

Abstract
Glycogen synthase kinase 3 (GSK3) is a serine-threonine kinase that regulates mammalian circadian rhythms at the behavioral, molecular and neurophysiological levels. In the central circadian pacemaker, the suprachiasmatic nucleus (SCN), inhibitory phosphorylation of GSK3 exhibits a rhythm across the 24h day. We have recently shown that GSK3 is capable of influencing both the molecular clock and SCN neuronal activity rhythms. However, it is not known whether GSK3 regulates the response to environmental cues such as light. The goal of the present study was to test the hypothesis that GSK3 activation mediates light-induced SCN excitability and photic entrainment. Immunofluorescence staining in the SCN of mice showed that late-night light exposure significantly increased GSK3 activity (decreased pGSK3β levels) 30–60 minutes after the light-pulse. Additionally, pharmacological inhibition of GSK3 blocked the expected light-induced excitability in SCN neurons; however, this effect was not associated with changes in resting membrane potential or input resistance. Behaviorally, mice with constitutively active GSK3 (GSK3-KI) re-entrained to a 6-h phase advance in the light-dark cycle in significantly fewer days than WT control animals. Furthermore, the behavioral and SCN neuronal activity of GSK3-KI mice was phase-advanced compared to WT, in both normal and light-exposed conditions. Finally, GSK3-KI mice exhibited normal negative-masking behavior and electroretinographic responses to light, suggesting that the enhanced photic entrainment is not due to an overall increased sensitivity to light in these animals. Taken together, these results provide strong evidence that GSK3 activation contributes to light-induced phase-resetting at both the neurophysiological and behavioral levels.
Keywords: electrophysiology, entrainment, light, circadian, mouse
Graphical Abstract
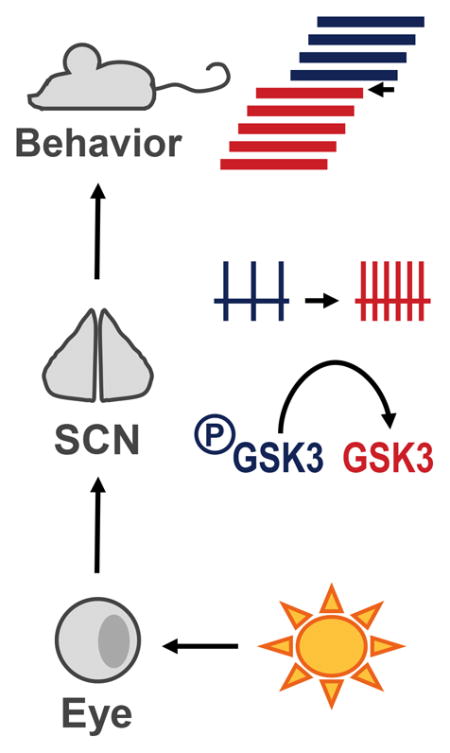
Exposure to late-night light pulse (LP) acutely increases GSK3 activation in the circadian pacemaker – the suprachiasmatic nucleus (SCN), increases SCN neuronal activity 3-5 hours later, and advances circadian behavior. Pharmacological inhibition of GSK3 blocks light-induced SCN activity. Mice with chronically active GSK3 exhibit phase-advanced behavioral rhythms and SCN excitability.
INTRODUCTION
Entrainment is the process by which internal circadian rhythms are synchronized to the external environment. Although there are many environmental factors that can influence circadian rhythmicity (i.e. food availability, novelty-induced exercise), light is the major entraining signal to the master circadian pacemaker, the suprachiasmatic nucleus (SCN; (Czeisler, 1995; Golombek & Rosenstein, 2010)). Exposure to light at specific times of the night shifts the phase of the molecular clock and behavioral activity rhythms (Golombek & Rosenstein, 2010). This process of phase-resetting in response to an acute light-pulse (LP) begins with glutamate release from the retinohypothalamic tract onto SCN neurons (Liou et al., 1986; Shibata et al., 1986; Hannibal, 2002) and activation of NMDA receptors (Colwell & Menaker, 1992), resulting in an influx of Ca2+ (Colwell, 2001) and subsequent activation of multiple signaling cascades. These cascades, which include activation of the mitogen activated protein kinases (MAPKs) extracellular signal-related kinases 1 and 2 (ERK1/2; (Obrietan et al., 1998; Butcher et al., 2003)), lead to the rapid induction of genes, including period1 (Per1; (Tischkau et al., 2003)). In the late-night, this upregulation of Per1 is followed by a persistent increase in SCN neuronal activity 3–5 hours later (Kuhlman et al., 2003).
Although many studies have examined the molecular events involved in the process of phase-resetting, very little is known about links connecting light-induced molecular events to neurophysiological excitability (Colwell, 2011). One candidate mediator is glycogen synthase kinase 3 (GSK3), a serine-threonine kinase that is an emerging regulator of mammalian circadian rhythms. Much of the previous research on the role of GSK3 in the circadian system has focused on its phosphorylation of multiple components of the core-molecular clock (Iitaka et al., 2005; Kurabayashi et al., 2010; Sahar et al., 2010). However, we have recently reported that chronic GSK3 activation increases SCN neuronal activity, whereas GSK3 inhibition decreases SCN neuronal activity, highlighting GSK3 as a regulator of neurophysiological rhythms as well (Paul et al., 2012; Paul et al., 2016). Thus, GSK3 is in a unique position to provide the link between molecular rhythms and neuronal excitability. Additionally, research examining the role of GSK3 in the process of phase-resetting has been limited. Therefore, the goal of the present study was to test the hypothesis that GSK3 activation mediates light-induced SCN excitability and photic entrainment.
MATERIALS AND METHODS
Animals
The experimental protocols in this study were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee (IACUC). All experiments were conducted in accordance with University of Alabama at Birmingham IACUC guidelines and the NIH Guide for the Care and Use of Laboratory Animals. All mouse strains were backcrossed on a C57BL6/J background for at least 10 generations. For the experiments in Figure 3, homozygous GSK3α/β21A/21A/9A/9A (GSK3-KI) mice (McManus et al., 2005) were compared with age-matched wildtype (WT) controls (as in (Paul et al., 2012)). For all other experiments, WT and GSK3-KI mice were homozygous for the Per1::GFP transgene reporter (Kuhlman et al., 2003). Mice (3 to 7 months old) were group-housed in a 12:12 light/dark (LD) cycle (unless stated otherwise) with food and water ad libitum. Due to the increased variability in activity onset seen in female mice (Iwahana et al., 2008), only male mice were used for behavioral measures; however, both male and female animals were used for all other experiments. All animals were euthanized by cervical dislocation and rapid decapitation. Mice were sacrificed between Zeitgeber Time (ZT) 22–23 (where ZT 0 equals time of lights-on) for all experiments.
Fig. 3. Chronic GSK3 activation enhances re-entrainment to advance in light-cycle.

(A–B) Representative double-plotted actograms of wheel-running behavior from WT (A) and GSK3-KI (B) mice undergoing a 6-hour advance in the LD cycle. Arrows indicated the first day each animal is considered re-entrained to the new LD cycle. (C) Daily activity onset (mean ± SEM) for days before and after advance in the LD cycle (day 0). Grey bars mark the times of lights-off. n = 10–12 animals per genotype.
Behavior
Mice were housed in individual wheel cages, and wheel-running activity was recorded and analyzed using ClockLab software (Actimetrics, Wilmette, IL, USA). The phase angle for each mouse was determined prior to shifting the light cycle by 6 h. Animals were considered to be entrained when the activity onset occurred within 20 min of the baseline phase angle. For the negative masking experiments, mice were maintained in LD, and exposed to 4-h LP of increasing intensity (~5-lux, dim red light; ~10-lux, dim white; and ~300-lux, bright white light) each night (ZT 14–18), with a 24-h period of normal LD in between. The amount of activity during the LP was compared to the activity at the same time on the previous night.
For acute LP experiments, we used a modified Aschoff type-II protocol (Jud et al., 2005) in which mice in LD were released into constant dark (DD) for 10 days (LP-), re-entrained to LD for 14 days, released into DD again, and exposed to a 1-hour LP (~300–500 lux; 3350K; F8T5/CW bulb) at ZT 22 on the first day in DD (LP+). To eliminate the non-photic effects of wheel-running, the running-wheel was locked during the 1-hour LP and during the equivalent time in the LP- condition (ZT 22–23 on first day in DD). Regression lines were fit to the activity onsets on the last 5 days in LD and on days 4–10 in DD. The magnitude of the phase-shift was calculated as the difference between the activity onsets predicted by each fit line on the second day in DD. Activity onsets were determined by eye by an observer blind to the genotype of each animal.
Electrophysiology
WT and GSK3-KI mice, crossed with homozygous Per1::GFP mice, were held in a 12:12 LD cycle and were exposed to a 15-min LP (~500 lux; 3350K; FO32/841/ECO bulb) at ZT 22 and sacrificed immediately following the LP. For no light controls, animals were sacrificed and enucleated in the dark with the aid of night-vision goggles at ZT 22.25. Fresh coronal brain slices (200 μm thickness) were prepared on a vibrating microtome (Campden 7000SMZ, World Precision Instruments, Lafayette, IN, USA) while bathed in ice-cold oxygenated high-sucrose saline (in mM: 250 sucrose, 26 NaHCO3, 1.25 Na2HPO4-7H2O, 1.2 MgSO4-7H2O, 10 glucose, 2.5 MgCl2, 3.5 KCl. Slices were allowed to rest for 20-min in a bath containing room-temperature, oxygenated solution of 50% high-sucrose saline and 50% normal extracellular solution (in mM: 130 NaCl, 20 NaHCO3, 1 Na2HPO4-7H2O, 1.3 MgSO4-7H2O, 10 glucose, 3.5 KCl, 2.5 CaCl2). For recordings involving a pharmacological treatment, slices were transferred to a heated bath (34 ± 0.5°C) of normal saline containing CHIR-99021 (CHIR; 1 μM; Stemgent, Lexington, MS, USA, as in (Paul et al., 2016)) or vehicle (DMSO; 0.002%) for 1-hour (ZT 23–24). Slices were then transferred to a recording chamber and continuously perfused (2ml/min) with oxygenated, normal saline heated to 34 ± 0.5°C. Cells were visualized using an Axio Examiner microscope (Zeiss) equipped for near-IR-DIC and epi-fluorescence (excitation spectrum BP 470/40). Glass pipettes were pulled at a resistance of 3–5MΩ and filled with a filtered internal solution (in mM: 135 K-gluconate, 10 KCl, 10 HEPES, 0.5 EGTA; pH 7.4). Electrophysiological signals were processed and controlled by a Multiclamp 700B amplifier and pClamp 10.02 software (Axon Instruments, Union City, CA, USA) in gap-free or current-clamp mode. Recordings were sampled at 20 kHz and filtered at 10 kHz. Spontaneous firing rate was measured as the average action potential frequency from a 2-min recording. Resting membrane potential (RMP) was determined at the half-width voltage between action potentials. All recordings were made between ZT 1–3 or ZT 0–3.
Immunohistochemistry
Per1::GFP mice were exposed to a 15-min light pulse (~300–500 lux) at ZT 22, returned to darkness, sacrificed and enucleated 30 or 60 minutes later. Brains were harvested and processed for IHC using rabbit anti-pGSK3β (1:1000; Cell Signaling, Danvers, MA, USA) and mouse anti-rabbit Alexafluor 488 (1:500; Invitrogen). Importantly, we confirmed in a separate cohort of animals that no fluorescence signal was detectable in the 488 nm excitation range in SCN from Per1::GFP mice to justify using the Alexa Fluor 488 secondary antibody for the detection of phospho-GSK3β (data not shown). GSK3α phosphorylation was not examined due to a lack of available IHC compatible antibody. All images were captured on a Leica confocal microscope at the same laser intensity, contrast and gain settings. Fluorescence intensity (arbitrary units) was measured in Image J software (NIH) by drawing a standard region of interest for the whole SCN. The background intensity, taken from the anterior hypothalamus just outside the SCN was subtracted from all values. Data are presented as mean intensity ± standard error.
Electroretinographic (ERG) Recordings
Standard electroretinography recording techniques were used (Viswanathan et al., 1999; Clark & Kraft, 2012). Following overnight dark adaptation, mice were anesthetized under dim red illumination with 100 mg/kg ketamine and 10 mg/kg xylazine. Under anesthesia, both eyes were treated with proparacaine HCl (0.5%) followed by a mixture of phenylephrine HCl (2.5%) and tropicamide (1%) for pupil dilation. A gold reference electrode was electrically connected to one eye and a platinum wire fiber-optic combination was connected to the other. Light stimuli were delivered directly onto the eye through the fiber-optic with a 100-W tungsten bulb light-source. Calibrated neutral density filters were used to control stimulus intensity, and the stimulus wavelength was set by a 505 nm band pass filter (35nm FWHM full width at half maximum). Stimulus flashes (2ms) were controlled by a computer-driven Uniblitz shutter. Electrical responses were amplified (CP122W; Astromed; DC 300 Hz) and digitized at 2 KHz (Real-Time PXI Computer; National Instruments). Data analysis was performed using IGOR software (WaveMetrics). The data in Fig. 4 were low-pass filtered at 40 Hz post-hoc.
Fig. 4. Chronic GSK3 activation does not alter negative masking behavior or ERG responses to light.

(A) Representative single-plotted actograms from WT and GSK3-KI mice exposed to increasing intensities of light for 4 hours during the dark phase (shown in box). Gray marks the time of lights off. (B) Means ± SEM of the percent of activity during LP relative to activity on the previous day. (C–D) Representative ERG recordings from WT (C) and GSK3-KI (D) dark-adapted mice elicited by increasingly intense flashes of light that delivered 2, 30, 5100, and 23,550 photons/μm2 to the cornea. Red trace is the b-wave elicited by flash intensity of 30 photons/μm2. (E) Means ± SEM of dark adapted and light adapted ERG amplitudes elicited by the highest intensity stimulus in (C–D). (F) The photopic negative response (PhNR) was elicited by bright flashes (5100 photons/μm2) that were delivered on a steady background (4,500 photons/μm2sec1). The amplitude and time-to-peak of the trough that follows the b-wave response represents the values of the PhNR reported.
Data Analysis and Availability
All statistics were calculated using SPSS 22 software. Data were analyzed with independent samples t-tests, two-way ANOVAs, two-way mixed-design ANOVAs, and linear-mixed model ANOVAs (using the Generalized Estimating Equations in SPSS). Where indicated, post hoc tests were conducted using Tukey’s HSD or Fisher’s LSD for planned comparisons. Significance was ascribed at P < 0.05. The data that support the findings of this study are available from the corresponding authors upon reasonable request.
RESULTS
Late-night light exposure induces GSK3β activation in SCN
In the early stages of photic-phase-shifting, signal cascades such as the MAPK (Obrietan et al., 1998) or the mammalian target of rapamycin (mTOR) pathways (Cao et al., 2008) are activated through kinase phosphorylation (e.g. induction of pERK1/2, p-p70 S6 kinase), initiating the molecular response to photic stimuli. To determine whether GSK3 was among the kinases involved in late-night phase-resetting, we exposed WT mice to a 15-min LP at ZT 22 and examined phosphorylation of GSK3β (pGSK3β) levels in the SCN of light-exposed and no light controls at 30 and 60 min after LP onset. In control animals, pGSK3β levels were high throughout the whole SCN (Fig. 1A) as expected, according to previously published research (Iitaka et al., 2005). Exposure to a LP significantly activated (dephosphorylated) GSK3β (two-way ANOVA, main effect of light, F1,8 = 25.268, P = 0.001; n = 3 mice per group per time point; Fig. 1B) at both time points, reducing pGSK3β levels by more than 50% overall.
Fig. 1. Acute light-pulse at ZT22 reduces GSK3β phosphorylation in SCN.

Representative images of anti-pGSK3β staining (A) and quantification of background subtracted fluorescence intensity (B) for whole SCN sections from mice 30- or 60-minutes after exposure to a 15-minute light pulse (LP+) or no light controls (LP-) taken at the same time. n = 3 mice per group per time point. Main effect of light **P < 0.005. a.u., arbitrary units.
GSK3 activation is necessary for light-induced SCN neuronal activity
Three to five hours following late-night light exposure, electrical activity is persistently elevated in the light-responsive neurons of the SCN which can be identified using the Per1::GFP reporter mouse model (Kuhlman et al., 2003). Recent work in the SCN has demonstrated that GSK3 activation promotes neuronal excitability (Paul et al., 2012), while GSK3 inhibition suppresses neuronal activity (Paul et al., 2016); therefore, we next sought to determine whether GSK3 activation was necessary for light-induced increase in SCN firing. To do this, a 15-min light pulse was administered at ZT 22, after which mice were immediately sacrificed for slice preparation and in vitro GSK3 inhibitor application. Specifically, acute brain slices from light-exposed (LP+) or no light control (LP-) Per1::GFP mice were treated with CHIR-99021 (CHIR; 1 μM) or vehicle (DMSO; 0.002%) for 1 hour (ZT 23–24), and fluorescence targeted loose-patch recordings on SCN neurons were performed 3–5 hours after light exposure (ZT 1–3). As expected, light-exposure significantly increased the spontaneous firing rate (SFR) in vehicle-treated slices, with LP+ cells spiking at a rate more than 2.5 times faster than LP- cells, on average (two-way ANOVA, light X treatment interaction, F1,165 = 7.849, P = 0.006; Tukey’s HSD post hoc test, P = 0.000; n = 41–43 cells, 3–4 mice per group; Fig. 2A, B). However, CHIR treatment significantly suppressed the SFR of LP+ neurons compared to vehicle treated LP+ cells (P = 0.008), such that the mean SFR of SCN CHIR treated LP+ neurons was essentially equal to that of the CHIR-treated LP- cells (Tukey’s HSD post hoc test, P = 0.997). Interestingly, there was no difference between vehicle and CHIR treated LP- groups, suggesting that the effect of CHIR was specific to light-induced excitability (Tukey’s HSD post hoc test, P = 0.88).
Fig. 2. GSK3 inhibition blocks light-induced increase in SCN neuronal activity.

(A) Spontaneous action potential frequencies (means ± SEM) of Per1::GFP expressing SCN neurons treated with vehicle (DMSO, 0.002%) or CHIR (1 μM) for 1 hour (ZT 23–24) following exposure to 15-min light-pulse (LP+) or no light (LP-) at ZT 22. Recordings were made 3–5 hours after onset of photic stimulus (ZT 1–3). (B) Representative cell-attached loose-patch traces (5 s) from each group in (A). ***P < 0.001, n = 41–43 cells, 3–4 slices per group. (C) Representative current clamp recordings from LP+ SCN neurons treated with vehicle or CHIR (as in A). (D–E) Means ± SEM of resting membrane potential (D) and input resistance (E) of cells represented in (C). n = 13–15 cells, 2–3 slices per group.
To examine if GSK3 inhibition during a shorter window of time would be sufficient to block light-induced SCN excitability, we treated SCN slices from LP+ animals with CHIR or vehicle for 15 minutes (ZT 23–23.25) and performed targeted loose-patch recordings as described above. Surprisingly, the SFR of LP+ neurons exposed to a 15-min CHIR treatment was significantly reduced compared to vehicle-treated neurons (means ± SEM: CHIR, 3.65 ± 0.63; vehicle, 5.18 ± 0.44; independent samples t-test, t50 = 2.053, P = 0.045; n = 22–30 cells per group, 2–3 mice per group).
The increase in SCN activity following a late-night LP has previously been associated with a depolarized resting membrane potential and increased input resistance (Kuhlman et al., 2003). To determine whether GSK3 inhibition blocked light-induced excitability by acting on the membrane properties of the cell, we performed whole-cell current clamp recordings in LP+ slices treated for 1 hour with either CHIR or vehicle using the same paradigm as above. There were no significant differences in the resting membrane potential (independent samples t-test, t27 = 0.434, P = 0.668; n = 13–15 cells, 2–3 mice per group; Fig. 2C, D) or input resistance (t25 = 1.066, P = 0.296; Fig. 2C, E) between LP+ vehicle- or CHIR-treated cells.
Chronic activation of GSK3 enhances behavioral and neurophysiological response to light
We next sought to determine the role of GSK3 activation in regulating the effect of light on circadian rhythms at the behavioral level by comparing WT mice to mice with constitutive activation of GSK3 (i.e., GSK3-KI mice) such that both α and β isoforms of GSK3 can no longer be phosphorylated and inactivated (McManus et al., 2005; Paul et al., 2012). To examine the effect of chronic GSK3 activity on photic entrainment, we first measured the rate of entrainment of GSK3-KI and WT mice after a 6-hour advance of the light cycle (Fig. 3A, B). In the 5 days prior to the shift, GSK3-KI mice showed a significantly earlier activity onset, compared to WT mice (means ± SEM: GSK3-KI, ZT 11.98 ± 0.08; WT, ZT 12.28 ± 0.09; two-way mixed design ANOVA, main effect of genotype, F1,20 = 5.99, P = 0.024; n = 10–12 mice per genotype). When the light cycle was advanced by 6 h, GSK3-KI animals entrained to the new LD cycle in significantly fewer days than WT mice (means ± SEM: GSK3-KI, 4.75 ± 0.41; WT, 7.4 ± 0.62 days; independent samples t-test, t20 = −3.677, P = 0.001; Fig. 3C). There was no difference between GSK3-KI and WT mice in the rate of entrainment to a 6 h delay in the light cycle (means ± SEM: GSK3-KI, 3.5 ± 0.43; WT, 4.8 ± 0.58 days; independent samples t-test, t9 = 1.836, P = 0.099; n = 5–6 mice per group).
To determine whether the enhanced photic entrainment of GSK3-KI mice was due to increased sensitivity to light outside of the circadian system (i.e., negative masking), we examined acute light-induced suppression of locomotor behavior in GSK3-KI and WT mice. While maintained in LD, mice were exposed to a 4-hour pulse of dim red, dim white, or bright white light in the middle of the night (ZT 14–18; Fig. 4A, B). Negative masking was measured as the percent of activity during the LP relative to amount activity during the same time on the previous day (Fig. 4B). When exposed to dim red light, the majority of WT mice (5/7) exhibited a slight increase in wheel-running activity, known as positive masking, whereas, in the GSK3-KI group, dim red light decreased activity in 4/7 animals; however, the percent of activity was not significantly different between groups. Furthermore, both genotypes exhibited similar levels of negative masking during the dim- and bright-white light pulses (two-way mixed design ANOVA, main effect of light intensity, F2,11 = 79.007, P = 0.000; Fig. 4B).
As an additional measure of non-circadian light processing, we investigated whether there were any gross retinal abnormalities in these mice. Specifically, we performed ERG analysis on GSK3-KI mice (n = 7; Fig. 4C) and WT controls (n = 6; Fig. 4D) and found that there were no significant differences between the two mouse lines in any of the parameters of dark-adapted retinal function that were tested (independent samples t-test: a-wave, t11 = 0.889, P = 0.39; b-wave, t11 = 0.651, P = 0.53; Fig. 4E). Light-adapted ERG, designed to isolate cone photoreceptor responses, also demonstrated similarity between WT and GSK3-KI mice (a-wave, t11 = 0.326, P = 0.75; b-wave, t11 = 0.359, P = 0.73; Fig. 4E). Likewise, there were no significant differences in the timing (time-to-peak, means ± SEM: WT, 124 ± 4; GSK3-KI, 116 ± 5 ms; t10 = 1.359, P = 0.20; n = 6 mice per genotype) or amplitude (means ± SEM: WT, 60.0 ± 14; GSK3-KI, 46.5 ± 5 μV; t6.1 = 0.913, P = 0.40; n = 6 mice per genotype) of the photopic negative response (PhNR), which represents ganglion cell activity (Fig. 4F). Overall, the retinas of each mouse line do not appear to be functionally different. Taken together with the negative-masking behavior, these results suggest that the effect of GSK3 on photic entrainment is likely not due to altered retinal function.
We also tested the phase-shifting effects of an acute light-pulse in WT and GSK3-KI mice (n = 7 mice per genotype) using a modified Aschoff type-II protocol (see methods). Briefly, we first measured the phase-angles of mice in two conditions: first, after release into DD with no light exposure (LP-; Fig. 5A, B) and second after re-entrainment and release into DD following a 60-min LP (LP+) at ZT 22 (Fig. 5C, D). As expected, exposure to a late-night LP caused a significant advance in both genotypes (mixed-design ANOVA, main effect of light, F1,12 = 70.227, P = 0.000; Fig. 5E). Surprisingly, the magnitude of the light-induced phase-shift was not different between genotypes (non-significant light x genotype interaction, P = 0.692). Instead, the activity onsets of GSK3-KI mice were significantly more advanced than WT mice in both LP- and LP+ conditions (main effect of genotype, F1,12 = 5.360, P = 0.039; Fig. 5E). Light exposure also significantly shortened the free-running period in DD similarly in both genotypes (mixed-design ANOVA, main effect of light, F1,12 = 12.242, P = 0.004; Fig. 5F).
Fig. 5. Phase-angle of entrainment is advanced in GSK3-KI mice and following acute LP in late-night.

(A–D) Representative single-plotted actograms of WT (A, C) and GSK3-KI (B, D) mice exposed to a 1-hr LP with accompanying wheel-clip (C–D; LP+) or wheel-clip alone (A–B; LP-) at ZT 22 on first day in DD. A line was fit to the activity onsets for the last 5 days in LD and days 3–9 in DD. The difference of these fit lines on day 1 in DD was defined as the phase shift. (E–F) Means ± SEM of phase-shift magnitude (E) and free-running period in DD (F) for animals represented in A–B. Panel E: Main effect of light (P < 0.001) and genotype (P < 0.05). Panel F: Main effect of light, (P < 0.005). n = 7 mice per genotype.
To determine whether the chronic GSK3 activity affected the SCN neurophysiological response to light, we performed targeted loose-patch recordings in SCN slices from light-exposed (LP+) or no-light control (LP-), GSK3-KI or WT mice, using the same paradigm as in the previous recordings (Fig. 2). Because the behavior of GSK3-KI mice was phase-advanced compared to WT mice in both LP- and LP+ conditions, we examined the spontaneous SCN activity 2–5 hours after light-exposure (ZT 0–3). Overall, SCN firing was significantly increased in GSK3-KI and light-exposed slices (linear mixed-model ANOVA; main effect of light, X2(1) = 24.717, P = 0.000; main effect of genotype, X2(1) = 9.568, P = 0.002; n = 61–92 cells, 3–6 mice per group; Fig. 6A); however, as shown in Fig. 6B and 6C, timing of light-induced excitability differed between genotypes (three-way interaction, X2(2) = 7.254, P = 0.027). In WT slices, light exposure did not increase SCN firing until the second hour of recording (LP- WT vs LP+ WT: first hour, P = 0.458; second hour, P = 0.023), whereas in LP+ GSK3-KI slices, the SFR was significantly increased as early as ZT 0 (P = 0.023; Fig. 6C). Furthermore, in the LP- groups, GSK3-KI cells were significantly more excited than WT neurons in the ZT 2–3 time window (P = 0.048). These results suggest that, similar to the advanced behavioral phase-angle (Fig. 3C and Fig. 5E), the early-morning increase in SCN excitability of GSK3-KI mice was advanced in both LP- and LP+ conditions.
Fig. 6. Light-induced increase in SCN excitability occurs earlier in GSK3-KI mice.

(A) Spontaneous AP frequencies (means ± SEM) of Per1::GFP expressing SCN neurons from WT or GSK3-KI mice following exposure to a 15-min light-pulse (LP+) or no light (LP-) at ZT 22. Recordings were made 2–5 hours after onset of photic stimulus (ZT 0–3). Main effect of light, P < 0.001; main effect of genotype, P < 0.005; n = 61, 92 cells, 3–6 slices per group. (B–C) Graph (means ± SEM) of data from (A) divided into 1-hr bins depicting WT and GSK3-KI SFR from LP- (B) or LP+ (C) groups. ***P = 0.001, *P < 0.05; Fisher’s LSD post hoc test.
DISCUSSION
In the present study, we present strong evidence that GSK3 activation serves a critical role in photic entrainment and light-induced SCN excitability. Specifically, we found that: 1) acute late-night light exposure activates GSK3β in the SCN, 2) GSK3 activation is necessary for light-induced neuronal excitability, 3) mice with constitutively active GSK3 exhibit enhanced photic entrainment despite having normal non-circadian light processing, and 4) locomotor activity onset and the early morning rise in SCN neuronal activity of GSK3-KI animals is phase-advanced in both LP- and LP-stimulated conditions. Together, these results support a role for GSK3 activation in transducing persistent increases in electrical activity in the early stages of light-induced phase resetting.
Our first result that GSK3 is activated by late-night light exposure is consistent with another recent report showing a light-induced decrease in the number of pGSK3β positive cells in rat SCN (Cervena et al., 2015). However, in that study, the change in GSK3β phosphorylation was not observed until two hours after light exposure as opposed to the data presented here which suggests the change in GSK3 activity occurs more rapidly, as early as 30 min post LP. These conflicting results are likely due to the numerous methodological differences, including animal species, LP timing and housing condition of the animals prior to experiment (DD vs. LD).
The light-induced reduction in pGSK3β (Fig. 1) appears to be concentrated in core regions of the SCN, with residual pGSK3β remaining in parts of the SCN that exhibit dense expression of vasopressin (Morin et al., 2006). The AVP-containing regions of the SCN have a considerably lower density of innervation from the retina than the core regions (Abrahamson & Moore, 2001; Lokshin et al., 2015), and light pulses induce Fos expression primarily in SCN core regions (Karatsoreos et al., 2004). These data therefore suggest that the observed reduction in GSK3β is largely confined to those parts of the cell that respond directly to retinal input.
The transient activation of other light responsive kinases such as ERK1/2 (Butcher et al., 2003; Cervena et al., 2015) and other MAPKs (Nakaya et al., 2003) parallels the acute molecular response to light (upregulation of Per transcription within 30–60 min; Kuhlman 2006), but then returns to pre-LP levels within an hour after light exposure. In contrast, GSK3β activation persists for at least one hour after the LP has ended (Fig. 1). Glutamate-induced phase-advances in SCN firing rhythms are attenuated when MAPK inhibitors and glutamate are applied simultaneously (Tischkau et al., 2000). However, we discovered that inhibition of GSK3 as late as 1–2 hours after the light stimulus was sufficient to block the increase in SCN firing during the early phase-resetting period. This result demonstrates a need for persistent GSK3 activation, long after other light-responsive kinases have returned to baseline activity levels. Given that light increases the spontaneous firing of Per1-induced neurons 3 to 5 h after stimulation, it appears that GSK3 activation begins in between the acute molecular phase and the persistent neural activity phase of the early phase shifting period.
The specific ionic mechanism by which GSK3 promotes light-induced excitability remains unclear. Our finding that GSK3 inhibition does not block light-induced changes in RMP or Rinput eliminate K+ leak currents as a potential target of GSK3. Furthermore, despite our previous finding that GSK3 regulates daily rhythms in SCN excitability through modulation of a persistent sodium current (INaP; (Paul et al., 2016)), we have found no difference in INaP magnitude between LP- and LP+ SCN neurons during the early phase-resetting period (ZT 1–3; unpublished results), suggesting that INaP is not involved in light-induced neuronal excitability. Therefore future work on the electrophysiological response to light could reveal additional ion currents that are regulated by GSK3 within the SCN.
Our finding that GSK3 activation promotes light-induced phase-advances is supported by the results that mice expressing chronically active forms of GSK3 exhibit rapid re-entrainment to an advance in the light-cycle. Interestingly, the enhanced response to light demonstrated in GSK3-KI mice was specific to circadian behavior, as GSK3-KI mice responded no differently from WT controls in negative masking behavior or in ERG responses under multiple lighting conditions. Conflicting with our data showing normal ERG responses in GSK3-KI mice, previous work has shown a decrease or increase in ERG b-wave amplitude in GSK3β-overexpressing or haploinsufficient mice, respectively (Lavoie et al., 2014). However, in the GSK3-KI mice used here, both isoforms of GSK3 are expressed at physiological levels suggesting that changes in the amount total GSK3 rather than levels of phosphorylation are necessary for changes in retinal function.
In addition to accelerating the rate of photic entrainment, chronic activation of GSK3 also advanced the phase angle of entrainment in wheel-running activity. This finding is consistent with past work showing significant delays in the phase angle of mice fed lithium, a known GSK3 inhibitor (Iwahana et al., 2004). Typically, the changes in the phase-angle of entrainment are associated with a change in the free-running period under constant conditions. Surprisingly, the free-running periods of GSK3-KI mice were not different from that of WT, despite exhibiting an advanced phase-angle. A recent study has demonstrated that optogenetic stimulation of SCN neurons alone is sufficient to shift both molecular and behavior rhythms, mimicking the effects of a light-pulse (Jones et al., 2015). Thus, one intriguing explanation for the advanced behavioral rhythms seen in GSK3-KI mice is the elevated SCN neuronal activity also seen in the GSK3-KI animals during the early day. This advanced increase in SCN activity could suggest that the full 24-h neurophysiological rhythm is shifted earlier, corresponding to an earlier behavioral onset. Future work in GSK3-KI animals examining SCN activity throughout the entire circadian cycle could provide more insight into the behavioral phenotype seen in these animals (Paul et al., 2012) as well as further elucidate the connection between neurophysiological and behavioral rhythms.
Taken together, the results of this study provide support for the hypothesis that GSK3 activity promotes light-induced SCN excitability. Previous work on GSK3 regulation of the circadian system has focused predominantly on daily rhythmicity; however, the results presented here strongly suggest that GSK3 is a previously unexplored regulator of photic entrainment as well. GSK3 dysregulation has also been implicated in numerous disorders that have also been associated with chronic light at night, such as depression (Bedrosian et al., 2013), bipolar disorder, obesity (Fonken et al., 2013; Fonken & Nelson, 2014), and cancer (Dauchy et al., 2014). Therefore, a better understanding the of effects of light on GSK3 and its role in regulating SCN neurophysiology could provide new treatment strategies for these disorders in the future.
Acknowledgments
This work was supported by NIH grants R01NS082413 (KLG) and F31NS086282.
ABBREVIATIONS
- CHIR
CHIR-99021
- DD
constant dark
- ERG
electroretinographic
- ERK1/2
extracellular signal-related kinase 1 and 2
- GSK3
glycogen synthase kinase 3
- GSK3-KI
GSK3α/β21A/21A/9A/9A
- LD
12:12 light/dark cycle
- LP
light-pulse
- MAPK
mitogen activated protein kinase
- Per1
period 1
- RMP
resting membrane potential
- SCN
suprachiasmatic nucleus
- SFR
spontaneous firing rate
- WT
wild-type
- ZT
Zeitgeber Time
References
- Abrahamson EE, Moore RY. Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res. 2001;916:172–191. doi: 10.1016/s0006-8993(01)02890-6. [DOI] [PubMed] [Google Scholar]
- Bedrosian TA, Weil ZM, Nelson RJ. Chronic dim light at night provokes reversible depression-like phenotype: possible role for TNF. Mol Psychiatry. 2013;18:930–936. doi: 10.1038/mp.2012.96. [DOI] [PubMed] [Google Scholar]
- Butcher GQ, Lee B, Obrietan K. Temporal regulation of light-induced extracellular signal-regulated kinase activation in the suprachiasmatic nucleus. J Neurophysiol. 2003;90:3854–3863. doi: 10.1152/jn.00524.2003. [DOI] [PubMed] [Google Scholar]
- Cao R, Lee B, Cho HY, Saklayen S, Obrietan K. Photic regulation of the mTOR signaling pathway in the suprachiasmatic circadian clock. Mol Cell Neurosci. 2008;38:312–324. doi: 10.1016/j.mcn.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervena K, Pacesova D, Spisska V, Bendova Z. Delayed Effect of the Light Pulse on Phosphorylated ERK1/2 and GSK3beta Kinases in the Ventrolateral Suprachiasmatic Nucleus of Rat. J Mol Neurosci. 2015;56:371–376. doi: 10.1007/s12031-015-0563-0. [DOI] [PubMed] [Google Scholar]
- Clark ME, Kraft TW. Measuring rodent electroretinograms to assess retinal function. Methods Mol Biol. 2012;884:265–276. doi: 10.1007/978-1-61779-848-1_19. [DOI] [PubMed] [Google Scholar]
- Colwell CS. NMDA-evoked calcium transients and currents in the suprachiasmatic nucleus: gating by the circadian system. Eur J Neurosci. 2001;13:1420–1428. doi: 10.1046/j.0953-816x.2001.01517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS. Linking neural activity and molecular oscillations in the SCN. Nat Rev Neurosci. 2011;12:553–569. doi: 10.1038/nrn3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Menaker M. NMDA as well as non-NMDA receptor antagonists can prevent the phase-shifting effects of light on the circadian system of the golden hamster. J Biol Rhythms. 1992;7:125–136. doi: 10.1177/074873049200700204. [DOI] [PubMed] [Google Scholar]
- Czeisler CA. The effect of light on the human circadian pacemaker. Ciba Found Symp. 1995;183:254–290. doi: 10.1002/9780470514597.ch14. discussion 290–302. [DOI] [PubMed] [Google Scholar]
- Dauchy RT, Xiang S, Mao L, Brimer S, Wren MA, Yuan L, Anbalagan M, Hauch A, Frasch T, Rowan BG, Blask DE, Hill SM. Circadian and melatonin disruption by exposure to light at night drives intrinsic resistance to tamoxifen therapy in breast cancer. Cancer Res. 2014;74:4099–4110. doi: 10.1158/0008-5472.CAN-13-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Aubrecht TG, Melendez-Fernandez OH, Weil ZM, Nelson RJ. Dim light at night disrupts molecular circadian rhythms and increases body weight. J Biol Rhythms. 2013;28:262–271. doi: 10.1177/0748730413493862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Nelson RJ. The effects of light at night on circadian clocks and metabolism. Endocr Rev. 2014;35:648–670. doi: 10.1210/er.2013-1051. [DOI] [PubMed] [Google Scholar]
- Golombek DA, Rosenstein RE. Physiology of circadian entrainment. Physiol Rev. 2010;90:1063–1102. doi: 10.1152/physrev.00009.2009. [DOI] [PubMed] [Google Scholar]
- Hannibal J. Neurotransmitters of the retino-hypothalamic tract. Cell Tissue Res. 2002;309:73–88. doi: 10.1007/s00441-002-0574-3. [DOI] [PubMed] [Google Scholar]
- Iitaka C, Miyazaki K, Akaike T, Ishida N. A role for glycogen synthase kinase-3beta in the mammalian circadian clock. J Biol Chem. 2005;280:29397–29402. doi: 10.1074/jbc.M503526200. [DOI] [PubMed] [Google Scholar]
- Iwahana E, Akiyama M, Miyakawa K, Uchida A, Kasahara J, Fukunaga K, Hamada T, Shibata S. Effect of lithium on the circadian rhythms of locomotor activity and glycogen synthase kinase-3 protein expression in the mouse suprachiasmatic nuclei. Eur J Neurosci. 2004;19:2281–2287. doi: 10.1111/j.0953-816X.2004.03322.x. [DOI] [PubMed] [Google Scholar]
- Iwahana E, Karatsoreos I, Shibata S, Silver R. Gonadectomy reveals sex differences in circadian rhythms and suprachiasmatic nucleus androgen receptors in mice. Horm Behav. 2008;53:422–430. doi: 10.1016/j.yhbeh.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JR, Tackenberg MC, McMahon DG. Manipulating circadian clock neuron firing rate resets molecular circadian rhythms and behavior. Nat Neurosci. 2015;18:373–375. doi: 10.1038/nn.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jud C, Schmutz I, Hampp G, Oster H, Albrecht U. A guideline for analyzing circadian wheel-running behavior in rodents under different lighting conditions. Biol Proced Online. 2005;7:101–116. doi: 10.1251/bpo109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatsoreos IN, Yan L, LeSauter J, Silver R. Phenotype matters: identification of light-responsive cells in the mouse suprachiasmatic nucleus. J Neurosci. 2004;24:68–75. doi: 10.1523/JNEUROSCI.1666-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman SJ, Silver R, Le Sauter J, Bult-Ito A, McMahon DG. Phase resetting light pulses induce Per1 and persistent spike activity in a subpopulation of biological clock neurons. J Neurosci. 2003;23:1441–1450. doi: 10.1523/JNEUROSCI.23-04-01441.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurabayashi N, Hirota T, Sakai M, Sanada K, Fukada Y. DYRK1A and glycogen synthase kinase 3beta, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol Cell Biol. 2010;30:1757–1768. doi: 10.1128/MCB.01047-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie J, Hebert M, Beaulieu JM. Glycogen synthase kinase-3 overexpression replicates electroretinogram anomalies of offspring at high genetic risk for schizophrenia and bipolar disorder. Biol Psychiatry. 2014;76:93–100. doi: 10.1016/j.biopsych.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Liou SY, Shibata S, Iwasaki K, Ueki S. Optic nerve stimulation-induced increase of release of 3H-glutamate and 3H-aspartate but not 3H-GABA from the suprachiasmatic nucleus in slices of rat hypothalamus. Brain Res Bull. 1986;16:527–531. doi: 10.1016/0361-9230(86)90182-6. [DOI] [PubMed] [Google Scholar]
- Lokshin M, LeSauter J, Silver R. Selective Distribution of Retinal Input to Mouse SCN Revealed in Analysis of Sagittal Sections. J Biol Rhythms. 2015;30:251–257. doi: 10.1177/0748730415584058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin LP, Shivers KY, Blanchard JH, Muscat L. Complex organization of mouse and rat suprachiasmatic nucleus. Neuroscience. 2006;137:1285–1297. doi: 10.1016/j.neuroscience.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Nakaya M, Sanada K, Fukada Y. Spatial and temporal regulation of mitogen-activated protein kinase phosphorylation in the mouse suprachiasmatic nucleus. Biochem Biophys Res Commun. 2003;305:494–501. doi: 10.1016/s0006-291x(03)00791-5. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- Paul JR, DeWoskin D, McMeekin LJ, Cowell RM, Forger DB, Gamble KL. Regulation of persistent sodium currents by glycogen synthase kinase 3 encodes daily rhythms of neuronal excitability. Nat Commun. 2016;7:13470. doi: 10.1038/ncomms13470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul JR, Johnson RL, Jope RS, Gamble KL. Disruption of circadian rhythmicity and suprachiasmatic action potential frequency in a mouse model with constitutive activation of glycogen synthase kinase 3. Neuroscience. 2012;226:1–9. doi: 10.1016/j.neuroscience.2012.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS One. 2010;5:e8561. doi: 10.1371/journal.pone.0008561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Liou SY, Ueki S. Influence of excitatory amino acid receptor antagonists and of baclofen on synaptic transmission in the optic nerve to the suprachiasmatic nucleus in slices of rat hypothalamus. Neuropharmacology. 1986;25:403–409. doi: 10.1016/0028-3908(86)90235-2. [DOI] [PubMed] [Google Scholar]
- Tischkau SA, Gallman EA, Buchanan GF, Gillette MU. Differential cAMP gating of glutamatergic signaling regulates long-term state changes in the suprachiasmatic circadian clock. J Neurosci. 2000;20:7830–7837. doi: 10.1523/JNEUROSCI.20-20-07830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkau SA, Mitchell JW, Tyan SH, Buchanan GF, Gillette MU. Ca2+/cAMP response element-binding protein (CREB)-dependent activation of Per1 is required for light-induced signaling in the suprachiasmatic nucleus circadian clock. J Biol Chem. 2003;278:718–723. doi: 10.1074/jbc.M209241200. [DOI] [PubMed] [Google Scholar]
- Viswanathan S, Frishman LJ, Robson JG, Harwerth RS, Smith EL., 3rd The photopic negative response of the macaque electroretinogram: reduction by experimental glaucoma. Invest Ophthalmol Vis Sci. 1999;40:1124–1136. [PubMed] [Google Scholar]