

Abstract
Restriction site‐associated DNA sequencing (RAD‐seq) was used to illuminate the genetic relationships among Eriobotrya species. The raw data were filtered, and 221 million clean reads were used for further analysis. A total of 1,983,332 SNPs were obtained from 23 Eriobotrya species and two relative genera. We obtained similar results by neighbor‐joining and maximum likelihood phylogenetic trees. All Eriobotrya plants grouped together into a big clade, and two out‐groups clustered together into a single or separate clade. Chinese and Vietnam accessions were distributed throughout the dendrogram. There was nonsignificant correlation between genotype and geographical distance. However, clustering results were correlated with leaf size to some extent. The Eriobotrya species could be divided into following three groups based on leaf size and phylogenetic analysis: group A and group B comprised of small leaves with <10 cm length except E. stipularis (16.76 cm), and group C can be further divided into two subgroups, which contained medium‐size leaves with a leaf length ranged from 10 to 20 cm and a leaf length bigger than 20 cm.
Keywords: cluster analysis, Eriobotrya genus, phylogenetic relationship, RAD‐seq, SNP
1. Introduction
High‐throughput sequencing technologies have revolutionized the genome research in recent years. The field of population genomics is rapidly expanding, and studies are now possible on unprecedented scales even in nonmodel organisms. Restriction site‐associated DNA (RAD‐tag) sequencing can simultaneously detect and genotype thousands of genome‐wide SNPs (Baird et al., 2008; Willing et al., 2011). It is one of the reduced representation methods that sampled a shared set of sites across the genome in many individuals or populations, making population‐scale sequencing possible at a fraction of the cost of whole genome sequencing (Davey et al., 2011). RAD‐Seq is suitable for fine‐scale linkage mapping (Scaglione et al., 2015; Wang, Fang, Xin, Wang, & Li, 2012), population genetics (Hohenlohe et al., 2011; Andersen et al. 2012), phylogenetics, and phylogeography (Cruaud et al., 2014; Rubin, Ree, & Moreau, 2012; Takahashi & Moreno, 2015; Valdisser et al., 2016). RAD‐Seq has also been used to generate large SNP datasets for many plants (Torres‐Martínez & Emery, 2016; Wang, Jin, Zhang, Shen, & Lin, 2015).
The genus Eriobotrya Lindl. Rosaceae, subfamily Maloideae, originated in China (Lin & Hu, 2000), and also found in Southeast Asian countries, such as Vietnam, Laos, and Burma. Previous studies showed that there are more than 30 species (varieties or forms) belong to Eriobotrya genus. Our research group have collected and conserved 25 species in loquat germplasm resources at South China Agricultural University. Yang, Liu, and Lin (2009) collected Eriobotrya germplasm from China and evaluated 18 accessions including 14 species and four varieties by using AFLP markers. ITS (Yang, Lin, & Hu, 2011) and ADH gene (Yang, Li, & Zhang, 2012) were used to analyze the phylogenetic relationships in Eriobotrya genus. These studies have got some common results, for example, Eriobotrya plants were grouped together and separated from other groups; similar species (varieties or forms) always clustered together; E. japonica, E. prinoides, and E. malipoensis were grouped together into the same clade, which showed a close relationship among these species. In addition, E. seguinii and E. henryi are very different species from other Eriobotrya species; they always clustered into the same clade. These previous studies revealed that species with the same morphological characteristics grouped together, while later these species clustered with other species. However, due to the lack of experimental materials and the limited polymorphic sites generated by molecular marker, ITS and ADH sequence, the available information is insufficient to evaluate the overall relationships in Eriobotrya genus. Especially, the relationships between the native species of Southeast Asian countries and the species originated in China are not yet clear. In this study, RAD‐seq is used to illuminate the genetic relationships between Eriobotrya genus, which will provide information on the origin and evolutionary history of different species in this genus, and enable us for the efficient utilization of germplasm resources and better future breeding strategies.
2. Materials and methods
2.1. Plant materials and DNA isolation
Twenty‐five accessions from 23 Eriobotrya species and two relative genera were collected from China, Vietnam, Burma, and Laos and used in this study (Table 1). All the samples were planted at the Loquat Germplasm Center, College of Horticulture, South China Agricultural University, P. R. China. DNA was extracted from young leaves by using a modified cetyltrime thylammonium bromide (CTAB) method as described by Doyle and Doyle (1990) with minor modifications (Liu et al., 2005). After quality assessment, DNA concentrations were adjusted to 100 ng/μl for RAD‐seq library preparation.
Table 1.
Scientific name, origin, and leaf length of the Eriobotrya accessions evaluated in the study
| Code | Scientific name | Origin location | The average leaf length(cm)a |
|---|---|---|---|
| A1 | Raphiolepis indica Lindl. | Huadu, Guangdong, China | 5.28 ± 0.52 |
| A2 | Photinia serrulata Lindl. | Kunming, Yunnan, China | 6.64 ± 0.68 |
| A3 | E. angustissima Hook. | Dalat, Vietnam, | 9.85 ± 0.24bc |
| A4 | E. stipularis Craib | Dalat, Vietnam, | 16.76 ± 0.47bc |
| A5 | E. seguinii Card. | Baishe Guangxi, China | 4.45 ± 0.93c |
| A6 | E. henryi Nakai | Chengjiang Yunnan, China | 9.27 ± 0.56bc |
| A7 | E. kwangsiensis Chun. | Xiangzhou Guangxi, China | 13.93 ± 1.23bc |
| A8 | E. prinoides var. laotica Vidal | Thong Hai Hin, Laos, | 11.06 ± 1.13bc |
| A9 | E. bengalensis f. angustifolia Vidal | Kunming Yunnan, China | 11.73 ± 0.16bc |
| A10 | E. deflexa f. koshunensis Nakai | Taiwan, China | 15.17 ± 2.64bc |
| A11 | E. fragrans Champ. | Ruyuan Guangdong, China | 16.24 ± 2.14bc |
| A12 | E. prinoides Rehd. and Wils. | Shiping Yunnan, China | 12.99 ± 1.03bc |
| A13 | E. deflexa f. buisanensis Nakai | Taiwan, China | 13.39 ± 0.21bc |
| A14 | E. cavaleriei Rehd. | Lianzhou Guangdong, China | 16.84 ± 1.69bc |
| A15 | Unknow speices | Jianfengling Hainan, China | 16.56 ± 1.95bc |
| A16 | E. × daduheensis H.Z.Zhang ex W.B.Liao, et al. | HanyuanSichuan, China | 14.96 ± 2.03bc |
| A17 | E. bengalensis f. Hook. | Lushui Yunnan, China | 13.36 ± 1.23bc |
| A18 | E. deflexa Nakai | Taiwan, China | 15.93 ± 1.78bc |
| A19 | E. petiolata Hook. | Pyi Oo Lwi, Burma, | 18.42 ± 1.66bc |
| A20 | E. salwinensis Hand‐Mazz | Pianma Yunnan, China | 17.21 ± 3.97bc |
| A21 | E. serrate Vidal | Jinhong Yunnan, China | 22.76 ± 0.30ab |
| A22 | E. japonica Lindl. | Yangshan Mountain Guangdong, China | 22.41 ± 2.51ab |
| A23 | E. elliptica var. petelotii Vidal | Lào Cai, Vietnam, | 33.49 ± 1.35a |
| A24 | E. elliptica Lindl. | Shiping Yunnan, China | 34.65 ± 1.10a |
| A25 | E. malipoensis Kuan | Malipo Yunnan, China | 35.78 ± 0.25a |
The data of leaf length represent 2‐year field investigations. Any two means not sharing a letter in common differ significantly at p ≤ .01.
2.2. The investigation of leaf length
Thirty mature leaves (from five to 10 individuals) were sampled to measure the leaf length. The leaf investigation was carried out for consecutive years. Significant difference analysis (SPSS) was performed at 0.01 level.
2.3. RAD‐seq library preparation
RAD‐seq library was prepared by using 5 units of NsiI and MseI (NEB, USA) to digest 1,000 ng genomic DNA per sample at 37°C for 2 hr in a 50 μl reaction volume and then inactivate enzyme at 80°C for 20 min (Zhang et al., 2012). The ligation reaction was performed with 8 μl of 0.1 μmol/L modified Solexa P1 Adaptor and 1 μl of 10 μmol/L Solexa P2 Adaptor (Illumina, USA), along with 30 μl of the digested DNA sample, 5 μl of 10 mmol/L ATP (Promega, USA), 10× NEB Buffer 3, 1.25 μl concentrated T4 DNA ligase (400 U/μl) (NEB, USA), and 2.75 μl H2O at room temperature for overnight. P1 and P2 adaptor sequences were as follows: P1 top: 5′‐GATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTxxxxxTGCA‐3′ (xxxx indicates barcode), P1 bottom: 5′‐yyyyAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATC‐3′ (yyyy indicates reverse complement of barcode); P2 top: 5′‐TAGATCGGAAGAGCACACGTCTGAACTCCAGTCACCTTGTAATCAGAACAA‐3′, P2 bottom:5′‐CAAGCAGAAGACGGCATACGAGATTACAAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC‐3′. After ligation, each DNA sample was heat inactivated at 65°C for 20 min and then purified using the QIA quick PCR purification kit (Qiagen, Germany). In order to enrich the adapter‐modified fragments, purified product was amplified with 25 μl Phusion Master Mix (NEB, USA), 2 μl of 10 μmol/L modified Solexa amplification primer mix (Illumina, USA), and add H2O to 50 μl. Phusion PCR preceded following product guidelines (NEB, USA) for 18 cycles. PCR products were electrophoresed on a 1% agarose gel (Sigma, USA), and DNA fragments between 200 and 500 bp were isolated using a Min Elute Gel Extraction kit (Qiagen, Germany) and diluted to 10 μmol/L for Illumina HiSeq2000 sequencing using single‐end sequence.
2.4. Quality filtering and SNP calling
Low‐quality reads (Q score < 20) and reads with contamination were filtered out; reads were trimmed to 84 nucleotides to remove flanking barcode sequences. All reads were pooled and used for a de novo assembly and SNP calling in ustacks (STACKS pipeline, Catchen, Amores, Hohenlohe, Cresko, & Postlethwait, 2011). We set a minimum stack size of 5 reads (‐m) and maximum distance between stacks (‐M) within a locus as 2. Population snps were filtered reserving more than half of the samples have snp information. The Illumina data set has been deposited in NCBI sequence read archive (SRA) under accession number PRJNA342569.
Neighbor‐joining and maximum likelihood phylogenetic trees were constructed by Treebest software, and bootstrap replicates were set to 1,000.
3. Results
3.1. The investigation of leaf length
The leaf length of Eriobotrya plants was ranged from 4.45 cm (E. seguinii.) to 35.78 cm (E. malipoensis) (Table 1). We found three groups: (1) Three species were found with <10 cm leaf length, including, E. seguinii., E. henryi, and E. angustissima, but only E. seguinii showed significant difference from other species; (2) A group of 15 species having leaf lengths between 10 and 20 cm was found, and there was nonsignificant difference among these species; (3) Five species exhibited >20 cm leaf length and grouped together. E. ellipticavar E. petelotii, E. elliptica, and E. malipoensis were found to be significantly different from the rest of species at the 0.01 significance level.
3.2. RAD‐tag sequencing and SNPs calling
We got 221 million clean reads by using Illumina HiSeq2000, after removing low‐quality reads (Q score < 20), and ambiguous reads with incorrect barcodes. The sequencing quality scores of 20 (Q20), which represent an error rate of 1 in 100, with a corresponding call accuracy of 99%, of all samples were more than 97.6%, indicating that the sequencing quality was good. Of these high‐quality reads, the highest reads (37.71 million reads) were detected in E. bengalensis f. angustifolia, and the lowest reads (1.96 million reads) were found in E. japonica, with an average read number of 8.84 million per accession.
We obtained a total of 1,983,332 SNPs, among them, 1,720,528 and 262,804 SNPs were homozygous and heterozygous, respectively (Table 2). The average number of detected SNPs was 79,333 per accession. The highest number of SNPs (123,089) was detected in E. bengalensis, while the lowest number of SNPs (47,351) was detected in Raphiolepis indica.
Table 2.
The SNPs number and information by RAD‐seq
| Codea | Clean reads number (M) | Q20 (%) | Homo SNPs | Hete SNPs | Total SNPs |
|---|---|---|---|---|---|
| A1 | 7.20 | 98.37 | 40,776 | 6,575 | 47,351 |
| A2 | 4.87 | 98.40 | 43,511 | 4,127 | 47,638 |
| A3 | 4.94 | 98.26 | 58,397 | 9,344 | 67,741 |
| A4 | 12.74 | 97.65 | 58,639 | 6,734 | 65,373 |
| A5 | 10.54 | 98.21 | 59,671 | 9,566 | 69,237 |
| A6 | 9.06 | 98.22 | 57,222 | 8,189 | 65,411 |
| A7 | 2.01 | 98.38 | 71,969 | 5,137 | 77,106 |
| A8 | 12.74 | 97.65 | 81,180 | 8,516 | 89,696 |
| A9 | 3.24 | 98.36 | 85,107 | 18,937 | 104,044 |
| A10 | 37.71 | 98.26 | 49,892 | 4,268 | 54,160 |
| A11 | 8.08 | 98.11 | 82,754 | 11,621 | 94,375 |
| A12 | 5.41 | 98.40 | 77,979 | 6,368 | 84,347 |
| A13 | 10.28 | 98.57 | 57,312 | 7,967 | 65,279 |
| A14 | 12.17 | 98.41 | 85,147 | 12,128 | 97,275 |
| A15 | 10.20 | 98.28 | 78,542 | 9,318 | 87,860 |
| A16 | 9.23 | 98.45 | 82,320 | 24,291 | 106,611 |
| A17 | 5.97 | 98.25 | 95,229 | 27,860 | 123,089 |
| A18 | 16.79 | 98.47 | 56,032 | 6,548 | 62,580 |
| A19 | 5.92 | 98.20 | 77,332 | 21,374 | 98,706 |
| A20 | 4.94 | 98.20 | 50,168 | 7,369 | 57,537 |
| A21 | 4.56 | 98.37 | 78,693 | 12,468 | 91,161 |
| A22 | 1.96 | 98.35 | 64,893 | 6,755 | 71,738 |
| A23 | 7.58 | 98.23 | 81,989 | 11,729 | 93,718 |
| A24 | 3.32 | 98.58 | 78,058 | 10,091 | 88,149 |
| A25 | 2.75 | 98.42 | 67,626 | 5,524 | 73,150 |
| Average | 8.84 | 98.26 | 68,821 | 10,512 | 79,333 |
| Total | 220.97 | 1,720,528 | 262,804 | 1,983,332 |
Please see Table 1 for scientific names of accessions.
3.3. Phylogenetic relationship revealed by RAD‐seq
Although two different approaches were used to construct the phylogenetic tree, similar results were obtained by both methods. All Eriobotrya plants grouped together and formed a bigger clade. In this clade, E. seguinii and E. henryi were always grouped together before clustering with other species. The same situation was found between E. stipularis and E. angustissima. The rest of 19 Eriobotrya plants grouped together. The major difference between two phylogenetic trees was the clustering of out‐groups. In NJ tree, two out‐groups, Rhaphiolepis indica and Photinia serrulata, grouped together and formed a clade, while they formed a separate clade in MJ tree. Interestingly, out‐groups were clearly separated from Eriobotrya plants.
The phylogenetic analysis divided 23 Eriobotrya plants into three major groups, A, B, and C, which were correlated with the size of leaves. Group A consisted of two species from China and both of them have the smallest leaves in Eriobotrya genus. Group B included two species from Vietnam and both have small, long, and narrow leaves. Group C was comprised of most of the Chinese species (16 of 19), and other three species were from Vietnam, Laos, and Burma. Group C could be divided into two subgroups, one subgroup included five species, and they have the biggest leaves. In contrast, other 14 species comprised the other subgroup, and they all have medium‐size leaves (Figure 1).
Figure 1.
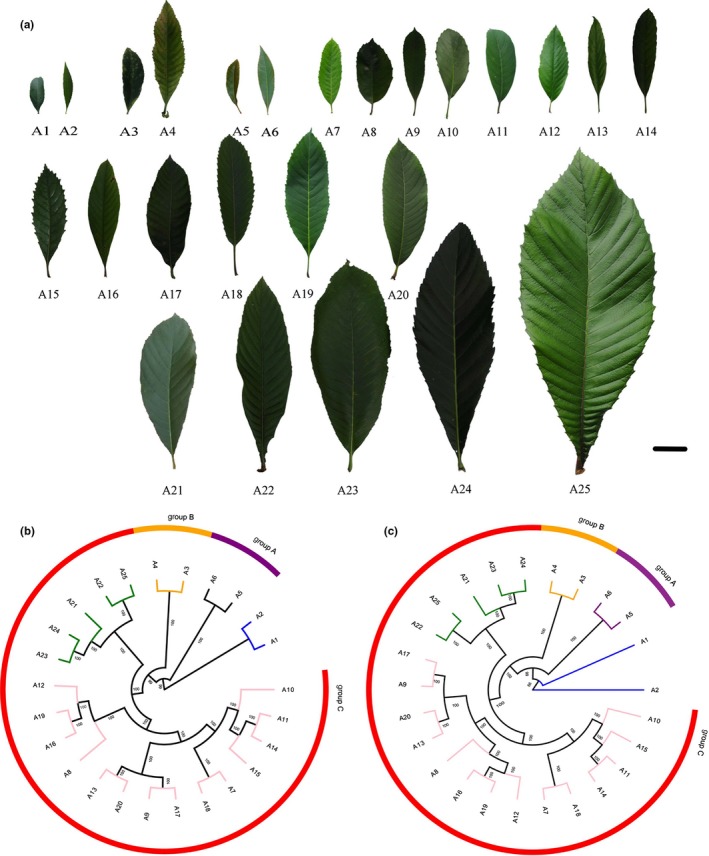
Leaf size and phylogenetic trees of Eriobotrya species and two relative genera. (a) The leaf size of 23 Eriobotrya species and two relative genera. Bar: 2 cm. (b,c) are neighbor‐joining and maximum likelihood phylogenetic trees of 23 Eriobotrya species and two relative genera by RAD‐seq. Node support is given as the maximum parsimony bootstrap value. Group C consists of two subgroups and marked as green and pink, respectively
Although we detected variations in the phylogenetic analysis, some results were consistent with the previous studies (Yang, Li, Liu, & Lin, 2009; Yang, Liu, et al., 2009; Yang et al., 2011, 2012). For example, the accessions belonging to the same species were classified into the same cluster, such as E. bengalensis and E. bengalensis f. angustifolia. Notably, E. japonica and E. malipoensis were always grouped together before clustering with other species. The same situation was found between E. seguinii and E. henryi and between E. fragrans and E. cavaleriei. However, E. defleax, E. deflexa f. buisanensis and E. defleax var. koshunensis belong to the same species, but clustered into different groups.
4. Discussion
Next‐generation sequencing technologies have facilitated the study of organisms on a genome‐wide scale. RAD‐seq allows sampling sequence information at reduced complexity across a target genome using the Illumina platform. Paired‐end RAD‐seq provides a large number of informative genetic markers in reference as well as nonreference organisms (Willing et al., 2011). In the present study, we detected 1,983,332 polymorphic SNPs through RAD‐seq technology, which was much higher than AFLP (282 polymorphic locus; Yang, Liu, et al., 2009) and RAPD (232 polymorphic bands; Yang, Li, et al., 2009).
There are more than 30 species in Eriobotrya genus, which are largely distributed in China and Southeast Asian countries. In order to analyze the genetic relationships between these species, a phylogeny was constructed using NJ and ML approach. Here, the 23 Eriobotrya plants were clustered into three major groups (A, B, and C). The results showed that the Chinese and Vietnam accessions were distributed throughout the dendrogram. We did not find a correlation between genotype and geographical distance However, it is worthwhile to mention that the clustering of accessions was highly correlated with the size of leaves, and the Eriobotrya plants were divided into three groups (A, B, and C). Cluster analysis showed that E. seguinii and E. henryi belong to the same group (group A), which was in agreement with the morphological analysis of these plants. These two species have the smallest leaves, which were quite different from other species. The average leaf length was 9.27 cm for E. henryi and 4.45 cm for E. seguinii. E. angustissima and E. stipular were clustered into another group (B), and the average leaf length was 9.85 cm for E. angustissima and 16.76 cm for E. stipularis. Most of the species (3 of 4) in group A and B have the smaller leaves (<10 cm) than other groups, except E. stipularis, which had leaves larger than 10 cm.
Group C was divided into two subgroups. Among these 19 species, E. japonica, E. malipoensis, E. serrate, E. elliptica, and E. elliptica var. petelotii were clustered together. All these species had larger leaves, >20 cm, especially E. malipoensis, which had leaves up to 35.78 cm. The rest of the 14 species had medium‐sized leaves (i.e., the average leaf length was ranged from 10 to 20 cm) and they clustered together. Previous studies carried out the classification of Eriobotrya plants according to the presence or absence of trichromes on adaxial surface of leaves (Yu, 1974) and flowering time (Zhang, 1996; ). The presence or absence of trichromes on adaxial surface of leaves can be the dichotomous characters or as one of the morphological characters of loquat but cannot be the criteria for the classification of Eriobotrya plants. However, the flowering time can be one of the main criteria for the classification of Eriobotrya plants, but this was performed with the little knowledge about Eriobotrya plants. Recently, the flowering time of some loquat species exhibited abundant variations under different geographical conditions (Lin & Liu, 2016). For example, spring is the flowering time of E. deflexa Nakai f. koshunensis (originated Taiwan), while the flowering appeared during autumn/winter season when introduced to Guangzhou, China. Therefore, the presence or absence of trichromes on adaxial surface of leaves and flowering time are not suitable criteria for the classification of the Eriobotrya plants. By RAD sequencing, the analysis results showed that the Eriobotrya plants may be classified according to the leaves size, with the combination of other characters. Here, Eriobotrya plants were divided into three categories, small leaves, medium leaves, and large leaves. These results were in accordance with the preliminary classification proposed by Yang and Lin (2007).
It has been universally recognized that common loquat (E. japonica) is native to China, and most of the Eriobotrya species are distributed in China. However, some Southeast Asian countries are also the distribution centers of Eriobotrya species. Our study clearly showed that two varieties native to Southeast Asian countries, E. elliptica var. petelottii and E. prioides var. laotica, were classified as Chinese species E. elliptica and E. prinoides, respectively. The results demonstrated that these two varieties may have close genetic relationship with Chinese species; however, whether these plants are derived from the Eriobotrya in China is still uncertain.
5. Conclusion
This study revealed the genetic relationships among Eriobotrya species by restriction site‐associated DNA sequencing (RAD‐seq). A total of 1,983,332 SNPs were obtained from 23 Eriobotrya species and two relative genera. We obtained similar results by neighbor‐joining and maximum likelihood phylogenetic trees. Our results are reliable, all Eriobotrya plants grouped together into a big clade, and two out‐groups clustered together into a single or separate clade. Chinese and Vietnam accessions were distributed throughout the dendrogram. The clustering results were correlated with leaf size, and the Eriobotrya species could be divided into three groups based on leaf size.
Conflict of interest
None declared.
Acknowledgments
This research was partially supported by Guangzhou Science and Technology Program key projects (201504010028).
Yang X, Najafabadi SK, Shahid MQ, et al. Genetic relationships among Eriobotrya species revealed by genome‐wide RAD sequence data. Ecol Evol. 2017;7:2861–2867. https://doi.org/10.1002/ece3.2902
References
- Andersen, E. C. , Gerke, J. R. , Shapiro, J. A. , Crissman, J. R. , Ghosh, R. , Bloom, J. S. , … Kruglyak, L. (2012). Chromosome‐scale selective sweeps shape caenorhabditis elegans genomic diversity. Nature Genetics, 44, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, N. A. , Etter, P. D. , Atwood, T. S. , Currey, M. C. , Shiver, A. L. , Lewis, Z. A. , … Johnson, E. A. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One, 3(10), e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. M. , Amores, A. , Hohenlohe, P. , Cresko, W. , & Postlethwait, J. H. (2011). Stacks:Building and genotyping loci de novo from short‐read sequences. G3, 1, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruaud, A. , Gautier, M. , Galan, M. , Foucaud, J. , Saune, L. , Genson, G. , … Rasplus, J. (2014). Empirical assessment of RAD sequencing for interspecific phylogeny. Molecular Biology and Evolution, 31, 1272–1274. [DOI] [PubMed] [Google Scholar]
- Davey, J. W. , Hohenlohe, P. A. , Etter, P. D. , Boone, J. Q. , Catchen, J. M. , & Blaxter, M. T. (2011). Genome‐wide genetic marker discovery and genotyping using next‐generation sequencing. Nature Reviews Genetics, 12, 499–510. [DOI] [PubMed] [Google Scholar]
- Doyle, J. J. , & Doyle, J. L. (1990). Isolation of plant DNA from fresh tissue. Focus, 12, 13–15. [Google Scholar]
- Hohenlohe, P. A. , Amish, S. K. , Catchen, J. M. , Allendorf, F. W. , Luikrt, G. (2011). Next‐generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and west slope cutthroat trout. Molecular Ecology Resources, 11, 117–122. [DOI] [PubMed] [Google Scholar]
- Lin, S. Q. , & Hu, Y. L. (2000). The species Eriobotrya Lindl. and the name for Eriobotrya hookeriana Decne in China. Journal of Fruit Science, 17, 300–304. [Google Scholar]
- Lin, S. Q. , & Liu, Y. X. (2016). Collection of illustration for Eriobotrya plants. Beijing: Science Press; pp. 25–27. [Google Scholar]
- Liu, Y. X. , Yang, X. H. , Lin, S. Q. , Hu, G. B. , & Liu, C. M. (2005). An improved procedure for nuclear DNA isolation from Eriobotrya plants and its application. Journal of Fruit Science, 22, 182–185. [Google Scholar]
- Rubin, B. E. , Ree, R. H. , & Moreau, C. S. (2012). Inferring phylogenies from RAD sequence data. PLoS One, 7, e33394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione, D. , Fornasiero, A. , Pinto, C. , Cattonaro, F. , Spadtto, A. , Infante, R. , … Testolin, R. (2015). A RAD‐based linkage map of kiwifruit (Actinidia chinensis Pl.) as a tool to improve the genome assembly and to scan the genomic region of the gender determinant for the marker‐assisted breeding. Tree Genet Genomies, 11, 115. [Google Scholar]
- Takahashi, T. , & Moreno, E. (2015). A RAD‐based phylogenetics for Orestias fishes from Lake Titicaca. Molecular Phylogenetics and Evolution, 93, 307–317. [DOI] [PubMed] [Google Scholar]
- Torres‐Martínez, L. , & Emery, N. C. (2016). Genome‐wide SNP discovery in the annual herb, Lasthenia fremontii (Asteraceae): Genetic resources for the conservation and restoration of a California vernal pool endemic. Conservation Genetics Resources, 8, 145–158. [Google Scholar]
- Valdisser, P. A. M. R. , Pappas, G. J. , de Menezes, I. P. P. , Müller, B. S. F. , Pereira, W. J. , Narciso, M. G. , … Vianello, R. P. (2016). SNP discovery in common bean by restriction‐associated DNA (RAD) sequencing for genetic diversity and population structure analysis. Molecular Genetics and Genomics, 291, 1277–1291. [DOI] [PubMed] [Google Scholar]
- Wang, N. , Fang, L. , Xin, H. , Wang, L. , & Li, S. (2012). Construction of a high‐density genetic map for grape using next generation restriction‐site associated DNA sequencing. BMC Plant Biology, 12(1), 148–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Jin, X. , Zhang, B. , Shen, C. , & Lin, Z. (2015). Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Research, 22, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing, E. M. , Hoffmann, M. , Klein, J. D. , Weigel, D. , & Dreyer, C. (2011). Paired‐end RAD‐seq for de novo assembly and marker design without available reference. Bioinformatics, 16, 2187–93. [DOI] [PubMed] [Google Scholar]
- Yang, X. H. , Li, P. , Liu, C. M. , & Lin, S. Q. (2009). Genetic diversity in Eriobotrya genus and its closely related plant species using RAPD markers. Journal of Fruit Science, 26(1), 55–59. [Google Scholar]
- Yang, X. H. , Li, P. , & Zhang, Z. K. (2012). A preliminarily phylogeny study of the Eriobotrya based on the nrDNA Adh sequences. Notulae botanicae Horti Agrobotanici Cluj‐Napoca, 40(2), 233–237. [Google Scholar]
- Yang, X. H. , & Lin, S. Q. (2007). New ideas on the classification of loquat. South China fruit, 36, 28–31. [Google Scholar]
- Yang, X. H. , Lin, S. Q. , & Hu, G. B. (2011). Preliminary study on ITS sequencing and characterization of Eriobotrya . Acta Horticulturae, 887, 85–88. [Google Scholar]
- Yang, X. H. , Liu, C. M. , & Lin, S. Q. (2009). Genetic relationships in Eriobotrya species as revealed by amplified fragment length polymorphism (AFLP) markers. Scientia Horticulturae, 122, 264–268. [Google Scholar]
- Yu, D. J. (1974). Delectis florae reipublicae popularis sinicae agendae academiae sinicae edita Flora Reipublicae Popularis Sinicae, Vol. 36 (pp. 260–275). Beijing: Science Press. [Google Scholar]
- Zhang, H. Z. (1996). Fruit flora of China Loquat (pp. 98–117). Beijing: China Forestry press. [Google Scholar]
- Zhang, Q. , Li, L. T. , Robert, V. B. , Liu, Y. L. , Yang, M. , Xu, L. M. , & Ray, M. (2012). Optimization of linkage mapping strategy and construction of a high‐density American lotus linkage map. BMC Genomics, 15(1), 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]