

Abstract
Oxidative and nitrosative stress are an umbrella term for pathophysiological processes that involve free radical generation during inflammation. In this review, the involvement of reactive oxygen and nitrogen species is evaluated during lung ischemia-reperfusion injury (LIRI) from a surgical point of view. The main biochemical and cellular mechanisms behind free radical generation are discussed, together with surgical procedures that may cause reperfusion injury. Finally, different therapeutic strategies are further explored. A literature search was performed, searching for “lung ischemia reperfusion injury”, “reperfusion injury”, “large animal model” and different search terms for each section: “surgery”, “treatment”, “cellular mechanism”, or “enzyme”. Although reperfusion injury is not an uncommon entity and there is a lot of evidence concerning myocardial ischemia-reperfusion injury, in the lung this phenomenon is less extensively described and studies in large animals are not easy to come by. With increasing number of patients on waiting lists for lung transplant, awareness for this entity should all but rise.
Keywords: Ischemia reperfusion injury, oxidative stress, nitrosative stress, reactive oxygen species (ROS), reactive nitrogen species (RNS)
Introduction
Definition of pulmonary ischemia-reperfusion injury
Pulmonary ischemia is defined as a temporary arrest of circulation and/or ventilation in at least one lung. Resupplying lung tissue with blood and air, i.e., reperfusion, causes an inflammatory reaction called acute lung injury (ALI) or lung ischemia-reperfusion injury (LIRI). In the setting of a lung transplantation, this process is defined as primary graft dysfunction (1). This might lead, in its most extreme form, to the possibly lethal adult respiratory distress syndrome (ARDS). Differential diagnosis between ALI and ARDS is made by evaluation of radiological criteria and oxygenation status of the patient in absence of any cardiac or chronic lung disease (Table 1) (2). Whereas ARDS is an acute pathology, LIRI can also compromise graft survival in the long run. There is now evidence that LIRI is a risk factor for development of the bronchiolitis obliterans syndrome, the most prevalent cause of long-term morbidity and mortality following lung transplantation (3).
Table 1. Criteria to differentiate between ALI and ARDS.
| ALI | ARDS | |
|---|---|---|
| Occurrence | Acute onset | Acute onset |
| Radiology | New patchy or homogeneous bilateral lung infiltrates | New patchy or homogeneous bilateral lung infiltrates |
| Cardiac status | PA wedge pressure <18 mmHg or absence of left atrial hypertension | PA wedge pressure <18 mmHg or absence of left atrial hypertension |
| Oxygenation status | PaO2:FiO2 <300 mmHg | PaO2:FiO2 <200 mmHg |
ALI, acute lung injury; ARDS, adult respiratory distress syndrome; PA, pulmonary artery; PaO2, alveolar air concentration; FiO2, oxygen concentration in inspired air.
Pulmonary IRI can be caused by either anoxic ischemia, in which the complete pulmonary hilum is occluded, or by ventilated ischemia, where the pulmonary artery (PA) is occluded selectively.
Anoxic ischemia typically occurs during cold ischemia storage of donor lungs, acute trauma surgery and in complete cardio-pulmonary bypass. It implies that PA, pulmonary vein, bronchus and bronchial artery are all temporarily occluded. The bronchial artery derives directly from the aorta and supplies the bronchial epithelium and connective tissue with oxygenated blood. It is occluded in anoxic ischemia by complete hilar clamping, but remains open during cardiac bypass. It is debated whether bronchial artery supply is sufficient to meet metabolic demand in the lung during cardiopulmonary bypass (4). Moreover, there is increasing evidence that failure to revascularize the bronchial artery following lung transplantation may lead to loss of airway microcirculation and increased risk of developing the bronchiolitis obliterans syndrome (5).
Ventilated ischemia occurs, for instance, during PA embolism. The alveolar tissue is still receiving oxygen supply because it directly uses inhaled oxygen, but as no erythrocytes pass through the pulmonary circulation, there is no normal gas exchange between the air in the alveoli and the blood in the pulmonary vessels (6).
With increasing numbers of lung transplantation procedures observed during the last years, pulmonary IRI is a clinically relevant phenomenon that warrants extensive study. Here we review the contribution of oxidative and nitrosative stress to LIRI pathogenesis, describe both intracellular and extracellular processes that lead to their production and provide information that is useful to a clinician.
Oxidative and nitrosative stress
Oxidative and nitrosative stress is caused by free radicals. These are molecules that contain an unpaired electron on their outer electron shell. Based on their central atom, they are defined as either reactive oxygen species [ROS, or as reactive nitrogen species (RNS)]. Oxidative and nitrosative stress is a natural byproduct of normal metabolism in the oxygen and nitrogen rich environment we live in (7). ROS and RNS are important in maintaining normal homeostasis as inter- and intracellular signal molecules, both in physiological as in pathophysiological conditions. For instance, phagocytic cells use ROS to destroy pathogens and combat infection, while nitric oxide (•NO) plays a pivotal role in flow-mediated vasodilation. However, ROS and RNS remain highly reactive molecules and can damage various cellular components when produced massively. Nucleic acids in DNA or RNA, amino acids in proteins and fatty lipids in the cell membrane are especially at risk. Because of their high reactivity, their half-life is only very short. A number of ROS and RNS with their respective half-life are summarized in Table 2.
Table 2. Common ROS and RNS in biological systems and their half-life.
| Name | Symbol | Half-life (s) |
|---|---|---|
| Hydroxyl radical | •OH | 1.10−6 |
| Superoxide anion | O2•− | 1 to 4.10−6 |
| Hydrogen peroxide | H2O2 | 1.10−3 |
| Peroxidized lipid | LOO• | 10.10−3 |
| Peroxynitrite | ONOO− | 1 |
| Nitric oxide | •NO | 2 to 6 |
| Ascorbyl radical | Asc• | 1.10−3 to days† |
†, dependent on temperature, presence of trace metals and oxygen concentration, ascorbyl radical is the most stabile biological radical that may remain detectable for days under low-temperature conditions. ROS, reactive oxygen species; RNS, reactive nitrogen species.
Intracellular sources of oxidative and nitrosative stress
NADPH oxidase
The NADPH oxidase enzyme complex (NOX) is a membrane-bound protein cluster that oxidizes its substrate nicotinamide adenine dinucleotide phosphate (NADPH) into NADP+, thereby transferring an electron from the cytoplasm into a phagocytic vacuole (8). In the lumen of the vacuole, also called phagosome, molecular oxygen is reduced by this electron into superoxide anion in massive quantities. This system is highly active in cells that are responsible for pathogen killing, such as neutrophils, macrophages and monocytes. The flip side of the medal is that ROS generated by activated NOX can aggravate certain pathologies, especially inflammatory diseases (9).
There are many NOX homologues which are not necessary involved in pathogen killing, but generate ROS for redox signaling purposes, to regulate cell growth and apoptosis (NOX2), angiogenesis (NOX1), kidney function (NOX3) and even thyroid hormone synthesis (DUOX2) (10). NOX4 is most predominant in smooth muscle cells in the PA and the airway epithelia where it serves as an oxygen sensor. In patients with idiopathic pulmonary hypertension, NOX4 induces vascular remodeling associated with this disease in response to chronic hypoxia (11).
Xanthine oxidase/dehydrogenase (XO/XDH) system
Xanthine oxidase/dehydrogenase (XO/XDH) is an enzyme that is pivotal in purine catabolism. It breaks down adenosine to hypoxanthine and xanthine with further degradation into uric acid, which can be excreted in urine. In physiological circumstances, the enzyme exists in its XDH form. It can be converted irreversibly by proteolysis or reversibly by sulfhydryl residue oxidation into XO (12). The main difference is that XDH mainly uses NAD+ as electron acceptor, whereas XO reduces oxygen to form superoxide anion, independent of NAD+. XO activity is enhanced by hypoxia and concentrations are increased in serum of patients with ARDS (13). During ischemia, XDH is increasingly converted into XO. Higher levels of cellular adenosine due to breakdown of ATP in ischemic cells therefore lead to an increase in substrate for XO, further inducing oxidative stress (14).
NO synthases (NOS)
•NO is an important intercellular messenger and neurotransmitter, responsible for destruction of pathogens, and flow-mediated vasodilation. It is generated through an enzymatic reaction in which arginine is converted into citrulline by NOS (15).
There are four major NOS isoforms: neuronal NOS (NOS I, nNOS) is primarily found in neuronal cells, inducible NOS (NOS II, iNOS) in cells of the immune system, endothelial NOS (NOS III, eNOS) in the endothelial lining of blood vessels, and mitochondrial NOS (mtNOS) in the inner mitochondrial membrane (16). iNOS is activated by an external trigger, as iNOS transcription is modulated by nuclear factor kappa bèta (NF-κB), and produces vast bursts of •NO when stimulated by pro-inflammatory cytokines (17). As iNOS induction mostly occurs in an environment with increased oxidative stress, the reaction of superoxide O2•− with •NO is also further propagated, leading to the production of peroxynitrite (ONOO−) which rapidly oxidizes fatty acids in the cell membrane, leads to cross-linking of proteins and other harmful reactions (18).
eNOS and nNOS are both constitutive types of NOS, generating a steady and constant amount of NO dependent on cofactors such as tetrahydrobiopterin (BH4), NADPH, calmodulin and flavins. There is evidence that an imbalance between these cofactors can change eNOS conformation, leading to both a functional and structural eNOS uncoupling (19). In absence of BH4, eNOS shifts from a dimeric into a monomeric state and catalyzes O2•- production instead of •NO, leading to increased oxidation of BH4 and a more oxidative-stress rich environment.
eNOS transcription appears to become upregulated under ischemic conditions, although the promoter of the enzymes mRNA sequence does not contain a specific HIF (hypoxia inducible factor) receptor. It is possible that an alteration in the redox balance, with higher concentrations oxidated NAD+ and NADP+ has greater influence on the eNOS promoter (20). The mechanisms described here are summarized in Figure 1.
Figure 1.

Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation in the endothelial cells by combined effects of eNOS uncoupling, xanthine dehydrogenase conversion and NOX induction.
Mitochondrial enzymatic chain disruption
Mitochondria are the energy suppliers of the cell. These organelles generate an electron flow during the oxidative phosphorylation reaction for the production of high-energy phosphates through a proton gradient. In physiological conditions, O2•− is a small but unavoidable byproduct of this respiratory chain (21). Electrons leak from the different chain complexes and are readily accepted by molecular oxygen, leading to O2•− and H2O2 formation. Mitochondrial complex I is the entry point for electrons in the electron transport chain, where NADH is oxidized to NAD+. Under specific circumstances, such as an abnormally high electrochemical proton gradient during ischemia, the direction of the electron transport chain may reverse (22). This implies that succinate is oxidized at complex II, where NAD+ is subsequently reduced to NADH at complex I. This reverse electron transfer, where NADH oxidation is replaced by succinate oxidation, generates massive amounts of ROS and leads to opening of the mitochondrial permeability transition pore (mPTP) complex in the inner mitochondrial membrane. This process is called uncoupling of mitochondrial function, which results in mitochondrial swelling, membrane rupture and initiation of cell death mechanisms (23). Induction of mtNOS by calcium influx leads to increasing •NO concentrations, which inhibit the cytochrome C reductase complex (complex IV) and causes cytochrome C release, leading to cell death. •NO further reacts with O2•− to form ONOO− which inhibits all mitochondrial complexes (24). There is some evidence from small animal models that cyclosporine treatment might prevent assembly of the mPTP, although a larger meta-analysis failed to show any significant effect (25). Other mitochondrial enzymes, apart from the respiratory chain, contribute to mitochondrial ROS generation as well. For instance, the NADPH oxidase NOX2 isoform is responsible for the formation of mitochondrial ROS in human endothelial cells (26). Mitochondrial cytochrome P450 and P2C can generate O2•− when electrons used to reduce the central heme iron are transferred to molecular oxygen (27,28). This mechanism of mitochondrial chain disruption and processes leading to assembly of the mPTP complex are illustrated in Figure 2.
Figure 2.

Dashed lines depict the pathways leading to mitochondrial uncoupling, which take place when increased amounts of reactive oxygen species (ROS) are produced, causing the assembly of the mitochondrial permeability transition pore (mPTP) complex and subsequent cell death. Full lines express normal physiological pathways.
Disruption of iron and calcium metabolism
The lactic acidosis that is associated with ischemia induces iron dissociation from its carrier proteins, such as transferrin, ferritin and different cytochromes. This free iron may serve as a catalyst of hydroxyl (•OH) radical formation in what is called the Fenton and Haber-Weisz reaction. In these reactions, O2•− and H2O2 react with free ferrous iron (Fe2+) to form the detrimental hydroxyl radical. This is further illustrated in Figure 3.
Figure 3.
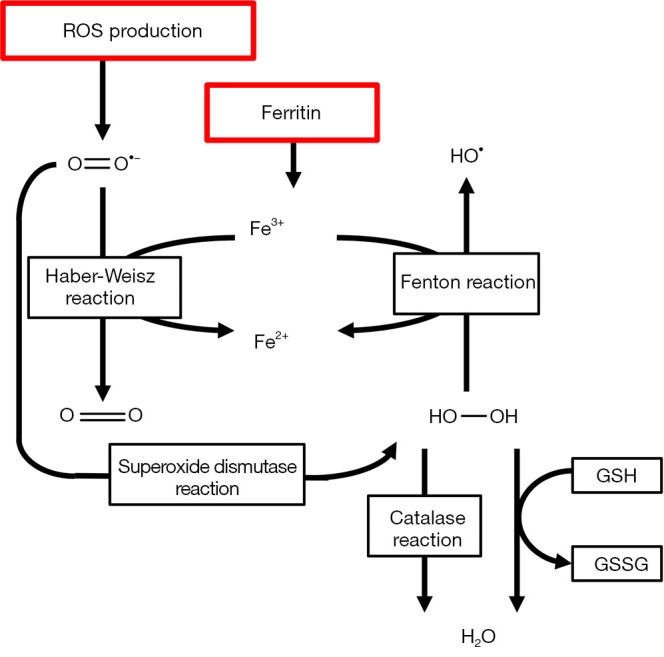
Biochemical reactions catalyzed by iron leading to increased oxidative stress formation and different enzyme systems responsible for neutralization of free radicals.
Iron also catalyzes tyrosine nitration by ONOO− (29). Iron is also responsible for glutathione (GSH) oxidation and may activate platelet aggregation in the microvasculature of the lung. Iron chelators such as deferoxamine have been proven to reduce acute pulmonary injury (30). Calcium is an intracellular messenger that is responsible for multiple biochemical pathways in the cell. Calcium stocks are maintained in the sarcoplasmatic or endoplasmic reticulum by calcium pumps and released through calcium channels. During IRI, lipid peroxidation alters Ca++ homeostasis leading to calcium overload and activation of Ca++ dependent proteases (31). Furthermore, because of ATP depletion, Ca++ pumps fail to work adequately which reduces active efflux and decreases reuptake by endoplasmatic and sarcoplasmatic reticulum. This leads to a netto increased calcium release from intracellular stocks during reperfusion. In the heart, this calcium overload may result in severe arrhythmias (32) that complicate post-operative recovery. Increases in cytosolic Ca++ also lead to failed Ca++ homeostasis in mitochondria, causing a massive influx of Ca++ ions and protons in the mitochondrial matrix. This ultimately leads to cell death by necrosis and further impairment of ATP production (33).
Elevated Ca++ levels also activate cytokines such as interleukin (IL)-1β that lead to increased iNOS transcription. Ca++ further increases eNOS activity through calmodulin activation (34) and pushes the XDH/XO balance further to XO, potentiating the effect of ROS on cellular damage (35).
Cellular mechanisms leading to pulmonary oxidative and nitrosative stress
Macrophages & macrophage polarization
Macrophages activate the innate immune response by producing cytokines and chemokines and assist in cleaning up cellular debris by phagocytosis. Currently, two phenotypes of macrophages are recognized: a Th-1 driven, pro-inflammatory M1 phenotype and a pro-angiogenic, debris-scavenging M2 phenotype. HIF, a transcription factor of which breakdown is inhibited by low oxygen concentrations can shift macrophages either toward the M1 or M2 phenotype (36). Apparently, HIF1-alpha shifts macrophages towards M1 polarization while HIF2-alpha causes a shift towards the M2 phenotype. Also, increased concentrations of ROS and iron overload (37) determine macrophages toward a M1 profile (38). M1 macrophages themselves produce ROS through NADPH oxidase and •NO through iNOS activation. Therefore, M1 macrophages aggravate the reaction to ischemia and are a major determinant of ROS and RNS production in response to hypoxia. M2 macrophages produce lower amounts of inflammatory cytokines and higher amounts of the anti-inflammatory cytokine IL-10. Higher expression of arginase I decreases the availability of the eNOS and iNOS substrate arginine in these cells, resulting in a decrease in inducible •NO production (39). Detection of pharmacological shifting of macrophages toward a M2-profile with flowcytometry or qPCR may therefore be an interesting approach to decrease the extent of IRI.
Neutrophils
Neutrophilic granulocytes expand and potentiate the immunologic reaction which is initiated by macrophages and activated endothelial cells (40). Neutrophils are responsible for the so-called respiratory burst that is part of an inflammatory reaction. This is the rapid and sudden release of ROS such as O2−, which is synthesized by NOX2, and OH, generated by the combined effect of the reaction catalyzed by superoxide dismutase and the Fenton reaction (41) as shown in Figure 3. In the neutrophil, these ROS are combined with Cl− by the enzyme myeloperoxidase to form highly toxic hypochlorous acid (HOCl) (42). Neutrophils are recruited during IRI by of macrophages toward a M2-profile with flowcytometry or qPCR may therefore be an interesting approach to decrease the extent of IRI.
Neutrophils
Neutrophilic granulocytes expand and potentiate the immunologic reaction which is initiated by macrophages and activated endothelial cells (40). Neutrophils are responsible for the so-called respiratory burst that is part of an inflammatory reaction. This is the rapid and sudden release of ROS such as O2•−, which is synthesized by NOX2, and •OH, generated by the combined effect of the reaction catalyzed by superoxide dismutase and the Fenton reaction (41) as shown in Figure 3. In the neutrophil, these ROS are combined with Cl− by the enzyme myeloperoxidase to form highly toxic hypochlorous acid (HOCl) (42). Neutrophils are recruited during IRI by increased expression of ICAM-1, CD-18 and P-selectin on the endothelial cells of the lung microvasculature. These proteins promote diapedesis and neutrophil activation. Activated macrophages and bronchial epithelia produce the neutrophil-chemoattractant IL-8 (43). All these signal molecules combined increase neutrophil diapedesis into the alveolar spaces, producing additional ROS, platelet activating factor (PAF) and tumor necrosis factor alpha (TNF-α). The importance of neutrophil activity on cellular damage during pulmonary ischemia and reperfusion is illustrated by the fact that increased IL-8 levels in donor lungs correlate with an elevated risk of early graft failure, development of ARDS and even increased mortality after transplantation (44).
Bronchial epithelia
Alveolar type II cells appear to be quite susceptible to the effect of ROS and RNS. These cells are responsible for the release of inflammatory mediators and cytokines such as lymphocyte activation by IL-1β, and increased acute phase protein synthesis via IL-6 (45). Type II cells also contribute to TNF-α production in response to oxidative and nitrosative stress and neutrophil recruitment through synthesis of IL-8. Transcription factors such as HIF1-alpha and NF-κβ further propagate the inflammatory responses that are activated in the bronchi and alveoli. Furthermore, there is increasing evidence that iNOS, NOX2 and NOX4 are also active in bronchial cells (46), which additionally marks the bronchial epithelium as a potential source of ROS and RNS during IRI.
Surgical procedures leading to pulmonary ischemia-reperfusion injury
Lung transplantation
Every transplant procedure, by definition, incorporates a period of ischemia, followed by reperfusion. Many methods have been developed to reduce reperfusion injury and keep the ischemia time as short as possible. Still, lung transplantation is a surgical procedure with many per- and postoperative risks. Death still occurs in 20% of patients undergoing lung transplantation procedure, most often following primary graft failure (47). Primary graft failure is a more specific term to describe the most severe and possibly lethal form of IRI, requiring more than 72 hours of continuous mechanical ventilation, development of radiologic shadowing on chest X-ray, pulmonary edema and occasionally extracorporeal membrane oxygenation. Moreover, severe IRI may also cause problems in the long term, as it is a determinant of graft dysfunction and may lead to the bronchiolitis obliterans syndrome after several years (48). Thorough assessment of donor organs (Table 3) also selects those lungs that can sustain a longer period of ischemia with minimal reperfusion injury. New techniques of donor lung preservation and organ management are being perfected, such as ex-vivo lung perfusion (EVLP) (49,50). EVLP allows the evaluation of donor lungs before transplantation, keeps the lung under physiological conditions for extended time, and maintains an active cellular metabolism. It is also possible to recondition less performing pulmonary grafts by reduction of pulmonary edema, remove toxic waste products such as cytokines, ROS and RNS, and potentially damaging graft inflammatory cells such as activated M1 macrophages and neutrophils (51).
Table 3. Pulmonary transplantation donor selection criteria (49).
| <55 years old |
| <20 pack-years tobacco use |
| No infiltrations on chest radiograph |
| No thoracic trauma |
| No earlier cardiopulmonary procedures |
| PaO2 /FiO2 ratio >300 mmHg; PEEP 5 cmH2O |
| Compatible blood group and HLA match |
| Compatible size of donor lung to recipient thoracic cavity |
| Absence of primary lung disease or active pulmonary infection |
| No aspiration pneumonia, patient not septic |
| Bronchoscopy specimens: non-purulent, with negative gram stain |
Cardiopulmonary bypass
During coronary artery bypass graft (CABG) procedure, circulation is temporarily derived from the right atrium directly into the aorta, with a perfusion pump in between. The pulmonary circulation is thus completely bypassed and the delivery of oxygenated blood to the lung parenchyma occurs solely through the bronchial arteries. If this bronchial circulation is not sufficient enough to provide enough blood flow, some degree of ischemia occurs (52). The effects of anesthesia, hypothermia, medication and peroperative transfusion may aggravate injury (53). Fortunately, ARDS only occurs in two to three percent of cardiac surgical patients. There is some controversy as to cardiopulmonary bypass is the main cause of lung dysfunction in patients undergoing CABG, as off-pump CABG produces a similar incidence of postoperative impaired pulmonary function (54).
Pulmonary thromboembolectomy
There are three different mechanisms that contribute to pulmonary ischemia-reperfusion injury in patients undergoing surgical treatment for PA embolism.
First, a thrombo-embolic event of the PA or any of its branches renders the lung segments that are perfused by that part of the PA ischemic. Second, cardiopulmonary bypass is part of the standard surgical treatment protocol, with the same possible influences on reperfusion injury as mentioned in “Cardiopulmonary bypass” section. Third, revascularization results in the diffusion of toxic mediators such as ROS and RNS in both the pulmonary and systemic circulation (55). The lung tissue itself may be protected from ischemic injury during pulmonary thrombo-embolic events when bronchial circulation remains intact. However, lack of sufficient oxygenation during embolism creates hypoxia in the entire circulation and subsequent reperfusion injury when oxygenation is adequately restored. An important risk during and after thromboembolectomy is massive endobronchial hemorrhage caused by reperfusion injury during re-establishment of the pulmonary blood flow (56-58).
Isolated lung perfusion
Isolated lung perfusion is an experimental technique for treatment of pulmonary metastases (59). Trials are currently running, investigating the applicability of this technique to treat pulmonary metastases from primary tumors such as sarcoma and colorectal carcinoma. High doses of chemotherapy, which would be toxic when administered systemically, are pumped through the pulmonary circulation for thirty minutes with the aid of a bypass pump. During the procedure, the treated lung is isolated from the circulation and oxygenation depends solely on the contralateral lung. The treated lung is thus completely isolated from the body’s circulation, as the bronchial artery and vein are temporarily occluded during the procedure, as well. This implies a period of 30 minutes of complete pulmonary ischemia in the treated lung. When the isolated lung perfusion procedure is terminated, reperfusion injury may occur. Recent findings show that this reperfusion injury can be prevented by addition of carbon dioxide to and removal of molecular oxygen from the perfusate. The type of pump used for the procedure may also contribute to the extent of injury (60). The addition to the perfusate of ROS and RNS scavengers or stabilizing enzymatic cofactors, such as BH4 to keep eNOS coupled, is still the subject of ongoing research.
Ischemia-reperfusion injury of another organ leading to lung injury
The lung is also prone to reperfusion injury originating from ischemia elsewhere in the body. There is an abundance of research models of intestinal (61), hepatic (62), renal (63) and hindlimb ischemia (64) that cause lung injury through oxidative and nitrosative stress mechanisms available in literature (65). It appears that the extension of lung injury is more dependent on the volume of tissue that is rendered ischemic, rather than on the duration of ischemia. The activation of intestinal, hepatic and renal inflammatory cascades induce oxidative and nitrosative stress that contributes to multiple organ failure, including the lung. Liver reperfusion injury, for instance, induces a downregulation of eNOS and increase of iNOS expression in lung tissue and an increase in •OH in the peripheral circulation (66). Clinical examples of procedures that cause peripheral ischemia and reperfusion injury are restoration of circulation after acute ischemic colitis due to superior mesenteric artery thrombosis, partial hepatectomy and liver or kidney transplantation.
Possible therapeutic strategies in tackling pulmonary ischemia-reperfusion injury
Dietary restrictions
Caloric restriction is not quite the same as malnutrition. It implies a dietary reduction of calory intake without a shortage of essential nutrients. It is thought to extend the lifespan of many species and is therefore considered an “anti-aging” strategy, as it may prevent senescent changes in the cell (67). A small, constant stressor for a longer period of time may “prime” the organism to better cope with larger stressors in the future.
Caloric restriction reduces production of oxidative and nitrosative stress, although the exact mechanisms still remain to be elucidated (68). Furthermore, it is not certain when, how and how long dietary modifications need to be initiated in order to prevent damage from ischemic events. In rats, caloric restriction for 2 weeks already showed a significant reduction in myocardial ischemia-reperfusion injury (67). Rats subjected to caloric restriction for 12 months showed an earlier reduction in superoxide dismutase and catalase expression during reperfusion compared to normally fed animals, indicating an earlier return to baseline oxidative stress levels (69).
Antioxidants & enzymatic cofactors
Antioxidants are molecules that terminate or alternate the chain reactions that are induced by free radicals and therefore may attenuate the effect of oxidative and nitrosative stress. Some antioxidants are enzymatic, such as superoxide dismutase and catalase, while others are small molecules like vitamins and GSH (70). Hydrophilic antioxidants, such as vitamin C, react with free radicals in the cell cytosol while lipid-soluble antioxidants, such as vitamin E, inhibit lipid peroxidation of the cell membrane. Iron binding enzymes such as transferrin and ferritin inhibit radical generation through the Fenton reaction (Figure 3) (14). Selenium, zinc and manganese ions are often mentioned as “antioxidant ions” but they do not have any anti-oxidative properties themselves. These ions are essential cofactors to cytosolic and mitochondrial SOD, catalase and the GSH reductase system. Other cofactors include arginine, BH4 and flavins which are essential to physiological NOS function and prohibit NOS uncoupling (15).
The “antioxidant paradox”
An important sidenote to the possible beneficial effect of vitamin and cofactor supplementation is that although free radicals appear to be involved in many inflammatory pathologies, extensive doses of dietary antioxidants do not seem to offer much preventative or therapeutic benefit. This observation is called the antioxidant paradox (71). The antioxidant barriers of normal cellular physiology are complex and interweaved, and free radicals may offer beneficial effects themselves, for example as a neurotransmitter. Free radicals are part of the communication system between cells, in a process dubbed redox-signaling (72). In vitro studies cannot be extrapolated immediately to in vivo situations. Flavonoids are plant-derived polyphenols that show radical-absorbing properties in vitro (73) but offer little benefit in vivo (74).
Medication
Pharmacological strategies to tackle damage caused by free radicals can consist of prevention, by inhibiting radical-generating systems, or treatment on a post-hoc basis, by attenuating the possibly detrimental effects of free radicals.
N-acetyl-cysteine (NAC)
NAC is a molecular precursor of reduced GSH, a thiol with intracellular and extracellular antioxidant capacity (70). GSH concentration in the lung rises significantly following intravenous NAC administration. GSH is the main cofactor of gluthathione peroxidase, an enzyme that prevents formation of hydroxyl radicals by catalyzing the reduction of hydrogen peroxide into water. GSH scavenges O2•−, too. In pigs undergoing isolated ventilation and reperfusion, NAC aerosol administration immediately before and after EVLP prevented warm ischemic damage compared to animals without treatment (75).
NO donors
•NO is a versatile molecule; it is the crucial molecular messenger in flow-mediated vasodilation, and an inhibitor of neutrophil activation, vascular adhesion and platelet aggregation. Nitroglycerin is a NO donor that has been frequently subscribed to patients suffering from transient myocardial ischemia. Adding nitroglycerin to lung preservation solutions results in better pulmonary function after transplantation (76), and continuous intravenous administration during the entire reperfusion period markedly improved gas exchange and reduced neutrophil activity in non-heart beating donor procedures in pigs (77). Nitroglycerin infusion is also more cost-effective than continuous •NO inhalation and immediately targets the endothelial cell, without the necessity for specialized devices.
Corticosteroids
Patients undergoing surgery that includes an episode of pulmonary ischemia-reperfusion injury, especially cardiopulmonary bypass, benefit from pre-treatment with methylprednisolone before the procedure. Glucocorticoids act on a transcriptional level and increase production of anti-inflammatory cytokines such as IL-10 while lowering levels of pro-inflammatory molecules such as Il-6, IL-8, and TNF-α. They also decrease injury by inhibiting neutrophil CD11b expression and neutrophil complement-induced chemotaxis, thereby decreasing neutrophil activation (53).
Enzymatic inhibitors
Specific inhibitors of the enzymes that were mentioned earlier may offer a pharmacological therapeutic strategy to combat reperfusion injury. NADPH oxidase inhibitors such as apocynin which have shown little effect on the rest of the immune system are useful candidates, because they avoid any “collateral damage”. The exact mechanism of action of apocynin is yet unknown, but there is evidence that it attenuates LIRI in sheep by lowering oxidative status and altering thromboxane metabolism while not interfering with leukocyte migration (78). Allopurinol is a well-known inhibitor of the XO/XDH system that is used to treat patients suffering from gout. Allopurinol does not seem to work solely by inhibition of XO/XDH and offer only partial protection from IRI. Moreover, it has more effect during anoxia and reoxygenation than when only ventilated ischemia is induced (6). Inhibiting iNOS may be another strategy, which reduces vascular dysfunction, oxidative stress and pulmonary edema following lung transplantation (79). Reducing iNOS mRNA expression with nicotinamide may equally result in a decrease in proinflammatory cytokines and a reduction in observed hydroxyl radical concentration (80).
Adenosine A2A receptor agonists
Adenosine has cytoprotective properties and attenuates organ ischemia-reperfusion injury. All subtype classes of this receptor (A1, A2A, A2B and A3) are expressed in lung tissue and work through adenyl cyclase or phospholipase C activation. Some studies attribute an anti-inflammatory role to A1, A2A and A3 (81) with reduction in oxidative stress by decreasing neutrophil-mediated O2•− production (82). A2B activation, on the contrary, may have a pro-inflammatory effect in the lung (83). Specific agonists of these adenosine receptors appear to offer more benefit than adenosine alone, probably because these agonists selectively activate the A1, A2A and A3 receptor without additional effect on the A2B receptor (84).
Surfactant therapy
The alveolar surfactant system reduces the surface tension that needs to be overcome before the alveoli can inflate completely. Surfactant therapy can compose of administration of animal-derived surfactants (typically a mixture of phospholipids and proteins) or synthetic surfactants which consist of synthetic lipids and recombinant peptides. Surfactant can be administered before ischemia (85), and before (86) or during (87) reperfusion. Inhalation of synthetic surfactant was shown to reduce LIRI, increase arterial oxygen tension and lower levels of oxidative stress after lung transplantation in pigs (88).
Perfusion liquids
EVLP is an attractive strategy to improve graft survival and even improve the quality of donor lungs that would previously be rejected. It might be an answer to meet the high need and long waiting lists with the current low availability of donor organs. EVLP reduces LIRI and may attenuate the oxidative stress response that is generated during brain death and ICU-related complications when compared to standard cold-ischemic storage. There is evidence that the perfusion solution developed by Steen et al. may provide antioxidant protection by inhibiting NOX2 activation in the donor lung during lung perfusion (89).
Anesthesia gases
NO
Adding NO to ventilation gases during 20 minutes of EVLP in pigs resulted in significantly better transplant oxygenation, reduced vascular resistance and reduced airway pressure. Ventilation with •NO also decreased neutrophil sequestration in the donor lung (90). In another porcine model, •NO inhalation in the first 4 hours of reperfusion after lung transplantation improved gas exchange, pulmonary vascular resistance and tissue oxygenation while lowering pulmonary neutrophil counts after 24 hours of reperfusion. These results demonstrate that even a short period of •NO administration in the immediate post-operative period might provide additional protective effects after lung surgery (91).
Noble gases
Anesthesias with noble gases such as xenon and argon have organoprotective effects and may provide a useful addition to the standard transplantation anesthesia protocol. These gases can also be added to the gas mixture used during EVLP (92). Attenuation of organ damage following ischemia and reperfusion has been attributed to an increase in apoptosis, an increase in HIF1-alpha or modification of the Akt-pathway (93). However, not all evidence supports this possibly beneficial role (92). There is still a lack of evidence concerning the pathways that are involved in the protective effect of these noble gases, and timing of administration and correct dosage are still uncertain. Moreover, these gases are quite expensive and have a low cost-benefit ratio.
Fluranes
Fluranes are nowadays commonly used for anesthesia during surgical procedures. Anesthetic preconditioning with isoflurane or sevoflurane may stabilize mitochondria and reduce the amount of oxidative stress that is generated during ischemia and reperfusion in the cell. It means that organ function is preserved and ischemic damage is reduced by exposing the target organ to volatile anesthetics before the ischemic insult. These anesthesia gases inhibit the electron transport chain at complex I and III (94), which causes tiny amounts of ROS to be formed. This small preconditioning impulse triggers different organoprotective intracellular pathways, such as tyrosine kinase phosphorylation and activation of mitochondrial KATP channels. Extra potassium ions stabilize membrane depolarization and decrease the risk of calcium overload in the mitochondria. Administration of sevoflurane appears to reduce oxidative stress but maintain NO production by eNOS and iNOS in a porcine autotransplant model (95).
Ischemic preconditioning
A brief period or a train of short periods of ischemia before the actual ischemic insult can significantly reduce ischemic damage and provide additional protection from additional hypoxic episodes. This was first demonstrated in myocardial tissue, but appears to be valid in lung tissue as well (96). There is currently no clear data available concerning the applicability of pulmonary ischemic preconditioning in the setting of a lung transplantation. Practical issues arise as to how pulmonary ischemia should be induced, and patients are often too weak to undergo such short periods of hypoxia. Many different cellular pathways have been suggested, such as increased transcription of heat shock proteins, triggering of a small burst of ROS as a “training” for further insult, or opening of KATP channels that stabilize the mitochondrial membrane. A more feasible approach is to apply remote ischemic preconditioning, for instance temporary occlusion of circulation in an arm or a leg with a blood pressure cuff. This technique has been applied successfully in patients undergoing a lobectomy and reduced oxidative stress levels in the peripheral circulation, decreased ALI incidence and resulted in earlier patient discharge (97).
Conclusions
Oxidative and nitrosative stress are umbrella terms for the highly reactive molecules called ROS and RNS that are generated during inflammatory processes. Free radicals are an important subset of these molecules. This review focuses on the involvement of free radicals in LIRI, or LIRI. Many surgical procedures can lead to LIRI. It is critical that clinicians are aware of the risks and have a notion of the pathophysiological mechanisms that lead towards it. It helps to provide adequate treatment and may even contribute to preventive strategies in the future.
Acknowledgements
Funding: This work was supported by a research grant (aspirant) awarded to JF Gielis by the Research Foundation Flanders—FWO (grant No. 11F6515N).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Christie JD, Kotloff RM, Ahya VN, et al. The effect of primary graft dysfunction on survival after lung transplantation. Am J Respir Crit Care Med 2005;171:1312-6. 10.1164/rccm.200409-1243OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994;149:818-24. 10.1164/ajrccm.149.3.7509706 [DOI] [PubMed] [Google Scholar]
- 3.Fiser SM, Tribble CG, Long SM, et al. Ischemia-reperfusion injury after lung transplantation increases risk of late bronchiolitis obliterans syndrome. Ann Thorac Surg 2002;73:1041-7; discussion 1047-8. 10.1016/S0003-4975(01)03606-2 [DOI] [PubMed] [Google Scholar]
- 4.Schlensak C, Doenst T, Preusser S, et al. Cardiopulmonary bypass reduction of bronchial blood flow: a potential mechanism for lung injury in a neonatal pig model. J Thorac Cardiovasc Surg 2002;123:1199-205. 10.1067/mtc.2002.121977 [DOI] [PubMed] [Google Scholar]
- 5.Nicolls MR, Zamora MR. Bronchial blood supply after lung transplantation without bronchial artery revascularization. Curr Opin Organ Transplant 2010;15:563-7. 10.1097/MOT.0b013e32833deca9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao G, al-Mehdi AB, Fisher AB. Anoxia-reoxygenation versus ischemia in isolated rat lungs. Am J Physiol 1997;273:L1112-7. [DOI] [PubMed] [Google Scholar]
- 7.Laskin DL. Oxidative/nitrosative stress and disease. Boston, Mass.: Blackwell Pub., 2010:167. [Google Scholar]
- 8.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313. 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- 9.Segal AW. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int J Biochem Cell Biol 2008;40:604-18. 10.1016/j.biocel.2007.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petry A, Djordjevic T, Weitnauer M, et al. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal 2006;8:1473-84. 10.1089/ars.2006.8.1473 [DOI] [PubMed] [Google Scholar]
- 11.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 2002;59:1428-59. 10.1007/s00018-002-8520-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waud WR, Rajagopalan KV. The mechanism of conversion of rat liver xanthine dehydrogenase from an NAD+-dependent form (type D) to an O2-dependent form (type O). Arch Biochem Biophys 1976;172:365-79. 10.1016/0003-9861(76)90088-6 [DOI] [PubMed] [Google Scholar]
- 13.Terada LS, Rubinstein JD, Lesnefsky EJ, et al. Existence and participation of xanthine oxidase in reperfusion injury of ischemic rabbit myocardium. Am J Physiol 1991;260:H805-10. [DOI] [PubMed] [Google Scholar]
- 14.den Hengst WA, Gielis JF, Lin JY, et al. Lung ischemia-reperfusion injury: a molecular and clinical view on a complex pathophysiological process. Am J Physiol Heart Circ Physiol 2010;299:H1283-99. 10.1152/ajpheart.00251.2010 [DOI] [PubMed] [Google Scholar]
- 15.Gielis JF, Lin JY, Wingler K, et al. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic Biol Med 2011;50:765-76. 10.1016/j.freeradbiomed.2010.12.018 [DOI] [PubMed] [Google Scholar]
- 16.Lacza Z, Snipes JA, Zhang J, et al. Mitochondrial nitric oxide synthase is not eNOS, nNOS or iNOS. Free Radic Biol Med 2003;35:1217-28. 10.1016/S0891-5849(03)00510-0 [DOI] [PubMed] [Google Scholar]
- 17.Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 1994;269:4705-8. [PubMed] [Google Scholar]
- 18.Gielis JF, Boulet GA, Briede JJ, et al. Longitudinal quantification of radical bursts during pulmonary ischaemia and reperfusion. Eur J Cardiothorac Surg 2015;48:622-9. 10.1093/ejcts/ezu518 [DOI] [PubMed] [Google Scholar]
- 19.Förstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 2006;113:1708-14. 10.1161/CIRCULATIONAHA.105.602532 [DOI] [PubMed] [Google Scholar]
- 20.Hoffmann A, Gloe T, Pohl U. Hypoxia-induced upregulation of eNOS gene expression is redox-sensitive: a comparison between hypoxia and inhibitors of cell metabolism. J Cell Physiol 2001;188:33-44. 10.1002/jcp.1092 [DOI] [PubMed] [Google Scholar]
- 21.Dröse S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145-69. 10.1007/978-1-4614-3573-0_6 [DOI] [PubMed] [Google Scholar]
- 22.Muntean DM, Sturza A, Danila MD, et al. The Role of Mitochondrial Reactive Oxygen Species in Cardiovascular Injury and Protective Strategies. Oxid Med Cell Longev 2016;2016:8254942. 10.1155/2016/8254942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: fixing a hole. Cardiovasc Res 2006;70:191-9. 10.1016/j.cardiores.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 24.Nowinski SM, Solmonson A, Rundhaug JE, et al. Mitochondrial uncoupling links lipid catabolism to Akt inhibition and resistance to tumorigenesis. Nat Commun 2015;6:8137. 10.1038/ncomms9137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song K, Wang S, Qi D. Effects of Cyclosporine on Reperfusion Injury in Patients: A Meta-Analysis of Randomized Controlled Trials. Oxid Med Cell Longev 2015;2015:287058. 10.1155/2015/287058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wosniak J, Jr, Santos CX, Kowaltowski AJ, et al. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal 2009;11:1265-78. 10.1089/ars.2009.2392 [DOI] [PubMed] [Google Scholar]
- 27.Fleming I. Cytochrome p450 and vascular homeostasis. Circ Res 2001;89:753-62. 10.1161/hh2101.099268 [DOI] [PubMed] [Google Scholar]
- 28.Lewis DF. Modelling human cytochrome P450-substrate interactions. Ernst Schering Res Found Workshop 2002(37):235-48. [DOI] [PubMed] [Google Scholar]
- 29.Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 1990;87:1620-4. 10.1073/pnas.87.4.1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ritter C, da Cunha AA, Echer IC, et al. Effects of N-acetylcysteine plus deferoxamine in lipopolysaccharide-induced acute lung injury in the rat. Crit Care Med 2006;34:471-7. 10.1097/01.CCM.0000199069.19193.89 [DOI] [PubMed] [Google Scholar]
- 31.Finkel T, Menazza S, Holmstrom KM, et al. The ins and outs of mitochondrial calcium. Circ Res 2015;116:1810-9. 10.1161/CIRCRESAHA.116.305484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talukder MA, Zweier JL, Periasamy M. Targeting calcium transport in ischaemic heart disease. Cardiovasc Res 2009;84:345-52. 10.1093/cvr/cvp264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 2006;34:232-7. 10.1042/BST0340232 [DOI] [PubMed] [Google Scholar]
- 34.Zhang B, Crankshaw W, Nesemeier R, et al. Calcium-mediated signaling and calmodulin-dependent kinase regulate hepatocyte-inducible nitric oxide synthase expression. J Surg Res 2015;193:795-801. 10.1016/j.jss.2014.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanduri J, Vaddi DR, Khan SA, et al. HIF-1alpha activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS One 2015;10:e0119762. 10.1371/journal.pone.0119762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004;104:2224-34. 10.1182/blood-2004-03-1109 [DOI] [PubMed] [Google Scholar]
- 37.Sindrilaru A, Peters T, Wieschalka S, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 2011;121:985-97. 10.1172/JCI44490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Choksi S, Chen K, et al. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res 2013;23:898-914. 10.1038/cr.2013.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlüter KD, Schulz R, Schreckenberg R. Arginase induction and activation during ischemia and reperfusion and functional consequences for the heart. Front Physiol 2015;6:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eppinger MJ, Jones ML, Deeb GM, et al. Pattern of injury and the role of neutrophils in reperfusion injury of rat lung. J Surg Res 1995;58:713-8. 10.1006/jsre.1995.1112 [DOI] [PubMed] [Google Scholar]
- 41.Babior BM. The respiratory burst of phagocytes. J Clin Invest 1984;73:599-601. 10.1172/JCI111249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Candeias LP, Patel KB, Stratford MR, et al. Free hydroxyl radicals are formed on reaction between the neutrophil-derived species superoxide anion and hypochlorous acid. FEBS Lett 1993;333:151-3. 10.1016/0014-5793(93)80394-A [DOI] [PubMed] [Google Scholar]
- 43.Nakamura H, Yoshimura K, Jaffe HA, et al. Interleukin-8 gene expression in human bronchial epithelial cells. J Biol Chem 1991;266:19611-7. [PubMed] [Google Scholar]
- 44.Fisher AJ, Donnelly SC, Hirani N, et al. Elevated levels of interleukin-8 in donor lungs is associated with early graft failure after lung transplantation. Am J Respir Crit Care Med 2001;163:259-65. 10.1164/ajrccm.163.1.2005093 [DOI] [PubMed] [Google Scholar]
- 45.Sharma AK, Fernandez LG, Awad AS, et al. Proinflammatory response of alveolar epithelial cells is enhanced by alveolar macrophage-produced TNF-alpha during pulmonary ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol 2007;293:L105-13. 10.1152/ajplung.00470.2006 [DOI] [PubMed] [Google Scholar]
- 46.Kobzik L, Bredt DS, Lowenstein CJ, et al. Nitric oxide synthase in human and rat lung: immunocytochemical and histochemical localization. Am J Respir Cell Mol Biol 1993;9:371-7. 10.1165/ajrcmb/9.4.371 [DOI] [PubMed] [Google Scholar]
- 47.Christie JD, Carby M, Bag R, et al. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part II: definition. A consensus statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2005;24:1454-9. 10.1016/j.healun.2004.11.049 [DOI] [PubMed] [Google Scholar]
- 48.Fiser SM, Kron IL, Long SM, et al. Influence of graft ischemic time on outcomes following lung transplantation. J Heart Lung Transplant 2001;20:1291-6. 10.1016/S1053-2498(01)00355-2 [DOI] [PubMed] [Google Scholar]
- 49.Van Raemdonck D, Neyrinck A, Verleden GM, et al. Lung donor selection and management. Proc Am Thorac Soc 2009;6:28-38. 10.1513/pats.200808-098GO [DOI] [PubMed] [Google Scholar]
- 50.Van Raemdonck D, Neyrinck A, Cypel M, et al. Ex-vivo lung perfusion. Transpl Int 2015;28:643-56. 10.1111/tri.12317 [DOI] [PubMed] [Google Scholar]
- 51.Sanchez PG, Mackowick KM, Kon ZN. Current state of ex-vivo lung perfusion. Curr Opin Organ Transplant 2016;21:258-66. 10.1097/MOT.0000000000000310 [DOI] [PubMed] [Google Scholar]
- 52.Young RW. Prevention of lung injury in cardiac surgery: a review. J Extra Corpor Technol 2014;46:130-41. [PMC free article] [PubMed] [Google Scholar]
- 53.Apostolakis E, Filos KS, Koletsis E, et al. Lung dysfunction following cardiopulmonary bypass. J Card Surg 2010;25:47-55. 10.1111/j.1540-8191.2009.00823.x [DOI] [PubMed] [Google Scholar]
- 54.Ng CS, Wan S, Yim AP, et al. Pulmonary dysfunction after cardiac surgery. Chest 2002;121:1269-77. 10.1378/chest.121.4.1269 [DOI] [PubMed] [Google Scholar]
- 55.Fard N, Saffari A, Emami G, et al. Acute respiratory distress syndrome induction by pulmonary ischemia-reperfusion injury in large animal models. J Surg Res 2014;189:274-84. 10.1016/j.jss.2014.02.034 [DOI] [PubMed] [Google Scholar]
- 56.Rice PL, Pifarre R, El-Etr A, et al. Management of endobronchial hemorrhage during cardiopulmonary bypass. J Thorac Cardiovasc Surg 1981;81:800-1. [PubMed] [Google Scholar]
- 57.Lyerly HK, Reves JG, Sabiston DC., Jr Management of primary sarcomas of the pulmonary artery and reperfusion intrabronchial hemorrhage. Surg Gynecol Obstet 1986;163:291-301. [PubMed] [Google Scholar]
- 58.Purut CM, Scott SM, Parham JV, et al. Intraoperative management of severe endobronchial hemorrhage. Ann Thorac Surg 1991;51:304-6; discussion 6-7. 10.1016/0003-4975(91)90808-4 [DOI] [PubMed] [Google Scholar]
- 59.Den Hengst WA, Van Putte BP, Hendriks JM, et al. Long-term survival of a phase I clinical trial of isolated lung perfusion with melphalan for resectable lung metastases. Eur J Cardiothorac Surg 2010;38:621-7. 10.1016/j.ejcts.2010.03.048 [DOI] [PubMed] [Google Scholar]
- 60.Reck Dos Santos P, Sakamoto J, Chen M, et al. Modified In Vivo Lung Perfusion for Local Chemotherapy: A Preclinical Study With Doxorubicin. Ann Thorac Surg 2016;101:2132-40. 10.1016/j.athoracsur.2015.12.043 [DOI] [PubMed] [Google Scholar]
- 61.Zhao B, Fei J, Chen Y, et al. Pharmacological preconditioning with vitamin C attenuates intestinal injury via the induction of heme oxygenase-1 after hemorrhagic shock in rats. PLoS One 2014;9:e99134. 10.1371/journal.pone.0099134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boeykens N, Ponsaerts P, Ysebaert D, et al. Biochemical Parameters for Longitudinal Monitoring of Liver Function in Rat Models of Partial Hepatectomy Following Liver Injury. PLoS One 2013;8:e66383. 10.1371/journal.pone.0066383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ysebaert DK, De Greef KE, De Beuf A, et al. T cells as mediators in renal ischemia/reperfusion injury. Kidney Int 2004;66:491-6. 10.1111/j.1523-1755.2004.761_4.x [DOI] [PubMed] [Google Scholar]
- 64.Chen LN, Yang XH, Nissen DH, et al. Dysregulated renin-angiotensin system contributes to acute lung injury caused by hind-limb ischemia-reperfusion in mice. Shock 2013;40:420-9. 10.1097/SHK.0b013e3182a6953e [DOI] [PubMed] [Google Scholar]
- 65.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008;295:L379-99. 10.1152/ajplung.00010.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin HI, Chou SJ, Wang D, et al. Reperfusion liver injury induces down-regulation of eNOS and up-regulation of iNOS in lung tissues. Transplant Proc 2006;38:2203-6. 10.1016/j.transproceed.2006.06.012 [DOI] [PubMed] [Google Scholar]
- 67.Shinmura K, Tamaki K, Ito K, et al. Indispensable role of endothelial nitric oxide synthase in caloric restriction-induced cardioprotection against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2015;308:H894-903. 10.1152/ajpheart.00333.2014 [DOI] [PubMed] [Google Scholar]
- 68.Melo DS, Costa-Pereira LV, Santos CS, et al. Severe Calorie Restriction Reduces Cardiometabolic Risk Factors and Protects Rat Hearts from Ischemia/Reperfusion Injury. Front Physiol 2016;7:106. 10.3389/fphys.2016.00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chandrasekar B, Nelson JF, Colston JT, et al. Calorie restriction attenuates inflammatory responses to myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2001;280:H2094-102. [DOI] [PubMed] [Google Scholar]
- 70.Inci I, Zhai W, Arni S, et al. N-acetylcysteine attenuates lung ischemia-reperfusion injury after lung transplantation. Ann Thorac Surg 2007;84:240-6; discussion 6. 10.1016/j.athoracsur.2007.03.082 [DOI] [PubMed] [Google Scholar]
- 71.Halliwell B. The antioxidant paradox: less paradoxical now? Br J Clin Pharmacol 2013;75:637-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Watson WH, Ritzenthaler JD, Roman J. Lung extracellular matrix and redox regulation. Redox Biol 2016;8:305-15. 10.1016/j.redox.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Halliwell B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch Biochem Biophys 2008;476:107-12. 10.1016/j.abb.2008.01.028 [DOI] [PubMed] [Google Scholar]
- 74.Halliwell B, Gutteridge JM. Free radicals in biology and medicine. USA: Oxford University Press, 2015. [Google Scholar]
- 75.Rega FR, Wuyts WA, Vanaudenaerde BM, et al. Nebulized N-acetyl cysteine protects the pulmonary graft inside the non-heart-beating donor. J Heart Lung Transplant 2005;24:1369-77. 10.1016/j.healun.2004.10.013 [DOI] [PubMed] [Google Scholar]
- 76.Kawashima M, Bando T, Nakamura T, et al. Cytoprotective effects of nitroglycerin in ischemia-reperfusion-induced lung injury. Am J Respir Crit Care Med 2000;161:935-43. 10.1164/ajrccm.161.3.9905003 [DOI] [PubMed] [Google Scholar]
- 77.Loehe F, Preissler G, Annecke T, et al. Continuous infusion of nitroglycerin improves pulmonary graft function of non-heart-beating donor lungs. Transplantation 2004;77:1803-8. 10.1097/01.TP.0000131155.81609.37 [DOI] [PubMed] [Google Scholar]
- 78.Dodd-O JM, Pearse DB. Effect of the NADPH oxidase inhibitor apocynin on ischemia-reperfusion lung injury. Am J Physiol Heart Circ Physiol 2000;279:H303-12. [DOI] [PubMed] [Google Scholar]
- 79.Wu JX, Zhu HW, Chen X, et al. Inducible nitric oxide synthase inhibition reverses pulmonary arterial dysfunction in lung transplantation. Inflamm Res 2014;63:609-18. 10.1007/s00011-014-0733-5 [DOI] [PubMed] [Google Scholar]
- 80.Su CF, Liu DD, Kao SJ, et al. Nicotinamide abrogates acute lung injury caused by ischaemia/reperfusion. Eur Respir J 2007;30:199-204. 10.1183/09031936.00025107 [DOI] [PubMed] [Google Scholar]
- 81.Gazoni LM, Laubach VE, Mulloy DP, et al. Additive protection against lung ischemia-reperfusion injury by adenosine A2A receptor activation before procurement and during reperfusion. J Thorac Cardiovasc Surg 2008;135:156-65. 10.1016/j.jtcvs.2007.08.041 [DOI] [PubMed] [Google Scholar]
- 82.van der Hoeven D, Wan TC, Auchampach JA. Activation of the A(3) adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Mol Pharmacol 2008;74:685-96. 10.1124/mol.108.048066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun CX, Zhong H, Mohsenin A, et al. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest 2006;116:2173-82. 10.1172/JCI27303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gazoni LM, Walters DM, Unger EB, et al. Activation of A1, A2A, or A3 adenosine receptors attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg 2010;140:440-6. 10.1016/j.jtcvs.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Novick RJ, MacDonald J, Veldhuizen RA, et al. Evaluation of surfactant treatment strategies after prolonged graft storage in lung transplantation. Am J Respir Crit Care Med 1996;154:98-104. 10.1164/ajrccm.154.1.8680706 [DOI] [PubMed] [Google Scholar]
- 86.Amital A, Shitrit D, Raviv Y, et al. The use of surfactant in lung transplantation. Transplantation 2008;86:1554-9. 10.1097/TP.0b013e31818a8418 [DOI] [PubMed] [Google Scholar]
- 87.Amital A, Shitrit D, Raviv Y, et al. Surfactant as salvage therapy in life threatening primary graft dysfunction in lung transplantation. European Journal of Cardio-Thoracic Surgery 2009;35:299-303. 10.1016/j.ejcts.2008.09.039 [DOI] [PubMed] [Google Scholar]
- 88.Sáenz A, Alvarez L, Santos M, et al. Beneficial effects of synthetic KL(4) surfactant in experimental lung transplantation. Eur Respir J 2011;37:925-32. 10.1183/09031936.00020810 [DOI] [PubMed] [Google Scholar]
- 89.Carnevale R, Biondi-Zoccai G, Peruzzi M, et al. New insights into the steen solution properties: breakthrough in antioxidant effects via NOX2 downregulation. Oxid Med Cell Longev 2014;2014:242180. 10.1155/2014/242180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aitchison JD, Orr HE, Flecknell PA, et al. Nitric oxide during perfusion improves posttransplantation function of non- heart-beating donor lungs. Transplantation 2003;75:1960-4. 10.1097/01.TP.0000067528.58552.34 [DOI] [PubMed] [Google Scholar]
- 91.Bacha EA, Herve P, Murakami S, et al. Lasting beneficial effect of short-term inhaled nitric oxide on graft function after lung transplantation. Paris-Sud University Lung Transplantation Group. J Thorac Cardiovasc Surg 1996;112:590-8. 10.1016/S0022-5223(96)70040-5 [DOI] [PubMed] [Google Scholar]
- 92.Martens A, Montoli M, Faggi G, et al. Argon and xenon ventilation during prolonged ex vivo lung perfusion. J Surg Res 2016;201:44-52. 10.1016/j.jss.2015.10.007 [DOI] [PubMed] [Google Scholar]
- 93.Rizvi M, Jawad N, Li Y, et al. Effect of noble gases on oxygen and glucose deprived injury in human tubular kidney cells. Exp Biol Med (Maywood) 2010;235:886-91. 10.1258/ebm.2010.009366 [DOI] [PubMed] [Google Scholar]
- 94.De Hert SG, Turani F, Mathur S, et al. Cardioprotection with volatile anesthetics: mechanisms and clinical implications. Anesth Analg 2005;100:1584-93. 10.1213/01.ANE.0000153483.61170.0C [DOI] [PubMed] [Google Scholar]
- 95.Casanova J, Simon C, Vara E, et al. Sevoflurane anesthetic preconditioning protects the lung endothelial glycocalyx from ischemia reperfusion injury in an experimental lung autotransplant model. J Anesth. 2016;30:755-62. 10.1007/s00540-016-2195-0 [DOI] [PubMed] [Google Scholar]
- 96.Gasparri RI, Jannis NC, Flameng WJ, et al. Ischemic preconditioning enhances donor lung preservation in the rabbit. Eur J Cardiothorac Surg 1999;16:639-46. 10.1016/S1010-7940(99)00335-8 [DOI] [PubMed] [Google Scholar]
- 97.Li C, Xu M, Wu Y, et al. Limb remote ischemic preconditioning attenuates lung injury after pulmonary resection under propofol-remifentanil anesthesia: a randomized controlled study. Anesthesiology 2014;121:249-59. 10.1097/ALN.0000000000000266 [DOI] [PubMed] [Google Scholar]