

PHD3 influences apoptotic cell survival decisions in macrophages by altered production of Angiopoietin-like protein 2.
Keywords: hypoxia, oxygen sensing, angiopoietin-like protein, HIF
Abstract
On a molecular level, cells sense changes in oxygen availability through the PHDs, which regulate the protein stability of the α-subunit of the transcription factor HIF. Especially, PHD3 has been additionally associated with apoptotic cell death. We hypothesized that PHD3 plays a role in cell-fate decisions in macrophages. Therefore, myeloid-specific PHD3−/− mice were created and analyzed. PHD3−/− BMDM showed no altered HIF-1α or HIF-2α stabilization or increased HIF target gene expression in normoxia or hypoxia. Macrophage M1 and M2 polarization was unchanged likewise. Compared with macrophages from WT littermates, PHD3−/− BMDM exhibited a significant reduction in TUNEL-positive cells after serum withdrawal or treatment with stauro and SNAP. Under the same conditions, PHD3−/− BMDM also showed less Annexin V staining, which is representative for membrane disruption, and indicated a reduced early apoptosis. In an unbiased transcriptome screen, we found that Angptl2 expression was reduced in PHD3−/− BMDM under stress conditions. Addition of rAngptl2 rescued the antiapoptotic phenotype, demonstrating that it is involved in the PHD3-mediated response toward apoptotic stimuli in macrophages.
Introduction
The primary defense of the immune response relies on the activation of the innate immune system. Inflamed lesions often become severely hypoxic; therefore, it is quite obvious that several cross-talk mechanisms exist between inflammatory and hypoxic signaling pathways [1,2]. Like other cells, innate immune cells adapt to the resulting hypoxic conditions via inducing a specific gene-expression program. This response is mediated by the oxygen-sensitive regulation of the α-subunits of HIF-1 and -2 [3]. The regulation of HIF-α occurs at the level of protein stability through the action of the PHD proteins that are also named EGLNs alternatively [4–6]. The three described PHDs, i.e. PHD1, PHD2, and PHD3, hydroxylate the HIF-α subunit in the presence of oxygen triggering the binding of the von Hippel-Lindau tumor suppressor protein to HIF-α, which results in ubiquitination and subsequent proteasomal degradation [7, 8]. Upon a decrease in the oxygen availability, PHD hydroxylation ceases [9], and HIF-α becomes stabilized, heterodimerizes with its β-subunit, and induces hypoxia-inducible gene expression responsible for hypoxia adaptation pathways [10].
Especially, PHD3 has been analyzed regarding its hypoxic regulation of the innate immune response previously [11]. PHD3−/− neutrophils reveal a decreased survival in hypoxia compared with WT neutrophils [12]. Whereas neutrophils are primarily circulating phagocytes recruited from the blood to sites of inflammation, macrophages are found resting in the tissues. In a previous report, Kiss et al. [13] have analyzed the role of PHD3 for macrophage function using BMDM isolated and differentiated from constitutive PHD3−/− mice. They found an eminent role of PHD3 for macrophage differentiation after 5 days of differentiation, which was followed by a change in macrophage polarization and consequently, proinflammatory behavior. We have generated myeloid-specific PHD3−/− mice using a conditional LysM Cre recombinase-mediated strategy. In the conditional PHD3−/− BMDM, we could verify a faster macrophage differentiation, up to 5 days of cell culture under differentiation conditions. Eight days after onset of differentiation, however, the difference was no longer detectable, and WT and PHD3−/− BMDM were equally differentiated. Similar to neutrophils, PHD3 had a significant impact on cell survival in fully differentiated macrophages. In the present study, this could be associated with an altered endogenous production of Angptl2.
MATERIALS AND METHODS
Myeloid-specific conditional PHD3−/− mice
The generation and detailed characterization of Phd3flox/flox mice are reported previously [14]. The mice were kindly provided by Prof. Guo-Hua Fong (Center for Vascular Biology, University of Connecticut Health Center, Farmington, CT, USA). All animals in this study were backcrossed to C57BL/6 mice, at least five times. Phd3flox/flox mice were crossed with LysMcre mice [15] to generate Phd3flox/flox × LysMcre+/− mice within two generations. Phd3flox/flox × LysMcre+/− mice were then crossed with Phd3flox/flox mice to obtain PHD3−/− mice (Phd3flox/flox×LysMcre+/−) and littermate control WT mice (Phd3flox/flox).
Differentiation and cultivation of BMDM
Mice were killed by cervical dislocation at the age of 8–12 weeks. Bone marrow cells were isolated from the femur and cultured on cell culture dishes in Pluznik medium [DMEM supplemented with 0.2 mM L-glutamine, 0.1 mM sodium pyruvate, 50 U/mL penicillin G, 50 μg/mL streptomycin, 10% heat-inactivated FCS, and 5% heat-inactivated horse serum (Pan Biotech, Aidenbach, Germany); 0.05% 1:1000 diluted β-ME (Carl Roth GmbH, Karlsruhe, Germany); and 15% L929 cell-conditioned medium] at 37°C and 5% CO2 in a humidified incubator [16]. After 24 h, nonadherent monocytes were harvested, seeded in Pluznik medium, and differentiated for 3–8 days. Adherent BMDM were detached with 3.5 mL accutase (PAA Laboratories, Cölbe, Germany) and resuspended in culture medium (DMEM supplemented with, 0.2 mM L-glutamine, 0.1 mM sodium pyruvate, 1 mM HEPES, 50 U/mL penicillin G, 50 μg/mL streptomycin, and 10% heat-inactivated FCS).
RNA extraction and qRT-PCR analyses
For RNA isolation, 0.7 × 106 BMDM were seeded in 2 mL culture medium in six-well plates and incubated for 4, 24, and 48 h at 37°C and 5% CO2 in normoxia (21% O2) or hypoxia (1% O2) using the INVIVO 400 work station (IUL Instruments, Königswinter, Germany). For M1/M2 polarization, cells were stimulated with 100 ng/mL LPS (Enzo Life Sciences, Lörrach, Germany) and 20 nM IFN-γ or 20 nM IL-4 (Peprotech, Hamburg, Germany). Cells were washed once with PBS and harvested in Trizol (Invitrogen, Darmstadt, Germany). RNA was isolated, according to the manufacturer's instructions, and 1 μg was transcribed using the First Strand cDNA Synthesis Kit (Fermentas, St. Leon-Rot, Germany). Transcript levels were analyzed by qRT-PCR, amplifying 1 μl cDNA with Brilliant II SYBR Green qPCR Master Mix in an MX3005Pro light cycler (Bioline, Luckenwalde, Germany). Primer sequences are listed in Supplemental Table 1.
Protein extraction and immunoblot analyses
BMDM were seeded at a density of 4 × 106 cells in 10 mL culture medium in 10 cm dishes and were cultivated for 4 or 24 h in 21% O2 or 1% O2. For Western blot analysis, proteins were extracted with 400 mM NaCl, 10 mM Tris HCl, pH 8, 1 mM EDTA, pH 8, 0.1% Triton X-100, supplemented with protease inhibitors (Roche Diagnostics, Mannheim, Germany). Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad, Munich, Germany). Proteins were separated via SDS-PAGE and transferred onto nitrocellulose membranes (GE Healthcare, Freiburg, Germany) by semidry blotting (Peqlab, Erlangen, Germany). Proteins were detected using the following primary and secondary HRP-labeled antibodies: anti-β-actin (#A5441; Sigma-Aldrich, Munich, Germany), anti-HIF-1α (#NB-100–479; Novus Biologicals, Littleton, CO, USA), anti-HIF-2α (AF 2997; R&D Systems, Minneapolis, MN, USA), anti-PHD2 (#NB 100–2219; Novus Biologicals), anti-PHD3 (#NB100-303; Novus Biologicals), HRP-labeled anti-mouse (sc-2005; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and HRP-labeled anti-rabbit (sc-2004; Santa Cruz Biotechnology). HRP-mediated chemiluminescence was induced by incubating the membranes in ECL (Merck Millipore, Darmstadt, Germany) for 1 min and subsequently, detected with LAS3000 (Fujifilm, Düsseldorf, Germany) or using chemiluminescence X-ray films (Sigma-Aldrich).
Calcein-AM cell viability assay
BMDM (0.5×105) were seeded in 24-well plates in 500 μl culture medium for 48 h, with or without serum. Subsequently, cell viability was determined by the cell‘s ability to hydrolyze calcein-AM using the calcein-AM cell viability assay (Trevigen, Gaithersburg, MD, USA), according to the manufacturer's recommendations.
TUNEL
BMDM (0.5×106) were seeded in six-well plates on coverslips in 2 mL culture medium for 48 h, with or without serum. After washing with PBS, cells were fixed using 4% paraformaldehyde for 20 min. Cells were permeabilized using 0.1% Triton X-100 in freshly prepared 0.1% sodium citrate (Carl Roth GmbH) for 2 min on ice. The cells were then stained for 60 min at 37°C in a dark and moist chamber using enzyme and labeling solution provided by the In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics). Cells treated with DNAse, served as positive control. The cells were then washed with PBS and were imaged using a fluorescent microscope (Axio Observer D1; Zeiss, Germany).
Annexin V staining
BMDM were seeded in 35-mm dishes in culture medium, with and without serum. After 48 h, the cells were detached using accutase (PAA Laboratories) for 15 min, and cells were counted. Cells (1.5×106)/sample were analyzed subsequently. After washing with PBS, cells were stained with Annexin V (BioLegend, London, UK) and propidium iodide (BioLegend) for 20 min at 4°C. Then, the cells were resuspended in Annexin V binding buffer (BioLegend) and were anlyzed using the BD FACSCanto flow cytometer (BD Biosciences, San Jose, CA, USA). In some experiments, cells were treated with 100 μM SNAP (sc-200319; Santa Cruz Biotechnology), 500 nM stauro (ALX-380-014-M001; Enzo Life Sciences), or 5–10 μg/mL LPS for 12 h.
Flow cytometry
After washing with PBS, BMDM were stained with anti-PirA/B, anti-F4/80, anti-integrin α5, and anti-integrin β1 (all BioLegend); anti-CD11b (BD Biosciences, Heidelberg, Germany); or the respective control antibodies, rat IgGk1, rat IgG2a, and IgG (all from BioLegend) for 20 min at 4°C. After washing with PBS, cells were resuspended and analyzed using the BD FACSCanto flow cytometer (BD Biosciences, San Jose, CA, USA).
Cytometric bead array
For analysis of IL-6 and TNF-α protein levels, supernatants were processed according to the BD Cytometric Bead Array Mouse Inflammation Kit instructions. Subsequently, the data were analyzed using BD FCAP Array software, version 3.0.
rAngptl2
The mature form of human rAngptl2 hexahisitidine-tagged protein was expressed in Escherichia coli Rosetta pLacI cells (Merck Millipore) as an inclusion body. The details of the preparation methods will be described elsewhere. In brief, the inclusion body was solubilized, reduced, and modified by TAPS-sulfonate (Wako Pure Chemicals, Osaka, Japan), according to the published procedure [17], with minor modifications. The TAPS-modified proteins were desalted on the Sephadex G-25 column (GE Healthcare, Tokyo, Japan). The desalted proteins were loaded onto a Talon column (Takarabio-Clontech, Otsu, Japan) and eluted with 0.15 M imidazole after washing the column with the solubilizing buffer. The eluted sample was desalted again, and a portion of the sample was diluted into a refolding buffer (2 μg/ml final concentration) containing 2 mM cysteine and 0.5 mM cystine at 4°C for 14 h. The proteins were adsorbed to a Source 30 reverse-phase matrix (GE Healthcare, Japan) and eluted with acetonitrile containing 0.04% trifluoracetic acid. The eluate was freeze-dried, dissolved in 0.1% acetic acid, and stored at −80°C. A part of the above-prepared Angptl2 protein was highly oligomerized with an extended conformation in solution; however, it showed better biological activity of a macrophage migration than that prepared from mammalian cells [18].
NF-κB activity
NF-κB activity was determined using the chemiluminescence NF-κB transcription factor assay kit from (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer's instructions.
Transcriptome screen
BMDM (0.7×106) were seeded in 2 mL culture medium in six-well plates and incubated for 48 h at 37°C and 5% CO2 in 21% O2 or 1% O2. Cells were washed once with PBS and harvested in Trizol (Invitrogen). RNA was isolated, according to the manufacturer's instructions, and 1 μg RNA was transcribed using the First Strand cDNA Synthesis Kit (Fermentas). RNA integrity was determined with the Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA was then added to the Spike mix, provided with the One-Color Microarray-Based Gene Expression analysis kit (Agilent Technologies). Samples were then labeled according to the Low Input Quick Amp Labeling kit one-color protocol (Agilent Technologies). cDNA was synthesized from the labeled mRNA. aRNA was then synthesized using the T7 RNA polymerase enzyme. Quantification of the labeled aRNA was done before hybridization onto the microarray slides. The hybridization was then carried out on the Agilent microarray hybridization slide at 65°C for 17 h. All subsequent steps were followed as instructed by the Low Input Quick Amp Labeling protocol.
Angptl2 ELISA
Angptl2 levels in cell culture supernatants were determined by ELISA (USCN Life Science, Wuhan, China), according to the manufacturer's instructions.
Statistical analyses
Statistical analyses were performed using Student‘s two-tailed t-test. Data are shown as means ± sd. Values of P < 0.05 were considered statistically significant.
RESULTS
Generation of myeloid-specific PHD3−/− mice
PHD3flox/flox mice, in which exon 2 is flanked by loxP sites, were crossed with LysM Cre mice to delete PHD3 in the myeloid cell lineage. Bone marrow was isolated from LysM Cre × PHD3flox/flox (referred to as PHD3−/−) and WT mice and differentiated for 8 days. Bone marrow cells of both genotypes differentiated to mature macrophages without any obvious difference in cell morphology (Fig. 1A). As demonstrated earlier by Kiss et al. [13], the macrophage differentiation process of bone marrow cells was altered in WT and PHD3−/− cells over time. PHD3−/− bone marrow cells showed under differentiation conditions at earlier time-points, i.e., Days 3–5, elevated levels for markers, indicative of macrophage differentiation, such as F4/80 and CD11b protein levels (Fig. 1B). Eight days after starting the differentiation, the difference was no longer detectable, and cells of both genotypes were fully differentiated. If not stated otherwise, for all follow-up experiments, cells that were differentiated for 8 days were used. PHD3−/− BMDM showed a 75% and 60% reduction of PHD3 RNA levels after culturing the cells for 24 h at normoxia or hypoxia, respectively (Fig. 1C). There was no compensatory change in the RNA levels of PHD1 or PHD2.
Figure 1. Characterization of myeloid-specific PHD3−/− mice.
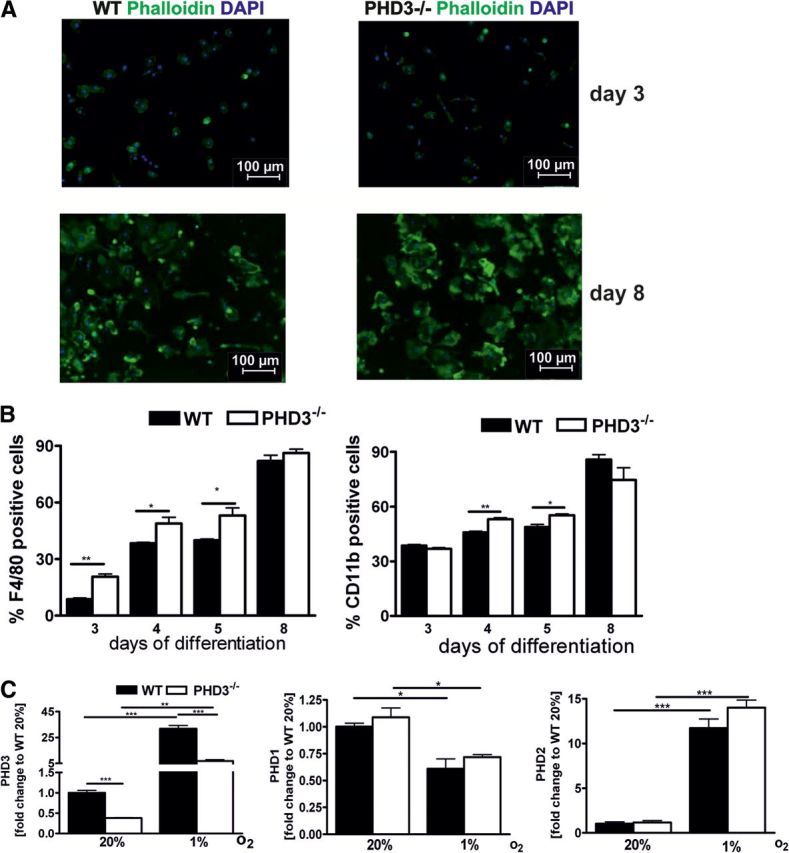
(A) Macrophages were differentiated from bone marrow cells of 8- to 12-week-old WT and PHD3−/− mice using M-CSF-containing conditioned medium over 3–8 days. PHD3−/− in myeloid cells did not affect the morphology of the cells, which was analyzed by staining the cells with phalloidin and 4′,6′-diamidino-2-phenylidole (DAPI). (B) PHD3−/− in myeloid cells promoted the differentiation of BMDM, as shown by F4/80 and CD11b staining, using FACS analysis. (C) PHD1, PHD2, and PHD3 RNA levels were quantified in BMDM isolated from WT and PHD3−/− mice and exposed to 20% or 1% O2 for 24 h; n = 3; mean ± sd; *P < 0.05, **P < 0.01, and ***P < 0.001.
A significant decrease of PHD3 was also seen at the protein level in the PHD3−/− BMDM (Fig. 2A). This difference was more obvious after incubating the cells for 24 h at hypoxia compared with normoxia, based on the hypoxic induction of PHD3. PHD3−/− did not result in a significant change in PHD2, HIF-1α, and HIF-2α protein levels. Further evidence that HIF-α stabilization and HIF activity were not affected by PHD3−/− was given by the fact that the normoxic expression and hypoxic induction of the HIF target genes pyruvate kinase M2 (PKM2), Pfk, or PDK-1 were unaltered in the PHD3−/− BMDM (Fig. 2B). In an unbiased transcriptome approach, we could indeed not find any significantly differentially expressed gene comparing WT with PHD3−/− BMDM in normoxia and hypoxia (Fig. 2C). There is evidence that apart from HIF-α, under specific circumstances, PHDs can additionally regulate NF-κB activity [19, 20]. In the PHD3−/− BMDM, however, there was no obvious difference in NF-κB activity compared with the WT cell under resting conditions or after serum starvation (Fig. 2D). After stimulation with LPS, however, the WT BMDM demonstrated higher NF-κB activity compared with the PHD3−/− BMDM, which is in line with the above-referenced work that has uncovered mechanisms by which hypoxia modulates the activation of NF-κB through decreased oxygen-dependent suppression of the key regulators of this pathway [19, 20].
Figure 2. HIF-dependent target gene expression is unchanged in PHD3−/− BMDM.

(A) HIF-1α, HIF-2α, PHD2, and PHD3 protein levels were analyzed after cultivating BMDM for 24 h at 20% or 1% O2. (B) The expression of HIF-1 target genes, such as PKM2, Pfk, and PDK-1, on mRNA level was analyzed after cultivating BMDM for 24 h at 20% O2 or 1% O2 using qRT-PCR. (C) PHD3−/− [knockout (KO)] and WT BMDM were cultivated for 48 h at 20% O2 and 1% O2. Subsequently, cells were lysed and RNA isolated. RNA samples were analyzed in a transcriptome microarray assay. The volcano blots were generated based on the microarray results. (D) PHD3−/− and WT BMDM were cultured with or without serum for 24 h or stimulated with 100 ng LPS for 12 h. Subsequently, NF-κB activities were analyzed. As a positive control for the NF-κB activity assay, a cell extract of HeLa cells, which were treated with LPS, was used; n = 3; mean ± sd; **P < 0.01, and ***P < 0.001. rel. LU, Relative light units.
Macrophage polarization is unaffected in fully differentiated PHD3−/− BMDM
Macrophages display remarkable plasticity and can change their phenotype upon stimulation [21]. The most prominent macrophage populations are the so-called M1- and M2-polarized macrophages. M1-polarized macrophages are proinflammatory and tissue-destructive, whereas M2-polarized macrophages are anti-inflammatory and involved in tissue remodeling, as well as angiogenesis. Macrophage polarization can be affected by HIF-1α and HIF-2α [22]. To analyze whether PHD3 affects macrophage polarization, we treated WT and PHD3−/− BMDM, which were differentiated for 8 days, with LPS and the T helper cell 1 cytokine IFN-γ or the Th2 cytokine IL-4 to characterize functionally M1 and M2 polarization, respectively (Fig. 3A and B). Successful M1 and M2 polarization after stimulation was verified by increased RNA levels of the M1 markers IL-6, iNOS, and TNF-α, as well as the M2 markers arginase, Ym-1, and FIZZ, respectively. Expression levels of the M1 and M2 marker RNAs were not different comparing WT with PHD3−/− BMDM. This was additionally verified by determining IL-6 and TNF-α protein levels in the cell culture supernatants after stimulation with LPS + IFN-γ (Fig. 3C). In line with a previous report [13] and the above-described, altered differentiation time course of PHD3−/− BMDM, Day 5-differentiated PHD3−/− BMDM, however, showed increased TNF-α and IL-6 RNA levels after stimulation with LPS compared with WT cells (Fig. 3D).
Figure 3. Macrophage polarization is unaffected in the PHD3−/− BMDM.

PHD3−/− and WT BMDM, which were differentiated for 8 days, were stimulated with LPS + IFN-γ (M1 polarization) or IL-4 (M2 polarization) for 6 h; subsequently, RNA levels for the (A) M1-polarization markers IL-6, iNOS, and TNF-α and (B) the M2-polarization markers arginase, Ym-1, and FIZZ were quantified by qRT PCR. (C) PHD3−/− and WT BMDM cell culture supernatants were harvested after stimulation of the cells with LPS + IFN-γ or IL-4 for 6 h. IL-6 and TNF-α protein levels were detected by FACS, as described in Materials and Methods. (D) BMDM, which were differentiated for 5 days, were stimulated with LPS for 6 h, and TNF-α and IL-6 protein levels were determined in the cell culture supernatants; n = 3; mean ± sd; *P < 0.05, **P < 0.01, and ***P < 0.001.
PHD3−/− BMDM are protected from apoptotic cell death
PHD3 has been associated with cell-survival decisions in the context of apoptosis in various cell and animal models [23–25]. Therefore, we next determined the role of PHD3 in macrophage cell-survival pathways. To analyze, if apoptosis is altered in the PHD3−/− BMDM, we deprived cells, which were differentiated for 8 days before starting the experiment, for 48 h from serum. The starvation of the cells induced apoptosis in WT BMDM, as determined by quantifying TUNEL and Annexin V-positive cells (Fig. 4A). PHD3−/− BMDM were partially protected. This was also reflected by an increased cell survival, 48 h after starvation, determined by calcein-AM assays (Fig. 4B). The antiapoptotic effect was not a result of the fact that we differentiated the BMDM with L929 conditioned medium. PHD3−/− BMDM, which were differentiated for 8 days with rM-CSF also demonstrated a significantly decreased PHD3 expression and an antiapoptotic effect after serum starvation (Supplemental Fig. 1). To analyze if the PHD3-mediated phenomenon is just limited to serum deprivation, macrophages were cultured in the presence of the known proapoptotic agents SNAP (100 μM), stauro (500 nM), or LPS (5 or 10 μg/mL), and rates of apoptosis were determined (Fig. 4C). Protection from apoptosis was not restricted to serum deprivation, as we also detected a lower number of Annexin V-positive PHD3−/− BMDM after treating the cells with SNAP or stauro and to a minor extent, LPS. To analyze the impact of hypoxia on serum starvation-induced apoptosis, we exposed WT and PHD3−/− BMDM to normoxia or hypoxia during serum starvation (Fig. 4D). In both conditions, PHD3−/− BMDM were protected significantly from apoptosis compared with WT macrophages. In hypoxia, however, the protective effect was slightly but not significantly weakened in the PHD3−/− BMDM. This correlates to the hypoxic induction of PHD3, as demonstrated above, which was present, albeit less significantly in the PHD3−/− macrophages.
Figure 4. PHD3−/− BMDM are protected from apoptotic cell death.

(A) PHD3−/− and WT BMDM were cultivated, with or without serum (starved). After 48 h, the number of apoptotic cells was quantified by TUNEL and Annexin V (AV) stainings. (B) Forty-eight hours after starvation, cell viability was determined using the calcein-AM viability assay. (C) The number of Annexin V-positive cells was quantified after cultivating PHD3−/− and WT BMDM, with or without serum, for 48 h or treating the cells with 100 μM SNAP, 500 nM stauro, or 5–10 μg/mL LPS for 12 h. (D) The number of Annexin V-positive cells was quantified after cultivating PHD3−/− and WT BMDM, with or without serum, for 48 h at 20% O2 or 1% O2; n = 5 for Annexin V stainings; all others, n = 3; mean ± sd; *P < 0.05, **P < 0.01, and ***P < 0.001.
To gain insight into the apoptotic phenotype, an unbiased transcriptome analysis was performed. With the use of a mouse genome array, we compared transcript abundance in WT with PHD3−/− BMDM deprived of serum for 48 h. Among the transcript changes, which differed in their extent in response to the serum deprivation between WT and PHD3−/− BMDM, we identified, in total, 19 transcripts that were down-regulated (Table 1) and 26 transcripts that were up-regulated (Table 2). The treatment of WT and PHD3−/− BMDM during serum deprivation with conditioned medium of serum-deprived PHD3−/− and WT cells, respectively, revealed a loss of the antiapoptotic phenotype in the PHD3−/− BMDM (Fig. 5A). This indicated that, at least in part, a secreted factor is involved in the PHD3-mediated response toward an apoptotic stimulus. Further gene cluster analysis of the 45 genes identified in the transcriptome array revealed a cluster of genes (i.e., Angptl2, Col8A1, Col5A1, Lys-Asp-Glu-Leu endoplasmic reticulum protein retention receptor 3, myosin regulatory light polypeptide 9, and peroxidasin homolog), which are known to be coregulated.
Table 1. Genes Reduced under Starvation and Differentially Regulated in PHD3−/− Versus WT BMDM.
| Gene ID | Symbol | Description |
|---|---|---|
| 20511 | Slc1a2 | Solute carrier family 1 (glial high-affinity glutamate transporter), member 2 |
| 69590 | Gpx8 | Glutathione peroxidase 8 (putative) |
| 12035 | Bcat1 | Branched chain aminotransferase 1, cytosolic |
| 17472 | Gbp4 | Guanylate-binding protein 4 |
| 64074 | Smoc2 | Secreted acidic cysteine-rich glycoprotein-related modular calcium-binding 2 |
| 20350 | Sema3f | Sema domain, Ig domain, short basic domain, secreted (semaphorin), 3F |
| 109264 | Me3 | Malic enzyme 3, NADP(+)-dependent, mitochondrial |
| 241230 | St8sia6 | ST8 α-N-acetyl-neuraminide α-2,8-sialyltransferase 6 |
| 105785 | Kdelr3 | Lys-Asp-Glu-Leu endoplasmic reticulum protein retention receptor 3 |
| 16429 | Itln1 | Intelectin 1 (galactofuranose-binding) |
| 12478 | Cd19 | CD19 antigen |
| 12837 | Col8a1 | Collagen, type VIII, α 1 |
| 69675 | Pxdn | Peroxidasin homolog (Drosophila) |
| 20692 | Sparc | Secreted acidic cysteine-rich glycoprotein |
| 20204 | Prrx2 | Paired, related homeobox 2 |
| 12831 | Col5a1 | Collagen, type V, α 1 |
| 22402 | Wisp1 | WNT1-inducible signaling pathway protein 1 |
| 98932 | Myl9 | Myosin, light polypeptide 9, regulatory |
| 18054 | Ngp | Neutrophilic granule protein |
Table 2. Genes Induced under Starvation and Differentially Regulated in PHD3−/− Versus WT BMDM.
| Gene ID | Symbol | Description |
|---|---|---|
| 56473 | Fads2 | Fatty acid desaturase 2 |
| 17392 | Mmp3 | Matrix metallopeptidase 3 |
| 14990 | H2-M2 | Histocompatibility 2, M region locus 2 |
| 26360 | Angptl2 | Angiopoietin-like 2 |
| 76009 | 5830415G21Rik | RIKEN cDNA 5830415G21 gene |
| 14858 | Gsta2 | GST, α 2 (Yc2) |
| 246049 | Slc36a2 | Solute carrier family 36 (proton/amino acid symporter), member 2 |
| 14238 | Foxf2 | Forkhead box F2 |
| 68027 | Tmem178 | Transmembrane protein 178 |
| 18548 | Pcsk1 | Proprotein convertase subtilisin/kexin type 1 |
| 17384 | Mmp10 | Matrix metallopeptidase 10 |
| 20408 | Sh3gl3 | Src homology 3 domain growth factor receptor-bound protein 2-like 3 |
| 19746 | Rhd | Rhesus blood group, D antigen |
| 13051 | Cx3cr1 | Chemokine (C-X3-C) receptor 1 |
| 19124 | Procr | Protein C receptor, endothelial |
| 15490 | Hsd17b7 | Hydroxysteroid (17-β) dehydrogenase 7 |
| 207683 | Igsf11 | Ig superfamily, member 11 |
| 16197 | Il7r | IL-7R |
| 79201 | Tnfrsf23 | TNFR superfamily, member 23 |
| 56018 | Stard10 | Steroidogenic acute regulatory protein-related lipid-transfer domain-containing 10 |
| 228543 | Rhov | Ras homolog gene family, member V |
| 217265 | Abca5 | ATP-binding cassette, subfamily A (ABC1), member 5 |
| 12614 | Celsr1 | Cadherin, epidermal growth factor LAG seven-pass G-type receptor 1 (flamingo homolog, Drosophila) |
| 627270 | Gm10055 | Predicted gene 10055 |
| 320121 | 6720473M08Rik | RIKEN cDNA 6720473M08 gene |
| 194126 | Mtmr11 | Myotubularin-related protein 11 |
Figure 5. Angptl2 is differentially expressed in PHD3−/− and WT BMDM and involved in the differential apoptotic response.

(A) PHD3−/− and WT BMDM were cultivated, with or without serum, in total for 48 h. After 24 h, the medium was exchanged across the genotypes and cultured for an additional 24 h. Induction of apoptosis was compared using Annexin V staining and FACS analysis; n = 5; mean ± sd. PHD3−/− and WT BMDM were cultivated, with or without serum, for 24 h. Subsequently Angptl2 mRNA (B) and protein (C) levels were analyzed; n = 6; mean ± sd. (D) PHD3−/− and WT BMDM were cultivated, with or without serum, for 24 h at 20% or 1% O2. Subsequently, Angptl2 mRNA levels were analyzed. (E) Angptl2−/− and WT BMDM were cultivated, with or without serum, for 48 h or exposed to 500 nM stauro for 12 h. Subsequently, induction of apoptosis was compared using Annexin V staining and FACS analysis; n = 4–5; mean ± sd; *P < 0.05, **P < 0.01, and ***P < 0.001.
Angptl are secreted proteins that like angiopoietin itself, have an N-terminal coiled-coil domain and a C-terminal fibrinogen-like domain. So far, seven Angptls have been identified. None of these bind to the angiopoietin receptor Tie2 or to the homologous Tie1 receptor. Angptl2 has been found to affect vascular cells and monocytes. In line, Angptl2 functions as a chronic inflammatory mediator in obesity [26], atherosclerotic disease [18], rheumatoid arthritis [27], and cancer [28]. Although the receptor signaling of Angptl2 is not fully understood, some of these biological effects seem to be, at least in part, mediated via binding to the α5β1 integrin and the PirB or human orthologue LILERB2 [26, 29]. The differential expression of Angptl2 in WT and PHD3−/− BMDM was verified by independent qRT-PCR and ELISA analyses (Fig. 5B and C). Exposure of macrophages to hypoxia during starvation significantly increased the expression of Angptl2 in the WT but not in the PHD3−/− BMDM (Fig. 5D). In addition, we found increased Angptl2 RNA levels after treating WT and PHD3−/− BMDM with SNAP or stauro. As in starvation, the expression was less in the PHD3−/− BMDM. In line with the antiapoptotic effect and diminished expression of Angptl2 in the PHD3−/− cells, BMDM, which were isolated and differentiated from previously described Angptl2−/− mice [26], exhibited less AV-positive cells after serum starvation or stauro treatment (Fig. 5E). Reconstitution of the diminished Angptl2 response by adding 2 or 4 μg rAngptl2 during starvation induced apoptosis in the PHD3−/− BMDM (Fig. 6A). This rescue of the antiapoptotic phenotype demonstrates that Angptl2 is, at least partially, involved in the PHD3-mediated response toward an apoptotic stimulus. Most interestingly, WT BMDM did not respond with increased apoptosis after adding 2 μg rAngptl2 under resting conditions or after starvation. After adding 4 μg rAngptl2, there was even an antiapoptotic effect after starvation and treatment with SNAP or stauro, which is in sharp contrast to the PHD3−/− BMDM (Fig. 6B). This was not a result of an altered expression of the up-to-now described receptors for Angptl2, i.e., PirB and α5β1 integrin, in the WT and PHD3−/− BMDM (Fig. 6C).
Figure 6. Treatment of BMDM with rAngptl2.

PHD3−/− and WT BMDM were (A) cultivated, with or without serum, in total for 48 h or (B) were treated with 100 μM SNAP or 500 nM stauro for 12 h. rAngptl2 was added as indicated. Induction of apoptosis was compared using Annexin V staining and FACS analysis. (C) PHD3−/− and WT BMDM were analyzed for α5β1 and PirB protein levels by FACS staining; n = 5; mean ± sd; **P < 0.01, and ***P < 0.001.
DISCUSSION
With the use of a conditional knockout strategy, we created myeloid-specific PHD3−/− mice. The knockout was not compensated by an altered expression of the two other HIF-regulating PHDs, i.e. PHD1 and PHD2, in BMDM. In addition, the knockout was not followed by increased HIF-1α or HIF-2α protein levels in normoxia and hypoxia. This finding was substantiated further by the fact that the HIF target gene expression was unaltered in the PHD3−/− BMDM compared with WT cells. PHD3 is strikingly induced by hypoxia in various models, including myeloid cells [12, 30, 31]. This could also be observed in the BMDM in our study. The hypoxic induction serves to limit the response to hypoxia by compensating the decreased oxygen levels [32, 33]. In a previous report, PHD3−/− BMDM, isolated from WT or constitutive PHD3−/− mice, demonstrated increased HIF-1α protein levels in hypoxia in the deficient macrophages [13]. HIF target gene expression was unfortunately not analyzed. The apparent difference to our results might relate to the constitutive versus conditional knockout models applied. The constitutive PHD3−/− macrophages demonstrated, as expected, a 100% knockout efficiency, whereas we detected in our conditional knockout strategy a PHD3−/− efficiency of ∼75%. PHD enzymes are currently tested for their potential to be used as drug targets to stimulate HIF-dependent gene expression [34]. This strategy is developed for applications in the context of ischemic tissue protection and severe anemia [35]. As pharmacological inhibition of the PHD enzymes will be similar to the conditional knockout efficiency, our data my help to predict the consequences of such a treatment regimen.
Most strikingly, the conditional PHD3−/− BMDM cells analyzed in the present study displayed a strong apoptotic phenotype. A major impact of PHD3 on cell survival of myeloid cells is in line with the findings that PHD3−/− neutrophils are not altered significantly regarding their proinflammatory function but regarding their apoptotic phenotype [12]. PHD3 has been linked to important regulatory, proapoptotic functions also in other cell types [36–38]. At the molecular level, this has been associated with a PHD3-dependent regulation of KIF1β, HCLK2, SIVA, and ATF-4 [12, 39–41]. None of these candidates were up- or down-regulated in the unbiased transcriptome screen comparing starved WT and PHD3−/− BMDM. Instead, we found a significant difference in the starvation-induced Angptl2 RNA and protein levels. Angptl represent a multifunctional family of secreted factors with structural similarity to angiopoietin, a molecule with proangiogenic function [42]. This family is characterized by a coiled-coil domain in the N-terminus and fibrinogen-like domain in the C-terminus. These factors probably do not signal through the Tie2 receptor, but it has been reported recently that the LILERB2 and PirB are receptors for several Angptl family members [29, 43]. Angptl2 has been reported to have myriad functions that are involved in inflammation, metabolism, angiogenesis, and cancer [18, 27, 44].
Our data demonstrate that PHD3 affects the production of Angptl2 and additionally influences the response toward this apoptosis-modulating factor. Addition of Angptl2, under serum starvation, resulted in an antiapoptotic effect in the WT BMDM. Taking into account the high induction of Angptl2 after serum starvation in WT BMDM, this can be interpreted as activation of a protective feedback mechanism. The antiapoptotic function of Angptl2 is in line with previous reports in endothelial cells [45]. Most interestingly, in PHD3−/− BMDM, the response was reverted to a proapoptotic effect. Angptl2 treatment increased the serum starvation-induced cell death of PHD3−/− macrophages but did not affect cell viability under normal cell culture conditions. This implies that Angptl2 is not, per se, a proapoptotic factor but interferes with the apoptosis pathway after the application of a stress stimulus. An impact of Angptl2 on serum-induced starvation was additionally proven using Angptl2−/− BMDM. Along this line, the decreased Angptl2 expression in PHD3−/− macrophages under serum starvation mirrors the shutdown of a death-supporting factor. Our data show, for the first time, a functional connection of PHD3 and Angptl2, as well as the underlying pathways.
Taken collectively, our data demonstrate that PHD3 plays a critical role for cell-survival decisions in macrophages. This includes apoptotic cell death induced by serum starvation, which mimics, in part, ischemic conditions but also treatment with stauro or the endogenous inducers of apoptosis, such as the NO donor SNAP or LPS. The antiapoptotic effect in the PHD3−/− BMDM is, at least partially, mediated by an altered production and response to Angptl2. As macrophages are responsible for the early immune response, our data imply that PHD3 may shape this response by altering the life expectancies of macrophages in inflamed and ischemic tissues.
Supplementary Material
ACKNOWLEDGMENTS
L.S. is a fellow of the International Research Training Group 1816 (GRK1816), funded by the Deutsche Forschungsgemeinschaft (DFG), and additionally funded by the Dorothea Schlözer program of the Georg August University Göttingen.
The authors thank the members of the Transcriptome Analysis Laboratory (TAL), Microarray and Deep Sequencing Core Facility, University Medical Center Göttingen for performing the microarray experiments.
SEE CORRESPONDING EDITORIAL ON PAGE 359
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- Angptl2
- angiopoietin-like protein 2
- Angptl2−/−
- Angptl2 knockout
- aRNA
- antisense RNA
- BMDM
- bone marrow-derived macrophages
- Col5a1
- collagen type V alpha1
- Col8a1
- collagen type VIII alpha1
- FIZZ
- found in inflammatory zone
- HIF
- hypoxia-inducible factor
- LILERB2
- leukocyte Ig-like receptor B2
- LysM
- lysozyme M
- PDK-1
- pyruvate dehydrogenase kinase-1
- Pfk
- phosphofructokinase
- PHD
- prolyl-4-hydroxylase domain
- PHD3−/−
- prolyl-4-hydroxylase domain 3 knockout
- PirB
- paired Ig-like receptor B
- PKM2
- pyruvate kinase M2
- qRT-PCR
- quantitative RT-PCR
- SNAP
- s-nitroso-N-acetylpenicillamine
- stauro
- staurosporine
- TAPS
- 3-trimethylammoniopropyl methanethiosulphonate bromide
- YM1
- secreted protein YM1
AUTHORSHIP
L.S. performed experiments, analyzed data, and wrote the manuscript. M.W. and K.F. performed experiments and analyzed data. A.H., A.B., H.O., K.T., and M.E. performed experiments. Y.O. performed experiments and provided important advice. D.M.K. analyzed data, directed the project, and wrote the manuscript.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Taylor C. T. (2008) Interdependent roles for hypoxia inducible factor and nuclear factor-κB in hypoxic inflammation. J. Physiol. 586, 4055–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Imtiyaz H. Z., Simon M. C. (2010) Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 345, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang G. L., Jiang B. H., Rue E. A., Semenza G. L. (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 92, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruick R. K., McKnight S. L. (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340. [DOI] [PubMed] [Google Scholar]
- 5. Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54. [DOI] [PubMed] [Google Scholar]
- 6. Lando D., Peet D. J., Gorman J. J., Whelan D. A., Whitelaw M. L., Bruick R. K. (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16, 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. [DOI] [PubMed] [Google Scholar]
- 8. Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J. M., Lane W. S., Kaelin W. G., Jr., (2001) HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. [DOI] [PubMed] [Google Scholar]
- 9. Hirsilä M., Koivunen P., Gunzler V., Kivirikko K. I., Myllyharju J. (2003) Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 278, 30772–30780. [DOI] [PubMed] [Google Scholar]
- 10. Wenger R. H., Stiehl D. P., Camenisch G. (2005) Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, re12. [DOI] [PubMed] [Google Scholar]
- 11. Thompson A. A., Binham J., Plant T., Whyte M. K., Walmsley S. R. (2013) Hypoxia, the HIF pathway and neutrophilic inflammatory responses. Biol. Chem. 394, 471–477. [DOI] [PubMed] [Google Scholar]
- 12. Walmsley S. R., Chilvers E. R., Thompson A. A., Vaughan K., Marriott H. M., Parker L. C., Shaw G., Parmar S., Schneider M., Sabroe I., Dockrell D. H., Milo M., Taylor C. T., Johnson R. S., Pugh C. W., Ratcliffe P. J., Maxwell P. H., Carmeliet P., Whyte M. K. (2011) Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J. Clin. Invest. 121, 1053–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kiss J., Mollenhauer M., Walmsley S. R., Kirchberg J., Radhakrishnan P., Niemietz T., Dudda J., Steinert G., Whyte M. K., Carmeliet P., Mazzone M., Weitz J., Schneider M. (2012) Loss of the oxygen sensor PHD3 enhances the innate immune response to abdominal sepsis. J. Immunol. 189, 1955–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeda K., Ho V. C., Takeda H., Duan L. J., Nagy A., Fong G. H. (2006) Placental but not heart defects are associated with elevated hypoxia-inducible factor a levels in mice lacking prolyl hydroxylase domain protein 2. Mol. Cell. Biol. 26, 8336–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Forster I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Trans. Res. 8, 265–277. [DOI] [PubMed] [Google Scholar]
- 16. Burgess A. W., Metcalf D., Kozka I. J., Simpson R. J., Vairo G., Hamilton J. A., Nice E. C. (1985) Purification of two forms of colony-stimulating factor from mouse L-cell-conditioned medium. J. Biol. Chem. 260, 16004–16011. [PubMed] [Google Scholar]
- 17. Terzyan S. S., Peracaula R., de Llorens R., Tsushima Y., Yamada H., Seno M., Gomis-Ruth F. X., Coll M. (1999) The three-dimensional structure of human RNase 4, unliganded and complexed with d(Up), reveals the basis for its uridine selectivity. J. Mol. Biol. 285, 205–214. [DOI] [PubMed] [Google Scholar]
- 18. Tazume H., Miyata K., Tian Z., Endo M., Horiguchi H., Takahashi O., Horio E., Tsukano H., Kadomatsu T., Nakashima Y., Kunitomo R., Kaneko Y., Moriyama S., Sakaguchi H., Okamoto K., Hara M., Yoshinaga T., Yoshimura K., Aoki H., Araki K., Hao H., Kawasuji M., Oike Y. (2012) Macrophage-derived angiopoietin-like protein 2 accelerates development of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 32, 1400–1409. [DOI] [PubMed] [Google Scholar]
- 19. Cummins E. P., Berra E., Comerford K. M., Ginouves A., Fitzgerald K. T., Seeballuck F., Godson C., Nielsen J. E., Moynagh P., Pouyssegur J., Taylor C. T. (2006) Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc. Natl. Acad. Sci. USA 103, 18154–18159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scholz C. C., Cavadas M. A., Tambuwala M. M., Hams E., Rodriguez J., Kriegsheim A., Cotter P., Bruning U., Fallon P. G., Cheong A., Cummins E. P., Taylor C. T. (2013) Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 110, 18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mosser D. M., Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda N., O'Dea E. L., Doedens A., Kim J. W., Weidemann A., Stockmann C., Asagiri M., Simon M. C., Hoffmann A., Johnson R. S. (2010) Differential activation and antagonistic function of HIF-a isoforms in macrophages are essential for NO homeostasis. Genes Dev. 24, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bishop T., Gallagher D., Pascual A., Lygate C. A., de Bono J. P., Nicholls L. G., Ortega-Saenz P., Oster H., Wijeyekoon B., Sutherland A. I., Grosfeld A., Aragones J., Schneider M., van Geyte K., Teixeira D., Diez-Juan A., Lopez-Barneo J., Channon K. M., Maxwell P. H., Pugh C. W., Davies A. M., Carmeliet P., Ratcliffe P. J. (2008) Abnormal sympatho-adrenal development and systemic hypotension in PHD3−/− mice. Mol. Cell. Biol. 28, 3386–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wottawa M., Köditz J., Katschinski D. M. (2010) Normoxic destabilization of ATF-4 depends on proteasomal degradation. Acta Physiol. (Oxf). 198, 457–463. [DOI] [PubMed] [Google Scholar]
- 25. Jaakkola P. M., Rantanen K. (2013) The regulation, localization, and functions of oxygen-sensing prolyl hydroxylase PHD3. Biol. Chem. 394, 449–457. [DOI] [PubMed] [Google Scholar]
- 26. Tabata M., Kadomatsu T., Fukuhara S., Miyata K., Ito Y., Endo M., Urano T., Zhu H. J., Tsukano H., Tazume H., Kaikita K., Miyashita K., Iwawaki T., Shimabukuro M., Sakaguchi K., Ito T., Nakagata N., Yamada T., Katagiri H., Kasuga M., Ando Y., Ogawa H., Mochizuki N., Itoh H., Suda T., Oike Y. (2009) Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell. Metab. 10, 178–188. [DOI] [PubMed] [Google Scholar]
- 27. Okada T., Tsukano H., Endo M., Tabata M., Miyata K., Kadomatsu T., Miyashita K., Semba K., Nakamura E., Tsukano M., Mizuta H., Oike Y. (2010) Synoviocyte-derived angiopoietin-like protein 2 contributes to synovial chronic inflammation in rheumatoid arthritis. Am. J. Pathol. 176, 2309–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Endo M., Nakano M., Kadomatsu T., Fukuhara S., Kuroda H., Mikami S., Hato T., Aoi J., Horiguchi H., Miyata K., Odagiri H., Masuda T., Harada M., Horio H., Hishima T., Nomori H., Ito T., Yamamoto Y., Minami T., Okada S., Takahashi T., Mochizuki N., Iwase H., Oike Y. (2012) Tumor cell-derived angiopoietin-like protein ANGPTL2 is a critical driver of metastasis. Cancer Res. 72, 1784–1794. [DOI] [PubMed] [Google Scholar]
- 29. Zheng J., Umikawa M., Cui C., Li J., Chen X., Zhang C., Huynh H., Kang X., Silvany R., Wan X., Ye J., Canto A. P., Chen S. H., Wang H. Y., Ward E. S., Zhang C. C. (2012) Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature 485, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Appelhoff R. J., Tian Y. M., Raval R. R., Turley H., Harris A. L., Pugh C. W., Ratcliffe P. J., Gleadle J. M. (2004) Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 279, 38458–38465. [DOI] [PubMed] [Google Scholar]
- 31. Rohrbach S., Simm A., Pregla R., Franke C., Katschinski D. M. (2005) Age-dependent increase of prolyl-4-hydroxylase domain (PHD) 3 expression in human and mouse heart. Biogerontology 6, 165–171. [DOI] [PubMed] [Google Scholar]
- 32. Stiehl D. P., Wirthner R., Köditz J., Spielmann P., Camenisch G., Wenger R. H. (2006) Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J. Biol. Chem. 281, 23482–23491. [DOI] [PubMed] [Google Scholar]
- 33. Hölscher M., Silter M., Krull S., von Ahlen M., Hesse A., Schwartz P., Wielockx B., Breier G., Katschinski D. M., Zieseniss A. (2011) Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J. Biol. Chem. 286, 11185–11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Myllyharju J. (2009) HIF prolyl 4-hydroxylases and their potential as drug targets. Curr. Pharm. Des. 15, 3878–3885. [DOI] [PubMed] [Google Scholar]
- 35. Katschinski D. M. (2009) In vivo functions of the prolyl-4-hydroxylase domain oxygen sensors: direct route to the treatment of anaemia and the protection of ischaemic tissues. Acta Physiol. (Oxf). 195, 407–414. [DOI] [PubMed] [Google Scholar]
- 36. Lee S., Nakamura E., Yang H., Wei W., Linggi M. S., Sajan M. P., Farese R. V., Freeman R. S., Carter B. D., Kaelin W. G., Jr., Schlisio S. (2005) Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 8, 155–167. [DOI] [PubMed] [Google Scholar]
- 37. Lipscomb E. A., Sarmiere P. D., Freeman R. S. (2001) SM-20 is a novel mitochondrial protein that causes caspase-dependent cell death in nerve growth factor-dependent neurons. J. Biol. Chem. 276, 5085–5092. [DOI] [PubMed] [Google Scholar]
- 38. Straub J. A., Lipscomb E. A., Yoshida E. S., Freeman R. S. (2003) Induction of SM-20 in PC12 cells leads to increased cytochrome c levels, accumulation of cytochrome c in the cytosol, and caspase-dependent cell death. J. Neurochem. 85, 318–328. [DOI] [PubMed] [Google Scholar]
- 39. Schlisio S., Kenchappa R. S., Vredeveld L. C., George R. E., Stewart R., Greulich H., Shahriari K., Nguyen N. V., Pigny P., Dahia P. L., Pomeroy S. L., Maris J. M., Look A. T., Meyerson M., Peeper D. S., Carter B. D., Kaelin W. G., Jr., (2008) The kinesin KIF1Bβ acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 22, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xie L., Pi X., Mishra A., Fong G., Peng J., Patterson C. (2012) PHD3-dependent hydroxylation of HCLK2 promotes the DNA damage response. J. Clin. Invest. 122, 2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Köditz J., Nesper J., Wottawa M., Stiehl D. P., Camenisch G., Franke C., Myllyharju J., Wenger R. H., Katschinski D. M. (2007) Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 110, 3610–3617. [DOI] [PubMed] [Google Scholar]
- 42. Kadomatsu T., Tabata M., Oike Y. (2011) Angiopoietin-like proteins: emerging targets for treatment of obesity and related metabolic diseases. FEBS J. 278, 559–564. [DOI] [PubMed] [Google Scholar]
- 43. Oike Y., Yasunaga K., Suda T. (2004) Angiopoietin-related/angiopoietin-like proteins regulate angiogenesis. Int. J. Hematol. 80, 21–28. [DOI] [PubMed] [Google Scholar]
- 44. Kanda A., Noda K., Oike Y., Ishida S. (2012) Angiopoietin-like protein 2 mediates endotoxin-induced acute inflammation in the eye. Lab. Invest. 92, 1553–1563. [DOI] [PubMed] [Google Scholar]
- 45. Kubota Y., Oike Y., Satoh S., Tabata Y., Niikura Y., Morisada T., Akao M., Urano T., Ito Y., Miyamoto T., Nagai N., Koh G. Y., Watanabe S., Suda T. (2005) Cooperative interaction of angiopoietin-like proteins 1 and 2 in zebrafish vascular development. Proc. Natl. Acad. Sci. USA 102, 13502–13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.