis VA, Slevin J, Pomerleau F, Huettl P, Gerhardt GA. Tonic glutamate in CA1 of aging rats correlates with phasic glutamate dysregulation during seizure. Epilepsia. 2014;55:1817–1825. doi: 10.1111/epi.12797. [DOI] [PubMed] [Google Scholar]
- Tellez-Zenteno JF, Matijevic S, Wiebe S. Somatic comorbidity of epilepsy in the general population in Canada. Epilepsia. 2005;46:1955–1962. doi: 10.1111/j.1528-1167.2005.00344.x. [DOI] [PubMed] [Google Scholar]