

Abstract
The tyrosine kinase encoded by the MET oncogene is activated by gene mutation or amplification in tumors, which in most instances maintain addiction, i.e., dependency, to MET activation. This makes MET an attractive candidate for targeted therapies. Here we show that, in 3/3 MET-addicted human gastric cancer cell lines, MET kinase inhibition resulted in a 3- to 4-fold increased expression of the antiapoptotic small heat-shock protein of 27 kDa (HSP27, HSPB1). HSP27 increase depended on the inhibition of the MEK/ERK pathway and on heat-shock factor 1 (HSF1) and hypoxia-inducible factor-1α (HIF-1α) regulation. Importantly, HSP27-silenced MET-addicted cells underwent 2- and 3-fold more apoptosis following MET inhibition in vitro and in vivo, respectively. Likewise, in human cancer cells susceptible to epidermal growth factor receptor (EGFR) inhibition, EGFR inhibitors induced HSP27 expression and were strengthened by HSP27 suppression. In control cell lines that were not affected by drugs targeting MET or EGFR, these drugs did not induce HSP27 increase. Therefore, in cancer therapies targeting the MET pathway, the induction of HSP27 might limit the efficacy of anti-MET agents. As HSP27 increase also impairs the effectiveness of EGFR inhibitors and is known to protect cells from chemotherapeutics, the induction of HSP27 by targeted agents might strongly affect the success of combination treatments.—Musiani, D., Konda, J. D., Pavan, S., Torchiaro, E., Sassi, F., Noghero, A., Erriquez, J., Perera, T., Olivero, M., Di Renzo, M. F. Heat-shock protein 27 (HSP27, HSPB1) is up-regulated by MET kinase inhibitors and confers resistance to MET-targeted therapy.
Keywords: hepatocyte growth factor receptor, EGFR
The MET receptor and its ligand hepatocyte growth factor (HGF) play important roles in oncogenesis and are thus among the most attractive candidates for targeted therapeutic intervention (2). Genetic amplification of the MET oncogene that encodes this receptor is present in gastric (3) and non-small-cell lung cancer; in the latter, MET is implicated in the resistance to inhibitors of the epidermal growth factor receptor (4). Activating MET mutations are found in hereditary and sporadic papillary renal cell carcinomas (5), in gastric carcinomas (6), in lymphnode metastases of head and neck squamous cell carcinomas (7), and in lung metastases of cancers of unknown origin (8). Besides cancer cells with mutations that cause dependence on MET sustained activity, i.e., MET addiction, many cancer cells are sensitive to MET inhibition because of the ability of MET to protect cells from death and to trigger cell motility and invasiveness (9). Therefore, an increasing number of trials are ongoing with inhibitors of either MET or its ligand HGF.
The members of the small heat-shock protein (sHSP) family are molecular chaperones that play roles in development, stress responses, and diseases, including cancer (10, 11). The sHSP of 27 kDa (HSP27) encoded by the HSPB1 gene is a molecular chaperone with antiaggregation property, as it participates in sequestering damaged proteins (12) and is involved in the proteasomal degradation of certain proteins under stress conditions (11). Besides its role in stressed cells, HSP27 plays crucial roles within the cell under unstressed conditions where it provides cytoskeletal structural stability (13). HSP27 also exerts an important antiapoptotic function by binding apoptotic proteins (14). HSP27 is regulated at both transcriptional and post-transcriptional levels. The synthesis of HSP27 can be induced not only by heat shock and other stress conditions but also by physiological stimuli such as those regulating differentiation (15). Clinically, HSP27 is highly expressed in many cancers, including breast (16), ovarian (17), prostate (18), and others (19), and is associated with aggressive tumor behavior, metastasis, poor prognosis, and resistance to chemotherapeutics. Moreover, HSP27 increases during the early phase of stem cell differentiation (15), and thus, it might play a role in sustaining cancer stem cell growth and survival. Hence, HSP27 might play important roles in cancer onset and progression and in its response to treatment. Here we show that the expression of HSP27 is up-regulated by MET inhibition through a pathway that depends on the mitogen-activated protein kinase MEK/ERK pathway and on heat-shock factor 1 (HSF1) and hypoxia-inducible factor-1α (HIF-1α). More important, we demonstrate that HSP27 up-regulation limits the effectiveness of MET-targeted therapies and that targeting HSP27 sensitizes cells to MET inhibitors.
MATERIALS AND METHODS
Cell lines and reagents
All cell lines but CAR1, CL14, and GTL-16 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The CAR1 cells were obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan), and the CL14 cells were obtained from the German Collection of Microorganisms and Cell Cultures (DMSZ; Braunschweig, Germany). All cell lines were cultured as suggested by the provider. GTL-16 cells were previously described (20). JNJ-38877605 (Janssen Pharmaceutical; Johnson & Johnson, New Brunswick, NJ, USA) and crizotinib, the latter purchased from Active Biochemicals Co. (Hong Kong, China), were used at the indicated doses. Recombinant human HGF was purchased from Raybiotech, Inc. (Norcross, GA, USA). Recombinant human epidermal growth factor (EGF) was from Sigma-Aldrich (St. Louis, MO, USA). AZD6244 (selumetinib), AS703026 (pimasertib), PD98059, SB203580, and NVP-BEZ235 inhibitors were used at the indicated doses and all purchased from Selleck Chemicals (Munich, Germany). Gefitinib (GFTB) and cetuximab (CTX) were purchased from Cayman Chemical (Ann Arbor, MI, USA) and Merck KGaA (Darmstadt, Germany), respectively. Cobalt chloride and MTT were from Sigma-Aldrich. Stable overexpression of the constitutively active K-Ras G12V (kindly provided by Silvia Giordano, Department of Oncology, University of Torino School of Medicine, Turin, Italy) and of the dominant negative p38MAPK mutant form was carried out with the respective mutant cDNA driven by lentiviral vectors. Cell transduction with lentiviral vectors is described in the relevant section.
Quantitative PCR
Quantitative PCR was carried out as described previously (21). Total cellular RNA was isolated using the SV Total RNA Isolation kit (Promega, Fitchburg, WI, USA). To quantify the expression levels of HSP encoding genes, equal amounts of cDNA were synthesized using the Moloney murine leukemia reverse transcriptase (Promega) and mixed with SsoFast EvaGreen Supermix (Bio-Rad, Hercules, CA, USA) and 300 μM of each of the respective forward and reverse primers. Quantitative real-time PCR was done on a MyiQ thermal cycler (Bio-Rad). Each target gene expression was evaluated using a relative quantification approach, with POLR2A (GenBank accession no. NM000937.4) as an internal reference. Primer sets used are as follows: POLR2A: forward TGCAAGGGCAAAAACATATGC, reverse AGCTCTAGGCCAGAACGCC; HSP27: forward GCGTGTCCCTGGATGTCAAC, reverse TGTATTTCCGCGTGAAGCAC; PDK1: forward CCAACCACGAGGCTGATGA, reverse TGTCTTTGGGTTCTCTCTGCTGG; HSP22: forward AAGCCAGAGGAGTTGATGGTG, reverse CTCTGGGGAAAGTGAGCAAA; αβ crystallin: forward GACTCTCAGAGATGCGCCTG, reverse AGGGTCTACATCAGCTGGGA. PCR cycling conditions were as follows: 30 s at 95°C 30, 5 s at 95°C plus 15 s at 60°C (40 cycles), 30 s at 95°C, and 10 s at 65°C plus 10 s at 0.5°C (60 cycles: melting curve).
Western blot analysis
Western blot analysis was carried out as described previously (22). The following antibodies were used: mouse monoclonal anti-vinculin from Sigma; rabbit anti-HIF-1α from Bethyl (Montgomery, TX, USA); mouse monoclonal anti-MET from Invitrogen (Camarillo, CA, USA); mouse monoclonal anti-Mcl-1 from EMD Millipore (Billerica, MA, USA); rabbit anti-p53, goat anti-P-p27 (T187), rabbit anti-β-tubulin, and rabbit anti-pan Ras from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal anti-HSP27, anti-P-Erk1/2 (Thr202/Tyr204), anti-p21 Waf1/Cip1, anti-cyclin D1, and rabbit polyclonal anti-lens epithelium-derived growth factor (LEDGF), anti-HSF1, anti-P-HSP27 (Ser82), anti-HSP70, anti-HSP90, anti-P-MET (Tyr1234/1235), anti-P-epidermal growth factor receptor (EGFR; Tyr1068), anti-P-Akt (Ser473), anti-Akt, anti-Erk1/2, anti-P-p38 MAPK (Thr180/Tyr182), anti-Bim, anti-Bmf, and anti-cleaved poly(ADP-ribose) polymerase (PARP; Asp214) antibodies were all obtained from Cell Signaling Technology (Beverly, MA, USA). Bound antibodies were detected using the appropriate peroxidase-conjugated secondary antibody and revealed by enhanced chemiluminescence (Pierce, Rockford, IL, USA). Where indicated, band intensities were quantified using ImageJ software (Wayne Rasband, U.S. National Institutes of Health, Bethesda, MD, USA).
RNA interference
Transient silencing of MET and HSP27 by RNA interference was performed using On-TargetPlus SmartPool (Dharmacon, Lafayette, CO, USA). In each experiment, the On-TargetPlus nontargeting pool (Dharmacon) was used as the negative control. The cell lines were plated at 30–40% confluency and transfected with the indicated siRNA pools (100 nM) using Oligofectamine (Invitrogen), according to the manufacturer's instructions. Stable silencing of HSP27, HSF1, LEDGF, HIF-1α, and p53 was achieved using specific shRNAs transduced into the cells by means of lentiviral vectors. The HIF-1α-specific shRNA was obtained from the Open Biosystem TRC lentiviral shRNA library (RHS3979-9572497); HSP27-specific shRNAs (TRCN0000008753 and TRCN0000342857), HSF1-specific shRNA (TRCN00286345), and LEDGF-specific shRNA (TRCN0000007480) were all obtained from the Sigma-Aldrich TRC lentiviral shRNA library (Sigma-Aldrich). The p53-specific shRNA was kindly provided by Dr. Davide Zecchin (Laboratory of Molecular Genetics, Candiolo Cancer Institute, FPO-IRCCS, Candiolo, Italy).
Cell transduction with lentiviral vectors
Cells were transduced using second-generation lentiviral vectors, whose stocks were produced by transient transfection of 293T cells with the packaging plasmid pCMV-DR8.74, the envelope plasmid pMD2G-VSVG, and the respective transfer gene-carrying vector. Serial dilutions of freshly harvested conditioned medium were used to infect 105 cells in a 6-well plate in the presence of polybrene (8 μg/ml).
Apoptosis assay and cell cycle analysis
Caspase activation was determined by labeling cells with anti-active caspase-3 antibody (BD Pharmingen, San Diego, CA, USA) followed by a PE-conjugated goat anti-rabbit Ig (BD Pharmingen). Cells were fixed and permeabilized with BD Cytofix/Cytoperm (Becton Dickinson, Franklin Lakes, NJ, USA), according to the manufacturer's instructions. For cell-cycle analysis, cells were harvested and fixed with 70% ethanol overnight, treated with RNase, and stained with propidium iodide (Invitrogen) for 3 h at 4°C. Apoptosis was also measured as staining with APC-conjugated annexin V (Bender MedSystems, Burlingame, CA, USA) and propidium iodide (Invitrogen), in accordance with the manufacturer's instructions. The samples were analyzed on CyAN-Adp flow cytometer (Dako, Carpinteria, CA USA). Data acquisition was performed using Summit software (Dako).
Viability assay
In vitro growth curve was performed using the MTT assay according to the manufacturer's instructions. Cell viability was determined using the CellTiter Glo proliferation assay (Promega) according to the manufacturer's protocol.
Tumorigenesis assay
For the assay, 1 × 106 control and HSP27 stably silenced cells were injected subcutaneously in the right posterior flank of 6-wk-old γ-null mice (Charles River Laboratories, Calco, Italy). When tumors were palpable (80–100 mm3), mice were randomized in experimental groups of 7 animals, and JNJ-38877605 (10 mg/kg) was administered orally by gavage every day for 2 wk. Tumors were measured using a caliper and volume calculated using the formula (D×d2)/2, where D is the major tumor axis and d is the minor tumor axis. All animal procedures were approved by the Veterinary Ethical Commission of the Candiolo Cancer Institute (Candiolo, Italy) and by the Italian Ministry of Health.
Immunohistochemistry
Tissues were processed using standard method. Cleaved caspase-3 staining was assessed using a rabbit monoclocal antibody specifically recognizing the activated form of caspase-3 (Cell Signaling Technology). The percentage of cells displaying active caspase-3 staining was quantitatively assessed using ImageJ.
Immunofluorescence
Cells were plated on glass coverslips for 24 h. Cells were fixed in 4% paraformaldehyde for 20 min at room temperature and permeabilized with 0.1% Triton-X100 in PBS for 2 min on ice. Then cells were incubated at room temperature with 1% BSA in PBS for 30 min and with an anti-HSF1 and LEDGF rabbit polyclonal antibody (Cell Signaling Technology) diluted 1:100 in PBS containing 1% donkey serum for 1 h. After being washed, cells were fluorescently labeled with an Alexa Fluor A488 donkey anti-rabbit antibody (Molecular Probes, Eugene, OR, USA) diluted 1:400 in PBS containing 1% donkey serum for 1 h. Nuclei were stained with DAPI. F-actin was stained with TRITC-conjugated phalloidin (50 μg/ml). The cells were analyzed using a Leica TCS SP2 AOBS confocal laser-scanning microscope (Leica Microsystems, Heerbrugg, Switzerland).
Statistical analysis
Statistical analysis of the data was performed using ANOVA (Microsoft Excel; Microsoft, Redmond, WA, USA).
RESULTS
MET receptor kinase regulates HSP27 expression
HSP27 expression was studied in the GTL-16, MKN-45, and HS-746-T human gastric carcinoma cell lines. These are MET-addicted cells lines (23), because of MET gene amplification, overexpression, and constitutive activation of the MET receptor in the absence of its ligand (Fig. 1A). In addition, the HS-746-T cells harbor a splice-site MET mutation (24). In these cell lines, the expression of HSP27 was induced by MET kinase inhibition, as shown by the 2- to 3-fold and 3- to 4-fold increase of HSP27, visible at the protein and mRNA level, respectively. This occurred after cell treatment with either the MET-specific small-molecule kinase inhibitor JNJ-38877605 (Fig. 1A, D, F) or the MET/ALK/ROS-1 inhibitor crizotinib (Fig. 1B, E) or MET-specific small interfering RNA (Fig. 1C). Interestingly, the phosphorylation of HSP27, i.e., activation (1, 25, 26), increased in parallel (Fig. 1F). The other heat-shock-responsive proteins HSP70 and HSP90 were not similarly induced (Fig. 1F). Together, these data suggested the transcriptional induction of HSP27 as a consequence of MET inhibition.
Figure 1.

The MET receptor kinase regulates HSP27 expression. A) Western blot analysis of MET receptor and HSP27 in the GTL-16, HS-746-T, and MKN-45 human gastric carcinoma cell lines. The MET 145 kD β chain (p145, arrow) and the 170 kD precursor were labeled with MET antibody. The 145 kD β chain was phosphorylated at tyrosine (P-p145, arrow) and labeled with the anti-phospho-MET (Tyr1234/1235). The MET inhibitor JNJ-38877605 (0.1 μM) was added for 48 h. B) Western blot analysis showing HSP27 increase on treatment of MKN-45 cells with crizotinib (0.3 μM) for the indicated time. C) Western blot analysis of MET and HSP27 in GTL-16 cells transfected with MET-specific siRNAs (siMET) or control siRNAs (siCTRL). D, E) Quantitative PCR (qPCR) of HSP27 mRNA levels following treatment of GTL-16 and MKN-45 cells with JNJ-38877605 (0.1 μM; D) and crizotinib (0.3 μM; E) for the indicated time. F) Western blot analysis of phospho-MET, HSP27, phospho-HSP27, HSP70, and HSP90 in GTL-16 cells on treatment with JNJ-38877605; the drug was added at the doses and times shown. G) Western blot of HSP27 levels in SK-OV-3 and TOV-21G ovarian carcinoma cells on treatment with JNJ-38877605 (0.1 μM) for 24 h. H) Western blot analysis of the MET receptor and HSP27 in SK-OV-3 and TOV-21G cells, treated with recombinant human HGF (rhHGF; 50 ng/ml) for the indicated time. I) qPCR of the HSP27 mRNA after treatment of ovarian carcinoma cells with rhHGF (50 ng/ml) and JNJ-38877605 (0.1 μM) for 24 h. Each blot was reprobed with either vinculin or tubulin antibody to confirm equal loading. Band intensities were quantified and normalized to either vinculin or tubulin using ImageJ software and are shown under each box.
To confirm that the HSP27 expression can be regulated by MET kinase, the SK-OV-3 and TOV-21G ovarian cancer cell lines were treated with the MET ligand HGF. These cell lines do not show either amplification of the MET gene or constitutive phosphorylation of the MET receptor and are not addicted to MET. In these cells, while MET inhibition did not induce HSP27 expression (Fig. 1G), the HGF-triggered MET activation was associated with the down-modulation of HSP27 at both the protein (Fig. 1H) and mRNA level (Fig. 1I). This down-modulation was reverted by MET inhibition (Fig. 1I).
Altogether, these data show that MET kinase activity regulates HSP27 expression.
MET inhibition induces HSP27 expression via the MEK/ERK pathway
In MET-addicted cells, both the MEK/ERK and PI3K/AKT pathways are constitutively active due to the activation of the MET receptor in the absence of ligand. MET inhibition resulted in the loss of ERK1/2 and AKT phosphorylation (Fig. 2A), as previously demonstrated (23). Conversely, p38MAPK might be activated by MET inhibition (Supplemental Fig. S1A and ref. 22). Using biochemical inhibitors of the above signal transducers (Fig. 2A), we investigated whether HSP27 induction associated with MET inhibition relied on MEK/ERK and/or PI3K/AKT pathways. MEK inhibition by AZD6244 (selumetinib) resulted in the increased expression of HSP27 at both protein (Fig. 2B) and mRNA level (Fig. 2E). HSP27 was also induced by 2 other structurally unrelated MEK inhibitors: PD98059 (Fig. 2C) or AS703026 (pimasertib; Fig. 2C). Conversely, the inhibition of the PI3K/mTOR pathway by the dual inhibitor NVP-BEZ235 (Fig. 2B) did not affect HSP27 levels. Neither the expression of a dominant negative form of p38MAPK (Supplemental Fig. S1A) nor cell treatment with the p38MAPK biochemical inhibitor SB203580 (Supplemental Fig. S1B, C) affected HSP27 induction.
Figure 2.

The MEK/ERK pathway mediates the regulation of HSP27 expression by MET kinase. A) Western blot analysis showing the effectiveness of AZD6244, NVP-BEZ235, and JNJ-38877605 inhibitors in GTL-16 cells in reducing Erk1/2, Akt and MET phosphorylation, respectively. Cells were treated with the inhibitors for 2 h at the indicated doses. B) Western blot analysis of HSP27 and HSP70 in GTL-16 cells treated with the indicated kinase inhibitors at the concentrations shown for 48 h. C) Western blot analysis showing HSP27 up-regulation on treatment of MKN-45 cells with the 2 indicated MEK1/2 kinase inhibitors (AS703026: 0.5 μM; PD98059: 10 μM) for the indicated time. Inset: effectiveness of the inhibitors in reducing Erk1/2 phosphorylation. D) Quantitative PCR (qPCR) of HSP27, HSP22, and αβ crystallin mRNA levels in response to AZD6244 (1μM) treatment for the indicated time in MKN-45 cells. E) qPCR of HSP27 mRNA in both wild-type (WT) and active K-Ras (G12V)-expressing GTL-16 cells treated for the indicated time with either AZD6244 (1 μM) or JNJ-38877605 (0.1 μM). F) Western blot analysis of HSP27 in GTL-16 cells where Erk1/2 phosphorylation (P-Erk1/2) is rescued in cells treated with JNJ-38877605 (0.1 μM) for the indicated time, because of the stable expression of an active K-Ras. G) HSP27 protein expression in wild-type and K-Ras G12V expressing GTL-16 cells, represented as the ratio between intensities of bands of normalized HSP27 in JNJ-38877605-treated vs. HSP27 detected in untreated wild-type cells from 3 independent experiments (means±sd). HSP27 protein levels in untreated wild-type cells were arbitrarily set to 1. All blots were reprobed with vinculin antibody to confirm equal loading. Band intensities were quantified and normalized to either vinculin or tubulin using ImageJ. Statistical significance was determined using ANOVA test. *P < 0.05.
While MEK inhibition did not affect the expression of HSP70 (Fig. 2B), it induced the increase of expression of the other small heat-shock proteins HSP22 and αβ crystallin (Fig. 2D). However, their expression level remained markedly lower than that of HSP27.
To further confirm the role of the MEK/ERK pathway in regulating HSP27 expression downstream of MET, a constitutively active Ras (K-Ras G12V) was expressed in MET-addicted cells. Active Ras almost completely rescued the inactivation of the MEK/ERK pathway due to MET inhibition and reduced the HSP27 increase at both mRNA (Fig. 2E) and protein (Fig. 2F, G) level. In the mirror experiment, the treatment of ovarian cancer cells with the MEK inhibitor impaired the HSP27 down-modulation by HGF (Supplemental Fig. S1D).
Altogether, these data show that HSP27 induction on MET inhibition relies on the MEK/ERK pathway.
MET-dependent HSP27 induction relies on the transcription factors HSF1 and HIF-1α
HSP27 expression is induced on heat shock by the transcription factors of the family of HSFs, which drive the expression of several heat-shock proteins (27). Moreover, another stress responsive factor, LEDGF, regulates the expression of HSP27 (28) and other sHSPs (29). Figure 3A shows that both HSF1 and LEDGF are expressed in the MET-addicted MKN-45 cells and localized in the nuclei.
Figure 3.
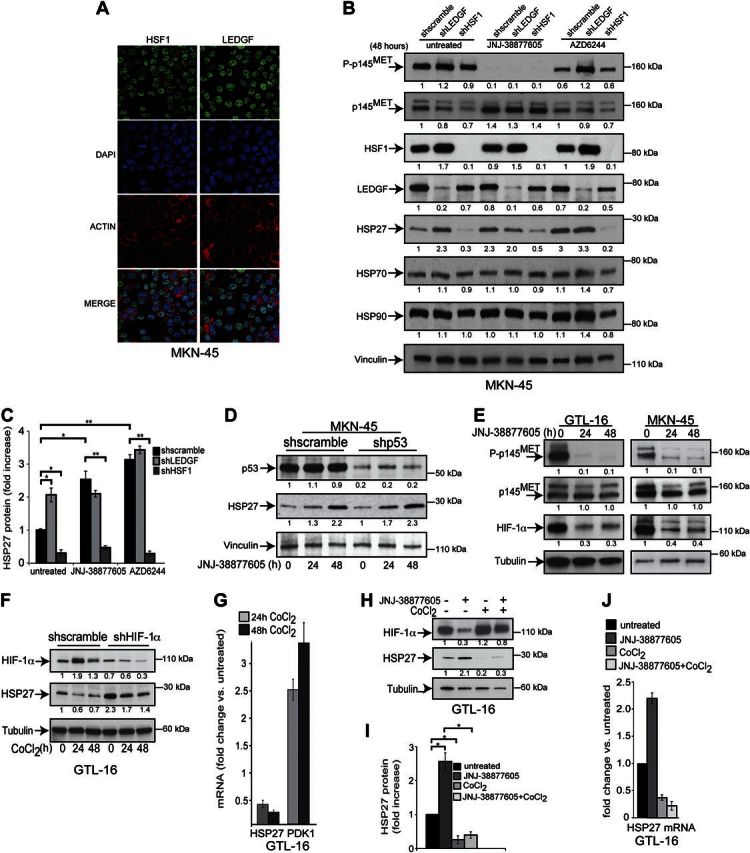
MET-dependent HSP27 induction relies on the transcription factors HSF1 and HIF-1α. A) Nuclear localization of HSF1 and LEDGF in MKN-45 cells, visualized with confocal microscopy using anti-HSF1 and anti-LEDGF polyclonal antibodies, respectively, revealed with an Alexa Fluor donkey anti-rabbit antibody. Cell nuclei were labeled with DAPI, and cell outlines were revealed with TRITC-conjugated phalloidin, which binds F-actin. B) Silencing of HSF1 and LEDGF with specific shRNAs in MKN-45 cells, as confirmed in Western blot analysis. HSF1 knockdown was sufficient to decrease both the basal and the MET- and MEK-dependent HSP27 protein expression, without affecting HSP70 and HSP90 levels. The inhibitors JNJ-38877605 (0.1 μM) and AZD6244 (1 μM) were used for 48 h. C) HSP27 protein expression in wild-type and MKN-45 cells, expressing either shLEDGF or shHSF1 or control sh (shscramble), and treated with JNJ-38877605 (0.1 μM) and AZD6244 (1μM) for 48 h, represented as the ratio between intensities of bands of normalized HSP27 in each experimental condition vs. HSP27 detected in untreated control cells from 3 independent experiments (mean±sd). HSP27 protein levels in untreated control cells were arbitrarily set to 1. D) Silencing of p53 with the specific shRNA in MKN-45 cells, as confirmed in Western blot analysis, does not impair JNJ-38877605-triggered HSP27 induction. Cells were treated with JNJ-38877605 (0.1 μM) for the indicated time. E) Western blot analysis of MET and HIF-1α after treatment of GTL-16 (left) and MKN-45 (right) cells with JNJ-38877605 (0.1 μM) for the indicated hours. F) Western blot analysis of HSP27 and HIF-1α in GTL-16 cells stably expressing HIF-1α-specific (shHIF-1α) or control (shscramble) shRNA with or without cobalt chloride (CoCl2; 200 μM) for the indicated hours. G) qPCR of HSP27 and PDK1 mRNA levels in response to CoCl2 (200 μM) for 24 and 48 h in GTL-16 cells. PDK1 was used a positive control as it is a known HIF-1α target gene. H) Western blot analysis of HSP27 and HIF- 1α in GTL-16 cells treated with CoCl2 (200 μM) and JNJ-38877605 (0.1 μM) for 24 h. I) Graph showing HSP27 protein expression in wild-type and GTL-16 cells treated with either JNJ-38877605 or cobalt chloride as in H, represented as the ratio between intensities of bands of normalized HSP27 in treated cells vs. HSP27 detected in untreated cells from 3 independent experiments (means±sd). HSP27 protein levels in untreated cells were arbitrarily set to 1. J) qPCR of HSP27 mRNA levels of GTL16 cells treated as in panel G for 24 h. All blots were reprobed with either vinculin or tubulin antibody to confirm equal loading. Band intensities were quantified and normalized to either vinculin or tubulin using ImageJ. Statistical significance was determined using ANOVA test. *P < 0.05.
Stable silencing of HSF1 using specific shRNA carrying lentiviral particles resulted in the decrease of the basal level of HSP27 and lack of its induction by MET and MEK inhibition (Fig. 3B). Surprisingly, LEDGF silencing induced HSP27 expression (Fig. 3B, C), likely by inducing the increase of HSF1 expression (Fig. 3B), making difficult appreciating further HSP27 induction on MET inhibition.
Transcription factors able to modulate HSP27 other than HSFs, such as the signal transducers and activators of transcription STAT3, are not affected by MET inhibition (23). Moreover, p53, which is known to regulate HSP27 expression (30), did not participate in its induction by MET inhibition (Fig. 3D).
We previously showed that the HIF-1α is stabilized by MET activation (31). In agreement, MET inhibition resulted in decreased level of the HIF-1α protein (Fig. 3E) that was rescued by the ectopic expression of an overactive K-Ras (G12V; Supplemental Fig. S1E). HIF-1α silencing using RNA interference was alone able to induce HSP27 expression (Fig. 3F). In the mirror experiments, HIF-1α stabilization with cobalt chloride (CoCl2), a hypoxia-mimicking agent, was accompanied by HSP27 decrease at both protein (Fig. 3F) and mRNA level (Fig. 3G); this decrease was reverted by HIF-1α silencing (Fig. 3F). Furthermore, CoCl2 counteracted both the HIF-1α decrease and HSP27 increase triggered by MET inhibition (Fig. 3H–J).
Silencing of HSP27 sensitizes MET-addicted gastric carcinoma cells to MET inhibition
To determine the role of HSP27 induction by MET inhibitors in functional assays, the protein was silenced using RNA interference in the MET-addicted GTL-16 and MKN-45 gastric carcinoma cell lines. First, HSP27 was silenced by means of the transient transfection of a pool of 4 siRNAs, each targeting the HSP27 mRNA (Fig. 4A). The use of this siRNA pool allows avoiding high siRNA concentrations and reduces off-target effects (32). As a control, cells were transfected with a nontargeting siRNA pool. In the GTL-16 cells, MET kinase inhibition by the selective MET kinase inhibitor JNJ-38877605 resulted in cell-cycle arrest, shown by the decreased percentage of cells in S phase (Fig. 4A), as also previously reported (23). Conversely, when HSP27 was silenced, the MET inhibitor induced cell death, as shown by the increased percentage of cells in the sub-G1 phase (Fig. 4A). It is noteworthy that HSP27 silencing alone provoked only a modest death rate of these MET-addicted gastric carcinoma cells, while targeting HSP27 results in the massive death of prostate (33) and pancreatic carcinoma cells (34). To rule out that in GTL-16 cells HSP27 induction was a general stress response due to cell cycle blockade, the cell cycle was blocked by serum withdrawal in wild-type GTL-16 cells. Supplemental Fig. S2A, B shows that the cell cycle blockade was not alone able to induce HSP27 expression. Moreover, in the same cells AZD6244 induced HSP27 expression because of MAPK pathway inhibition without affecting the cell cycle (Supplemental Fig. S2C).
Figure 4.

The combination of HSP27 silencing and MET inhibition induces the apoptotic death of MET-addicted gastric carcinoma cells. A) Cell cycles of GTL-16 cells transfected with either control (siCTRL) or HSP27-specific (siHSP27) siRNA pools for 72 h and, when indicated, treated with JNJ-38877605 (0.1 μM) for 24 h; percentages of cells in sub-G1 and S phase of the cell cycle are shown; a representative experiment of 3 independent replicates is shown. Inset: Western blot analysis of HSP27 protein levels 72 h after transfection with either siCTRL or siHSP27 pools. B) Apoptotic death of GTL-16 cells expressing either of 2 HSP27-specific shRNAs and treated with JNJ-38877605 (0.1 μM) for 36 h, evaluated as percentage of cells labeled with an active caspase-3 antibody. Inset: Western blot analysis of HSP27 protein levels 96 h after infection with either control (shscramble) or HSP27-specific (shHSP27) shRNA delivered by lentiviral particles. C, D) Silencing of either HSP27 (C) or HSF1 (D) using specific shRNAs lentiviral particles sensitized MKN-45 cells to apoptotic death following MET inhibition by either JNJ-38877605 (0.1 μM) or crizotinib (0.3 μM) for 48 h; apoptosis was evaluated as percentage of cells labeled with an active caspase-3 antibody. E, F) Western blot analysis of the listed proteins in HSP27-silenced and control MKN-45 cells treated with JNJ-38877605 (0.1 μM; E) or crizotinib (0.3 μM; F) for 36 h, where indicated. Each blot was reprobed with vinculin antibody to confirm equal loading; representative blots are shown. Percentages of apoptotic cells are the means ± sd of ≥3 independent experiments. Statistical significance was determined using ANOVA test. *P < 0.05.
To dissect the functional effects of HSP27 silencing and MET inhibition, HSP27 was also stably silenced in the same cells using shRNAs carried by lentiviral vectors (Fig. 4B). In cells where HSP27 was silenced with shRNAs, the MET inhibitor induced cell death as above (compare Fig. 4A and Supplemental Fig. S3). Increased caspase-3 activation (Fig. 4B, C) and PARP cleavage (Fig. 4E, F) further demonstrated that HSP27 silencing potentiated the cytotoxic effects of the MET inhibitors JNJ-38877605 and crizotinib. In agreement with the role of HSF1 in inducing HSP27 expression after MET inhibition, the silencing of HSF1 similarly sensitized the cells to these targeted drugs (Fig. 4D). Moreover, while cell treatment with JNJ-38877605 alone resulted in the down-regulation of cyclin D1, in line with its role in cell entry in S phase (Fig. 4E), the combination of MET inhibition and HSP27 silencing was necessary to activate the degradation of the cyclin-dependent kinase inhibitor (CDKI) p21 and the phosphorylation at threonine of the CDKI p27, that makes it susceptible to degradation (Fig. 4E). The MET inhibitor JNJ-38877605 alone increased the level of Bim (Fig. 4E), like crizotinib, as reported by others (35). Conversely, HSP27 silencing was able to increase the proapoptotic BMF protein and to decrease the antiapoptotic Mcl-1 protein (Fig. 4E). Notably, HSP27 silencing did not affect either the constitutive activation of ERK1/2 and AKT or its inhibition by crizotinib (Fig. 4F). Altogether, these data show that cell death of HSP27-silenced cells caused by MET inhibition is accompanied by the combined regulation of cell cycle-associated proteins and BH3-only effectors of apoptosis. It is noteworthy that the loss of HSP27 per se did not affect the constitutive phosphorylation, i.e., the basal activation of the MET receptor (Fig. 4E, F), ruling out a stabilizing role of HSP27 toward this receptor.
Cell viability assay confirmed that the MET-addicted gastric carcinoma cells treated with JNJ-38877605 did not proliferate but did regrow after drug washout (Fig. 5A). Conversely, HSP27-silenced cells treated with JNJ-38877605 did not regrow (Fig. 5A).
Figure 5.

Silencing of HSP27 potentiated the cytotoxic effect of the MET inhibitor in vitro and in vivo. A) In vitro growth of MKN-45 cells expressing a HSP27-specific shRNA (shHSP27) or control shRNA (shscramble), assessed using MTT viability assay; cells were treated with JNJ-38877605 (0.1μM) from d 1 to 4 followed by drug washout; values represent the means ± sd of 3 independent experiments carried out in triplicate; in parallel, at the end of the experiment, cells were also stained with crystal violet (inset). B) Growth as xenografts of HSP27-silenced (shHSP27) and control (shscramble) MKN-45 cells; where indicated, mice were treated daily with JNJ-38877605 (10 mg/kg), started after 1 wk growth (first arrow) and suspended after 2 wk treatment (second arrow). C) Immunohistochemical detection of active caspase-3 in the xenografts at the end of the experiment. Scale bar = 20 μM. D) Quantification of the cleaved caspase-3 positive cells (mean±se percentage) carried out using ImageJ. Statistical significance was determined using ANOVA test. *P < 0.05; **P < 0.005.
In vivo, MET-addicted cells formed tumors that grew less when either HSP27 was suppressed or MET was inhibited by JNJ-38877605 (Fig. 5B). Furthermore, after suspension of anti-MET treatment, tumors regrew. Conversely, the combined treatment was more effective, as even after suspension of the MET-targeted therapy, HSP27-silenced tumors did not regrow (Fig. 5B). Indeed, 2 wk after suspension of the treatment with JNJ-38877605, HSP27-silenced tumors showed an increased percentage of active caspase-3 positive, i.e., apoptotic, cells (Fig. 5C, D).
Altogether, these data show that HSP27 silencing allowed the MET inhibitor to exert a full cytotoxic effect in MET-addicted gastric carcinoma cell lines.
HSP27 expression is induced by EGFR inhibitors and limits their effectiveness
To assess whether HSP27 induction on MEK/ERK inhibition was a MET-specific effect, the expression of HSP27 following inhibition of the EGFR was assessed in the A431 squamous carcinoma and SW48 colorectal carcinoma cells, which are both susceptible to EGFR inhibitors. In the A431 cells, dependency on EGFR is due to EGFR gene amplification (36). In the SW48 cells, the EGFR gene is not amplified nor mutated; however, these cells are susceptible to treatment with the anti-EGFR antibody CTX (37). As shown in Fig. 6A, in SW48 cells HSP27 was induced by cell treatment with CTX, as well as in A431 cells treated with the small molecule EGFR inhibitor GFTB (Fig. 6C). Moreover, HSP27 silencing sensitized both the SW48 (Fig. 6B) and A431 (Fig. 6D) cells to EGFR inhibition. It is noteworthy that HSP27 silencing was alone able to commit SW48 cells to death. As reported previously (36), in the EGFR-dependent cell lines, inhibition of this receptor resulted in Erk1/2 inhibition (Fig. 6E). In the mirror experiments, EGFR activation by its ligand EGF in the SK-OV-3 ovarian cancer cells resulted in the decreased expression of HSP27 (Fig. 6F) while it did not affect HSP70. Control cell lines, i.e., colorectal cancer cell lines resistant to CTX (Fig. 6G), did not show induction of HSP27 after drug treatment (Fig. 6H) and were not affected by HSP27 silencing (Fig. 6G).
Figure 6.

HSP27 is induced by EGFR inhibition and limits the effectiveness of inhibitors. A) Western blot analysis of HSP27 in SW48 cells expressing either HSP27-specific shRNA (shHSP27) or control shRNA (shscramble) and treated with CTX (5 nM for 24 h). B) Viability assay of control and HSP27-silenced SW48 cells treated with CTX as in panel A. C) Western blot analysis of HSP27 in A431 cells expressing either HSP27-specific shRNA or control shRNA and treated with GFTB (0.5 μM) for 24 h). D) Viability assay of control and HSP27 silenced, A431 cells treated with GFTB as in panel C. E) Western blot analysis of Erk1/2 phosphorylation and HSP27 in A431 treated as in C. F) Western blot analysis of HSP27 and HSP70 in the SK-OV-3 ovarian carcinoma cells treated with EGF (50 ng/ml) for the indicated times, that resulted in the EGFR phosphorylation (P-EGFR). G) Survival of CAR1, CL-14, and COLO320-DM colorectal cancer cells treated with CTX (5 nM for 24 h) after and before HSP27 knockdown using specific HSP27 shRNA (shHSP27). Graph shows the percentage of DAPI and annexin V negative (i.e., alive) cells. H) Western blot analysis of HSP27 expression in the CTX-resistant colorectal cancer cells CAR1, CL-14 and COLO320-DM on treatment with CTX (5 nM for 24 h). Blots were reprobed with vinculin antibody to confirm equal loading. Statistical significance was determined using ANOVA test. *P < 0.05.
Altogether, these data show that EGFR inhibition in susceptible cells results in increased expression of HSP27 that in turn can reduce the efficacy of EGFR-targeted therapies.
DISCUSSION
This study shows that the inhibition of the MET kinase results in increased expression of HSP27 protein and that, more notably, this increase might limit the efficacy of MET-targeted therapies, as suppression of HSP27 made MET-addicted cells more vulnerable to MET inhibition.
Data show that MET signaling regulates the transcription of the HSPB1 gene encoding HSP27 and that this regulation occurs through the MEK/ERK pathway. Either MET inhibition in MET-addicted gastric carcinoma cells or MET activation by HGF in ovarian cancer cells resulted in up- and down-regulation of HSP27, respectively. Both up- and down-regulation of HSP27 relied on the MEK/ERK pathway. In agreement, an overactive Ras impaired the up-regulation of HSP27 by rescuing the activation of the MEK/ERK pathway. Here we show also that HSP27 levels increased after the inhibition of EGFR in cells susceptible to this inhibition, through a MEK/ERK-dependent mechanism. This suggests that HSP27 induction by inhibition of the MEK/ERK pathway might be more widespread than expected and thus might interfere with different RTK-targeted therapies. Interestingly, it has been shown in HER2-overexpressing breast cancer cells that, on the one hand, the development of resistance to the anti-HER2 monoclonal antibody trastuzumab is associated with HSP27 increase and, on the other hand, HSP27 knockdown increases the susceptibility of the resistant cells to trastuzumab (38).
The induction of HSPs in response to various stresses mostly depends on the activation of specific members of the family of HSFs, which bind to the heat-shock element (HSE) in the promoters of the genes encoding HSPs (27). Here we show that inhibition of the MET kinase in MET-addicted cells resulted in the up-regulation of HSP27 and other sHSPs, but not of HSP70 and HSP90, through HSF1. Indeed, data show that in these cells, HSF1 controls HSP27 basal expression, and it is also required for HSP27 induction downstream of MET/MEK/ERK axis inactivation. Moreover, we show that HIF-1α also participates in the induction of HSP27 following MET inhibition. Indeed, HSP27 increased expression was associated with a HIF-1α decrease that depended on inactivation of the MEK/ERK pathway; in agreement, HIF-1α expression was rescued by an overactive K-Ras. It has been previously reported (36) that EGFR inhibition similarly results in HIF-1α down-modulation. Conflicting reports suggest that either HIF-1α is a transcriptional activator of HSP27 (39) or that HSP27 is negatively regulated by hypoxia (40). Here we show that cell treatment with cobalt chloride, a hypoxia-mimicking agent, counteracted the up-regulation of HSP27 via HIF-1α stabilization. Taken together, data show that in MET-addicted cells HIF-1α is likely involved in HSP27 induction downstream of MET inactivation.
Here we also show that other cytoprotective sHSPs, such as HSP22 and αβ crystallin, are induced following inhibition of the MET/MEK/ERK signaling axis, suggesting that MET inhibition triggers a stress-like response in MET-addicted cells. However, functional data reveal that HSP27 suppression is sufficient to make these cells more susceptible to MET inhibition. This is likely due to the markedly higher expression level of HSP27 in comparison with that of the other sHSPs. Furthermore, HSP27 does not act as a molecular chaperone of MET, as in HSP27-silenced cells neither the phosphorylation nor the expression of the receptor was affected. More likely, other cytoprotective roles played by HSP27 (41, 42) are impaired after HSP27 loss. In fact, cell death after HSP27 silencing is associated with an increase of the apoptotic marker active caspase 3, which is a known HSP27 client (43).
Here we show that, on deprival of MET signals, MET-addicted cells become quiescent but are not committed to death. Interestingly, silencing of HSP27 turns the antiproliferative effect of MET inhibition toward cell death. Indeed, we observed that the CDKI p21 and p27 undergo degradation on JNJ-38877605 treatment of HSP27-silenced cells. It is likely that the loss of these CDKI could reflect the abrogation of the cell-cycle blockade triggered by MET inhibition. Furthermore, we found that HSP27 loss per se weakened the antiapoptotic machinery of the MET-addicted cells, as shown by the increased and decreased levels of the proapoptotic Bmf and the antiapoptotic Mcl-1, respectively. This implies that the combination of HSP27 silencing and MET kinase inhibition could more steadily control the growth of MET-addicted tumors.
Altogether, data suggest that the up-regulation of HSP27 triggered by MET inhibition might strongly limit the success of MET-targeted therapies. Furthermore, as HSP27 expression is also triggered by EGFR inhibitors and its suppression increased the efficacy of EGFR inhibitors as well as that of chemotherapeutics (44), we can infer that HSP27 might limit the effectiveness of both MET- and EGFR-targeted agents and conventional chemotherapeutics when these drugs are simultaneously administered in combination therapies. Notably, MEK inhibition was alone able to induce HSP27 expression, which, in turn, could limit the efficacy of MEK inhibitors in cancer cells otherwise addicted to this pathway.
HSP27 is overexpressed in many cancers and frequently related to adverse prognosis (10) so that HSP27 has been proposed as a target for therapy. Antisense (33, 34) and peptide aptamer (45) strategies have shown that targeting HSP27 increases cancer cell death in vitro and in vivo in preclinical models. More important, an antisense drug inhibiting HSP27 is available for human therapy and is currently being tested in phase II clinical trials (http://clinicaltrials.gov/ct2/results?term=OGX427&Search=Search). As we show here that HSP27 expression increased in cells treated with targeted agents, HSP27 knockdown in combination with targeted therapies can be envisaged as a viable and effective therapeutic approach for clinical application.
Supplementary Material
Acknowledgments
The authors thank Livio Trusolino for the helpful discussion and critical reading of the manuscript, Dr. Luca Cirillo for enthusiastic help in performing experiments, Dr. Davide Zecchin [Laboratory of Molecular Genetics, Candiolo Cancer Institute, Fondazione del Piemonte per l'Oncologia (FPO)–Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Candiolo, Italy] for providing p53 shRNA, and Silvia Giordano (Department of Oncology, University of Torino School of Medicine, Turin, Italy) for providing K-Ras G12V.
This work has been supported by the following grants to M.F.D.: 2012 IG grant 13050 and 2010 Special Program Molecular Clinical Oncology 5xMille of the Italian Association of Cancer Research (AIRC) Project 9970, a Cariplo Foundation grant, and Progetto di Ateneo-Compagnia di San Paolo grant ORTO11RKTW.
Conflicts of interest: T.P. is a full-time employee and shareholder of Janssen Research and Development (Beerse, Belgium). The other authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- CTX
- cetuximab
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- GFTB
- gefitinib
- HGF
- hepatocyte growth factor
- HIF-1α
- hypoxia-inducible factor-1α
- HSF1
- heat-shock factor-1
- HSP
- heat-shock protein
- HSP27
- heat-shock protein of 27 kDa
- LEDGF
- lens epithelium-derived growth factor
- PARP
- poly(ADP-ribose) polymerase
- sHSP
- small heat-shock protein
REFERENCES
- 1. Arrigo A. P., Gibert B. (2012) HspB1 dynamic phospho-oligomeric structure dependent interactome as cancer therapeutic target. Curr. Mol. Med. 12, 1151–1163 [DOI] [PubMed] [Google Scholar]
- 2. Comoglio P. M., Giordano S., Trusolino L. (2008) Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 7, 504–516 [DOI] [PubMed] [Google Scholar]
- 3. Kuniyasu H., Yasui W., Kitadai Y., Yokozaki H., Ito H., Tahara E. (1992) Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem. Biophys. Res. Commun. 189, 227–232 [DOI] [PubMed] [Google Scholar]
- 4. Engelman J. A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J. O., Lindeman N., Gale C. M., Zhao X., Christensen J., Kosaka T., Holmes A. J., Rogers A. M., Cappuzzo F., Mok T., Lee C., Johnson B. E., Cantley L. C., Janne P. A. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 5. Schmidt L., Duh F. M., Chen F., Kishida T., Glenn G., Choyke P., Scherer S. W., Zhuang Z., Lubensky I., Dean M., Allikmets R., Chidambaram A., Bergerheim U. R., Feltis J. T., Casadevall C., Zamarron A., Bernues M., Richard S., Lips C. J., Walther M. M., Tsui L. C., Geil L., Orcutt M. L., Stackhouse T., Zbar B. (1997) Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 16, 68–73 [DOI] [PubMed] [Google Scholar]
- 6. Lee J. H., Han S. U., Cho H., Jennings B., Gerrard B., Dean M., Schmidt L., Zbar B., Vande Woude G. F. (2000) A novel germ line juxtamembrane met mutation in human gastric cancer. Oncogene 19, 4947–4953 [DOI] [PubMed] [Google Scholar]
- 7. Di Renzo M. F., Olivero M., Martone T., Maffe' A., Maggiora P., De Stefani A., Valente G., Giordano S., Cortesina G., Comoglio P. M. (2000) Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 19, 1547–1555 [DOI] [PubMed] [Google Scholar]
- 8. Stella G. M., Benvenuti S., Gramaglia D., Scarpa A., Tomezzoli A., Cassoni P., Senetta R., Venesio T., Pozzi E., Bardelli A., Comoglio P. M. (2011) MET mutations in cancers of unknown primary origin (CUPs). Hum. Mutat. 32, 44–50 [DOI] [PubMed] [Google Scholar]
- 9. Trusolino L., Bertotti A., Comoglio P. M. (2010) MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 11, 834–848 [DOI] [PubMed] [Google Scholar]
- 10. Zoubeidi A., Gleave M. (2012) Small heat shock proteins in cancer therapy and prognosis. Int. J. Biochem. Cell Biol. 44, 1646–1656 [DOI] [PubMed] [Google Scholar]
- 11. Lanneau D., Wettstein G., Bonniaud P., Garrido C. (2010) Heat shock proteins: cell protection through protein triage. Sci. World J. 10, 1543–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garrido C., Paul C., Seigneuric R., Kampinga H. H. (2012) The small heat shock proteins family: the long forgotten chaperones. Int. J. Biochem. Cell Biol. 44, 1588–1592 [DOI] [PubMed] [Google Scholar]
- 13. Wettstein G., Bellaye P. S., Micheau O., Bonniaud P. (2012) Small heat shock proteins and the cytoskeleton: an essential interplay for cell integrity? Int. J. Biochem. Cell Biol. 44, 1680–1686 [DOI] [PubMed] [Google Scholar]
- 14. Acunzo J., Katsogiannou M., Rocchi P. (2012) Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death. Int. J. Biochem. Cell Biol. 44, 1622–1631 [DOI] [PubMed] [Google Scholar]
- 15. Mehlen P., Mehlen A., Godet J., Arrigo A. P. (1997) hsp27 as a switch between differentiation and apoptosis in murine embryonic stem cells. J. Biol. Chem. 272, 31657–31665 [DOI] [PubMed] [Google Scholar]
- 16. Conroy S. E., Sasieni P. D., Amin V., Wang D. Y., Smith P., Fentiman I. S., Latchman D. S. (1998) Antibodies to heat-shock protein 27 are associated with improved survival in patients with breast cancer. Br. J. Cancer 77, 1875–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arts H. J., Hollema H., Lemstra W., Willemse P. H., De Vries E. G., Kampinga H. H., Van der Zee A. G. (1999) Heat-shock-protein-27 (hsp27) expression in ovarian carcinoma: relation in response to chemotherapy and prognosis. Int. J. Cancer 84, 234–238 [DOI] [PubMed] [Google Scholar]
- 18. Cornford P. A., Dodson A. R., Parsons K. F., Desmond A. D., Woolfenden A., Fordham M., Neoptolemos J. P., Ke Y., Foster C. S. (2000) Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res. 60, 7099–7105 [PubMed] [Google Scholar]
- 19. Bruey J. M., Paul C., Fromentin A., Hilpert S., Arrigo A. P., Solary E., Garrido C. (2000) Differential regulation of HSP27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 19, 4855–4863 [DOI] [PubMed] [Google Scholar]
- 20. Giordano S., Ponzetto C., Di Renzo M. F., Cooper C. S., Comoglio P. M. (1989) Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 339, 155–156 [DOI] [PubMed] [Google Scholar]
- 21. Bardella C., Olivero M., Lorenzato A., Geuna M., Adam J., O'Flaherty L., Rustin P., Tomlinson I., Pollard P. J., Di Renzo M. F. (2012) Cells lacking the fumarase tumor suppressor are protected from apoptosis through a hypoxia-inducible factor-independent, AMPK-dependent mechanism. Mol. Cell. Biol. 32, 3081–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coltella N., Rasola A., Nano E., Bardella C., Fassetta M., Filigheddu N., Graziani A., Comoglio P. M., Di Renzo M. F. (2006) p38 MAPK turns hepatocyte growth factor to a death signal that commits ovarian cancer cells to chemotherapy-induced apoptosis. Int. J. Cancer 118, 2981–2990 [DOI] [PubMed] [Google Scholar]
- 23. Bertotti A., Burbridge M. F., Gastaldi S., Galimi F., Torti D., Medico E., Giordano S., Corso S., Rolland-Valognes G., Lockhart B. P., Hickman J. A., Comoglio P. M., Trusolino L. (2009) Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci. Signal. 2, er11. [DOI] [PubMed] [Google Scholar]
- 24. Asaoka Y., Tada M., Ikenoue T., Seto M., Imai M., Miyabayashi K., Yamamoto K., Yamamoto S., Kudo Y., Mohri D., Isomura Y., Ijichi H., Tateishi K., Kanai F., Ogawa S., Omata M., Koike K. (2010) Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem. Biophys. Res. Commun. 394, 1042–1046 [DOI] [PubMed] [Google Scholar]
- 25. Mehlen P., Arrigo A. P. (1994) The serum-induced phosphorylation of mammalian hsp27 correlates with changes in its intracellular localization and levels of oligomerization. Eur. J. Biochem. 221, 327–334 [DOI] [PubMed] [Google Scholar]
- 26. Kostenko S., Moens U. (2009) Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell. Mol. Life Sci. 66, 3289–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akerfelt M., Morimoto R. I., Sistonen L. (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bhargavan B., Fatma N., Chhunchha B., Singh V., Kubo E., Singh D. P. (2012) LEDGF gene silencing impairs the tumorigenicity of prostate cancer DU145 cells by abating the expression of Hsp27 and activation of the Akt/ERK signaling pathway. Cell Death Dis. 3, e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shinohara T., Singh D. P., Fatma N. (2002) LEDGF, a survival factor, activates stress-related genes. Prog. Retin. Eye Res. 21, 341–358 [DOI] [PubMed] [Google Scholar]
- 30. Gao C., Zou Z., Xu L., Moul J., Seth P., Srivastava S. (2000) p53-dependent induction of heat shock protein 27 (HSP27) expression. Int. J. Cancer 88, 191–194 [PubMed] [Google Scholar]
- 31. Costa B., Dettori D., Lorenzato A., Bardella C., Coltella N., Martino C., Cammarata C., Carmeliet P., Olivero M., Di Renzo M. F. (2010) Fumarase tumor suppressor gene and MET oncogene cooperate in upholding transformation and tumorigenesis. FASEB J. 24, 2680–2688 [DOI] [PubMed] [Google Scholar]
- 32. Jackson A. L., Linsley P. S. (2010) Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 9, 57–67 [DOI] [PubMed] [Google Scholar]
- 33. Zoubeidi A., Zardan A., Beraldi E., Fazli L., Sowery R., Rennie P., Nelson C., Gleave M. (2007) Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 67, 10455–10465 [DOI] [PubMed] [Google Scholar]
- 34. Baylot V., Andrieu C., Katsogiannou M., Taieb D., Garcia S., Giusiano S., Acunzo J., Iovanna J., Gleave M., Garrido C., Rocchi P. (2011) OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2, e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okamoto W., Okamoto I., Arao T., Kuwata K., Hatashita E., Yamaguchi H., Sakai K., Yanagihara K., Nishio K., Nakagawa K. (2012) Antitumor action of the MET tyrosine kinase inhibitor crizotinib (PF-02341066) in gastric cancer positive for MET amplification. Mol. Cancer Ther. 11, 1557–1564 [DOI] [PubMed] [Google Scholar]
- 36. Lu Y., Liang K., Li X., Fan Z. (2007) Responses of cancer cells with wild-type or tyrosine kinase domain-mutated epidermal growth factor receptor (EGFR) to EGFR-targeted therapy are linked to downregulation of hypoxia-inducible factor-1alpha. Mol. Cancer 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dunn E. F., Iida M., Myers R. A., Campbell D. A., Hintz K. A., Armstrong E. A., Li C., Wheeler D. L. (2011) Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 30, 561–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang S. H., Kang K. W., Kim K. H., Kwon B., Kim S. K., Lee H. Y., Kong S. Y., Lee E. S., Jang S. G., Yoo B. C. (2008) Upregulated HSP27 in human breast cancer cells reduces Herceptin susceptibility by increasing Her2 protein stability. BMC Cancer 8, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whitlock N. A., Agarwal N., Ma J. X., Crosson C. E. (2005) Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest. Ophthalmol. Vis. Sci. 46, 1092–1098 [DOI] [PubMed] [Google Scholar]
- 40. Eguchi R., Naitou H., Kunimasa K., Ayuzawa R., Fujimori Y., Ohashi N., Kaji K., Ohta T. (2008) Proteomic analysis of hypoxia-induced tube breakdown of an in vitro capillary model composed of HUVECs: potential role of p38-regulated reduction of HSP27. Proteomics 8, 2897–2906 [DOI] [PubMed] [Google Scholar]
- 41. Garrido C., Brunet M., Didelot C., Zermati Y., Schmitt E., Kroemer G. (2006) Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle 5, 2592–2601 [DOI] [PubMed] [Google Scholar]
- 42. Arrigo A. P. (2007) The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv. Exp. Med. Biol. 594, 14–26 [DOI] [PubMed] [Google Scholar]
- 43. Pandey P., Farber R., Nakazawa A., Kumar S., Bharti A., Nalin C., Weichselbaum R., Kufe D., Kharbanda S. (2000) Hsp27 functions as a negative regulator of cytochrome c-dependent activation of procaspase-3. Oncogene 19, 1975–1981 [DOI] [PubMed] [Google Scholar]
- 44. Andrieu C., Taieb D., Baylot V., Ettinger S., Soubeyran P., De-Thonel A., Nelson C., Garrido C., So A., Fazli L., Bladou F., Gleave M., Iovanna J. L., Rocchi P. (2010) Heat shock protein 27 confers resistance to androgen ablation and chemotherapy in prostate cancer cells through eIF4E. Oncogene 29, 1883–1896 [DOI] [PubMed] [Google Scholar]
- 45. Gibert B., Hadchity E., Czekalla A., Aloy M. T., Colas P., Rodriguez-Lafrasse C., Arrigo A. P., Diaz-Latoud C. (2011) Inhibition of heat shock protein 27 (HspB1) tumorigenic functions by peptide aptamers. Oncogene 30, 3672–3681 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.