

Abstract
Aim
The objective was to evaluate in treated heart failure (HF) patients whether multidrug therapy interferes with the cardiovascular autonomic response to postural stress.
Methods and results
Blood pressure (BP; Finapres), heart rate (HR), stroke volume, and total peripheral resistance (TPR) responses to standing up were measured in 33 HF patients and 10 healthy age‐matched controls. Ten hypertensive (HT) patients treated with a similar combination of drugs but without heart failure served as reference subjects to account for use of medication. Frequency domain measures of HR and BP variability were calculated as correlates of cardiovascular autonomic function. Postural hypotension was found in 16 out of 33 HF patients independently from New York Heart Association functional class. In HF patients vs. HT patients and healthy controls the haemodynamic postural response was abnormal with a large initial BP fall and a slackened reflex increase in TPR resulting in inadequate BP recovery. HR and BP variability were normal in HT patients and healthy controls but attenuated in HF patients. The magnitude of the postural HR, stroke volume, and TPR responses as well as HR and BP variability was inversely related to the New York Heart Association class.
Conclusions
In HF patients, the autonomic vasomotor response to postural stress is abnormal, more pronounced with increasing disease severity, and frequently associated with overt postural hypotension. These phenomena appear related to the cardiac condition rather than treatment.
Keywords: Chronic heart failure, Haemodynamic stress, Orthostasis, Autonomic function, Hypotension
Introduction
The pathophysiological hallmark in patients with chronic heart failure (HF) is chronically elevated sympathetic activation by neurohormonal excitation in response to a decreased cardiac output (CO) and subsequent tissue hypoperfusion.1 Activation of the sympathetic nervous system in HF is presumed to maintain tissue perfusion, but the adverse effects dominate and contribute to further deterioration of myocardial function.2 Significant advances in HF pharmacotherapeutics, aiming to offset the adverse neurohormonal response, include neurohormonal blockade by angiotensin‐converting enzyme (ACE) inhibition, angiotensin II receptor type 1 antagonists, and beta adrenergic receptor blockade. This therapeutic approach has considerably improved morbidity and mortality in HF patients.3
Assuming the upright posture triggers reflex sympathetic activation with profound haemodynamic changes, most of them developing within the first 30 s of standing.4 In short, the postural reduction in venous return reduces CO but mean arterial pressure is maintained by vasoconstriction and increases in heart rate (HR). In the late sixties of the last century, Abelmann and Fareeduddin demonstrated an abnormal postural response in decompensated HF patients characterized by maintained or even enhanced CO and thus blood pressure, and no reflex increases in HR and total peripheral resistance (TPR).5, 6 This so‐called HF response was attributed to volume overload and correlated with enhanced orthostatic tolerance. In addition to this response, Kubo and Cody showed in a minority of patients another two postural response patterns (normal respectively analogous to idiopathic orthostatic hypotension) underlining the heterogeneity of the postural cardiovascular response in HF patients.7 In those days, pharmacological treatment of HF was limited to salt restriction, ethacrynic acid, and digitalis.8 Nowadays, a low blood pressure in HF patients is more regularly encountered with an estimated prevalence of up to 30% and associated with new multidrug neurohormonal blockade strategies.3, 9, 10 Although orthostatic hypotension is a major risk factor for recurrent falls and syncope, and may reduce quality of life substantially,11, 12 this has not been mentioned as important comorbidity in HF patients both in the European and American guidelines for the management of HF.12
We wondered whether multidrug neurohormonal blockade interferes with the cardiovascular autonomic response to postural stress in HF patients. To that purpose, the present study was set up to evaluate the postural haemodynamic response in compensated HF patients treated with neurohormonal blockade agents in an outpatients setting. Hypertensive patients treated with a similar combination of drugs in absence of HF served as reference subjects.
Methods
Subjects
Patients with haemodynamically stable HF [classified according to the New York Heart Association (NYHA)] due to (ischemic) dilated cardiomyopathy with a left ventricular ejection fraction <35% as confirmed by echo or gated single‐photon emission computed tomography (SPECT) were recruited from the Heart Failure Outpatients Clinic. All HF patients were on constant doses of AII receptor antagonists and/or ACE inhibitory agents, beta adrenergic receptor blockade, digitalis, nitrates, and diuretics for at least 4 weeks prior to the study and had not been admitted to the hospital in the preceding 3 months. Hypertension (HT) patients with echocardiographically confirmed normal left ventricle ejection fraction (>60%) and treated with ACE inhibitory agents, beta adrenergic blockade, and/or diuretics were recruited from the Internal Medicine Outpatients Clinic. They served as reference subjects using neurohormonal blockade in absence of HF. Age‐matched volunteers served as healthy controls. The study protocol was approved by the institutional Medical Ethics Committee and conforms to the principles outlined in the Declaration of Helsinki. Informed consent was obtained from all subjects prior to the study.
Measurements
Beat‐to‐beat arterial blood pressure was non‐invasively measured (Finometer, FMS, Amsterdam, the Netherlands) using the volume clamp technique.13 A finger cuff was applied on the mid‐phalanx of the third finger and held at heart level to avoid hydrostatical errors. Left ventricular stroke volume (SV) was determined by pulse contour (CO‐trek, Edwards Lifesciences BMEYE, Amsterdam, the Netherlands). Pulse contour SV from both invasive and non‐invasive arterial pressure tracks a thermodilution‐based estimate.14 CO was determined from SV times HR, and systemic vascular resistance (TPR) from the ratio of mean arterial pressure and CO.
Blood pressure and heart rate variability
Frequency domain low‐frequency (0.07–0.15 Hz; Lf) and high‐frequency (>0.15 Hz; Hf) indices of HR and BP variability (HRV and BPV, respectively) were quantified as physiological correlates of fluctuations in autonomic cardiovascular output.15 Efferent vagal activity contributes importantly to the Hf component, whereas the Lf component of variability, although being considered as a marker of sympathetic modulation, does include both sympathetic and vagal influences. This also holds true for the Lf/Hf ratio.16, 17
Three‐minute periods (last 3 min of supine rest and last 3 min of standing) of R‐R interval and systolic BP recordings were selected for analysis and manually inspected for artefacts. The HRV and BPV spectra were then obtained by a fast Fourier transform with a maximal smoothing window of 0.04 Hz. Lf and Hf components were expressed in normalized units (n.u.) that represent the relative value of each power component in proportion to total spectral power.
Protocol
After at least 5 min of supine rest, subjects were asked to stand up swiftly and remain in the standing position for 4 min. The last 10 s of each minute was used for analysis. Reproducibility of the haemodynamic response to postural stress was verified in four HF patients by repeating this protocol two times.
Statistical analysis
Data were statistically analysed using SigmaPlot (11.0, Systat Software Inc., USA) and presented as mean ± SD. The postural stress responses within and between groups were assessed using one‐way (repeated measures) analysis of variance with pairwise multiple comparisons (Holm–Sidak). When data were not normally distributed, non‐parametric statistical tests were used. A P‐value less than 0.05 was considered to indicate a statistically significant difference.
Results
Forty‐three HF patients, 10 HT patients, and 10 healthy controls were included in the study. Data from 10 HF patients were excluded from analysis because of cardiac arrhythmia and/or insufficient signal quality, leaving 33 HF patients for analysis. Baseline subject characteristics are given in Table 1.
Table 1.
Subject characteristics
| HF patients | HT patients | Healthy controls | |
|---|---|---|---|
| n (male/female) | 33 (24/9) | 10 (6/4) | 10 (6/4) |
| Mean age (years) | 60 ± 11 | 58 ± 10 | 61 ± 10 |
| Body mass (kg/m2) | 27 ± 4 | 28 ± 4 | 24 ± 3a , b |
| Ejection fraction (%) | <35% | >60% | |
| NYHA class | |||
| I | 10 | ||
| II | 15 | ||
| III | 8 | ||
| Medication, n (%) | |||
| AIIR1 antagonists | 7 (21) | 0 | |
| ACE inhibitors | 23 (70) | 5 (50) | |
| β‐adrenergic receptor blocker | 27 (82) | 6 (60) | |
| Nitrates | 13 (39) | 0 | |
| Digitalis | 9 (27) | 0 | |
| Diuretics | 24 (73) | 2 (20) | |
| Vitamin K antagonist | 11 (33) | 0 | |
ACE, angiotensin‐converting enzyme; AIIR1, angiotensin II receptor type 1; HF, heart failure; HT, hypertensive; NYHA, New York Heart Association.
P < 0.05 vs. HF patients.
P < 0.05 vs. HT patients.
Supine resting position
Supine systolic and diastolic arterial pressure, HR, SV, and TPR were comparable for HF patients, HT patients, and healthy controls (Tables 2 and 3).
Table 2.
Haemodynamic postural response in HF patients vs. HT patients and healthy controls
| HF patients | HT patients | Healthy controls | |
|---|---|---|---|
| (n = 33) | (n = 10) | (n = 10) | |
| SAP (mmHg) | |||
| Supine | 119 ± 26 | 127 ± 14 | 131 ± 16 |
| Δ% 60 s upright | −9 ± 15a | +2 ± 8b | +4 ± 7b |
| Δ% 240 s upright | +3 ± 14 | +3 ± 11 | +7 ± 15 |
| DAP (mmHg) | |||
| Supine | 55 ± 13 | 61 ± 9b | 60 ± 12 |
| Δ% 60 s upright | −8 ± 21a | +7 ± 14a , b | +8 ± 6a , b |
| Δ% 240 s upright | +11 ± 19a | +12 ± 19a | +15 ± 13a |
| HR (beats/min) | |||
| Supine | 66 ± 7 | 65 ± 9 | 69 ± 12 |
| Δ% 60 s upright | +16 ± 12a | +10 ± 15a | +17 ± 8a |
| Δ% 240 s upright | +11 ± 10a | +11 ± 13a | +17 ± 10a |
| SV (mL) | |||
| Supine | 89 ± 21 | 105 ± 19 | 85 ± 17 |
| Δ% 60 s upright | −13 ± 12a | −21 ± 15a | −16 ± 8a |
| Δ% 240 s upright | −15 ± 11a | −19 ± 13a | −19 ± 10a |
| TPR (dyn·s/cm5) | |||
| Supine | 1316 ± 458 | 1114 ± 547 | 1403 ± 503 |
| Δ% 60 s upright | −9 ± 17a | +19 ± 21a , b | +3 ± 9b |
| Δ% 240 s upright | +7 ± 16 | +20 ± 29a | +12 ± 14a |
DAP, diastolic arterial pressure; HF, heart failure; HR, heart rate; HT, hypertensive; SAP, systolic arterial pressure; SV, stroke volume; TPR, total peripheral resistance.
P < 0.05 supine vs. upright.
P < 0.05 vs. HF patients.
Table 3.
Haemodynamic postural response in NYHA class I, II, and III HF patients
| HF patients | |||
|---|---|---|---|
| NYHA I (n = 10) | NYHA II (n = 15) | NYHA III (n = 8) | |
| SAP (mmHg) | |||
| Supine | 116 ± 21 | 127 ± 30 | 109 ± 21 |
| ∆% 60 s upright | −9 ± 20 | −10 ± 14a | −6 ± 7 |
| ∆% 240 s upright | +4 ± 16 | +2 ± 15 | +4 ± 12 |
| DAP (mmHg) | |||
| Supine | 54 ± 12 | 56 ± 17 | 52 ± 7 |
| ∆% 60 s upright | −9 ± 26 | −5 ± 21 | −12 ± 15a |
| ∆% 240 s upright | +14 ± 14a | +12 ± 24 | +2 ± 10 |
| HR (beats/min) | |||
| Supine | 63 ± 8 | 64 ± 4 | 70 ± 8 |
| ∆% 60 s upright | +18 ± 9a | +17 ± 15a | +10 ± 9a |
| ∆% 240 s upright | +14 ± 9a | +11 ± 10a | +9 ± 11 |
| SV (mL) | |||
| Supine | 96 ± 18 | 85 ± 25 | 88 ± 15 |
| ∆% 60 s upright | −18 ± 9a , b | −16 ± 12a , b | −1 ± 9 |
| ∆% 240 s upright | −20 ± 10a , b | −16 ± 10a , b | −6 ± 7 |
| TPR (dyn·s/cm5) | |||
| Supine | 1211 ± 423 | 1503 ± 510 | 1095 ± 244 |
| ∆% 60 s upright | −6 ± 20 | −9 ± 18a | −12 ± 14a |
| ∆% 240 s upright | +12 ± 15b | +7 ± 19 | +1 ± 9 |
DAP, diastolic arterial pressure; HF, heart failure; HR, heart rate; NYHA, New York Heart Association; SAP, systolic arterial pressure; SV, stroke volume; TPR, total peripheral resistance.
P < 0.05 supine vs. upright.
P < 0.05 vs. NYHA class III HF patients.
BPV Lf/Hf ratio was lower in class II and III HF patients, and BPV Hf power higher in class III Hf patients compared with healthy controls and HT patients (Table 4). HRV Lf power was lower in class III HF patients compared with healthy controls and HT patients with a higher HRV Lf/Hf ratio in healthy controls vs. class III HF patients.
Table 4.
Heart rate and blood pressure variability
| HF patients | HT patients | Healthy controls | ||||
|---|---|---|---|---|---|---|
| NYHA I (n = 10) | NYHA II (n = 15) | NYHA III (n = 8) | (n = 10) | (n = 10) | ||
| Heart rate variability | ||||||
| Lf (n.u.) | Supine | 0.17 ± 0.09 | 0.14 ± 0.08 | 0.10 ± 0.06 | 0.19 ± 0.09d | 0.19 ± 0.10d |
| Upright | 0.17 ± 0.13 | 0.14 ± 0.09 | 0.07 ± 0.05 | 0.22 ± 0.12d | 0.27 ± 0.15d | |
| Hf (n.u.) | Supine | 0.25 ± 0.16 | 0.37 ± 0.19 | 0.38 ± 0.23 | 0.33 ± 0.16 | 0.22 ± 0.12 |
| Upright | 0.24 ± 0.12 | 0.30 ± 0.15 | 0.33 ± 0.19 | 0.28 ± 0.20 | 0.23 ± 0.20 | |
| Lf/Hf | Supine | 1.03 ± 0.95d | 0.53 ± 0.41 | 0.41 ± 0.41 | 0.72 ± 0.47 | 1.05 ± 0.80d |
| Upright | 0.82 ± 0.59d | 0.59 ± 0.51 | 0.31 ± 0.38 | 1.34 ± 1.25a,d | 2.15 ± 1.66a,c,d | |
| Blood pressure variability | ||||||
| Lf (n.u.) | Supine | 0.09 ± 0.06 | 0.10 ± 0.05 | 0.08 ± 0.04 | 0.17 ± 0.13 | 0.16 ± 0.10d |
| Upright | 0.14 ± 0.09 | 0.14 ± 0.07 | 0.10 ± 0.09 | 0.27 ± 0.14b , c , d | 0.30 ± 0.15a , b , c , d | |
| Hf (n.u.) | Supine | 0.09 ± 0.05c , d | 0.15 ± 0.08 | 0.26 ± 0.20 | 0.11 ± 0.13d | 0.10 ± 0.10d |
| Upright | 0.15 ± 0.16 | 0.19 ± 0.15 | 0.28 ± 0.17 | 0.14 ± 0.09d | 0.14 ± 0.09d | |
| Lf/Hf | Supine | 1.61 ± 1.52d | 0.91 ± 0.63 | 0.43 ± 0.22 | 2.55 ± 2.19c , d | 2.85 ± 2.53c , d |
| Upright | 1.61 ± 1.28d | 1.08 ± 0.75 | 0.47 ± 0.35 | 2.92 ± 3.41c , d | 3.44 ± 3.63c , d | |
Lf/Hf, low/high frequency; HF, heart failure; HT, hypertensive; n.u., normalized units (power in the respective bands is normalized by division of total variance); NYHA, New York Heart Association.
P < 0.05 supine vs. upright.
P < 0.05 vs. NYHA class I HF patients.
P < 0.05 vs. NYHA class II HF patients.
P < 0.05 vs. NYHA class III HF patients.
Postural stress
Orthostatic hypotension defined according to the American Autonomic Society guidelines18 was only found in HF patients (16 out of 33). They were equally divided across NYHA classes [five patients (50%) in class I, eight patients (53%) in class II, and three patients (38%) in class III].
Heart failure patients vs. hypertensive patients and healthy controls
The initial BP fall upon standing was larger in HF patients compared with HT patients and healthy controls, with insufficient recovery at 60 s upright (supine systolic pressure: −9 ± 15% vs. 4 ± 7% and 2 ± 8% (P < 0.001); diastolic arterial pressure: −8 ± 21% vs. 8 ± 6% and 7 ± 14% (P = 0.003), respectively) (Figure 1). In HF patients, the initial peak in HR was lower compared with HT patients and healthy controls (Figure 2), but no differences in HR response were found during continued standing. The postural reduction in SV did not differ between groups and neither between HF patients with vs. without orthostatic hypotension (−14 ± 12% vs. −12 ± 13%; P = 0.748). The postural increase in TPR observed in both healthy controls and HT patients was absent in HF patients (3 ± 9% and 19 ± 21% vs. −9 ± 17% (P < 0.001), respectively).
Figure 1.
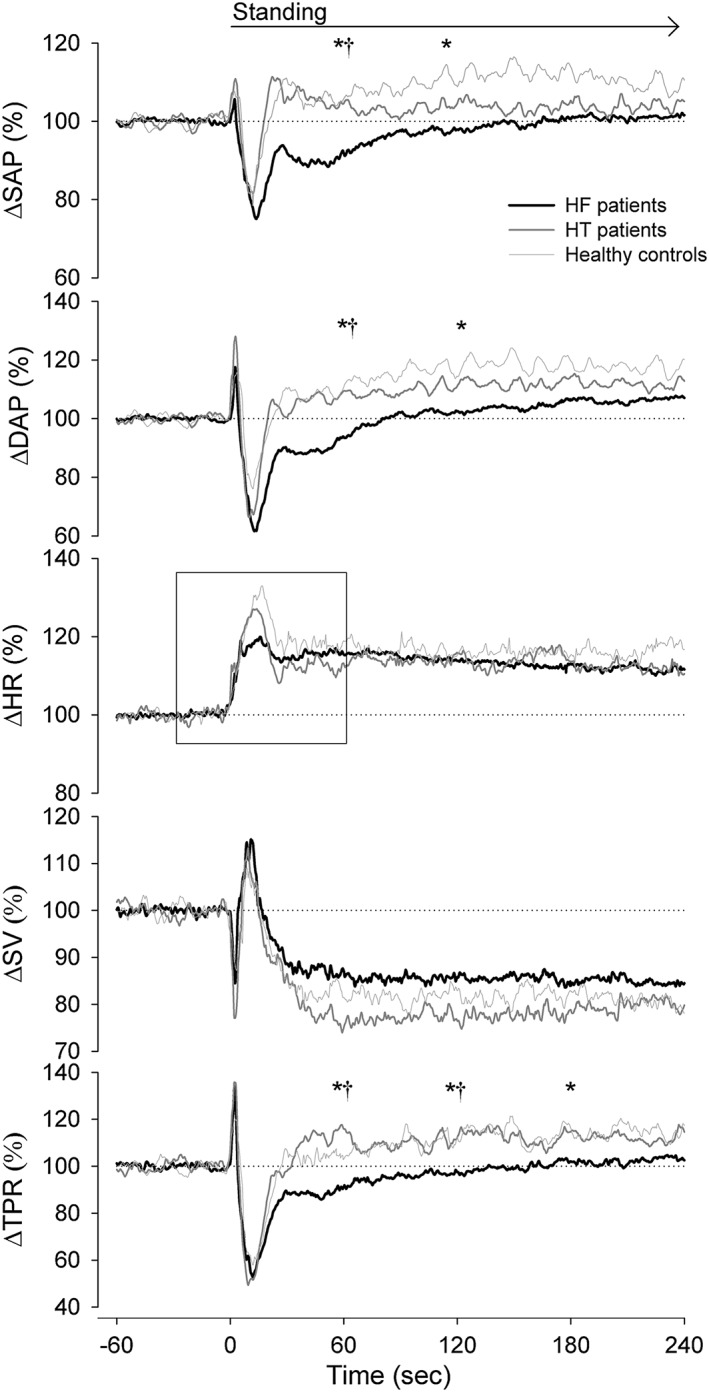
Postural response in HF patients (black lines), HT patients (dark grey lines), and healthy controls (light grey lines). The box represents a zoom in of the HR response (Figure 2). DAP, diastolic arterial pressure; HF, heart failure; HR, heart rate; HT, hypertensive; SAP, systolic arterial pressure; SV, stroke volume; CO, cardiac output; TPR, total peripheral resistance.*P < 0.05 HF patients vs. HC; †P < 0.05 HF patients vs. HT patients.
Figure 2.
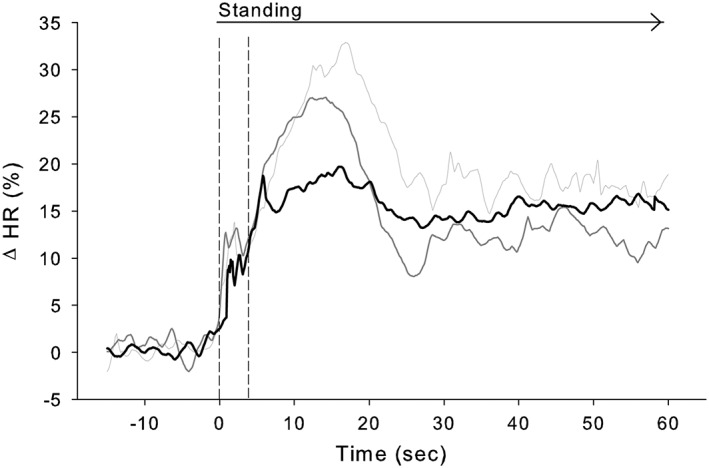
Average heart rate (HR) response to standing in heart failure patients (black line), hypertensive patients (dark grey line), and healthy controls (light grey line). Note the similarity among groups of the vagally mediated initial (first ~4 s) HR response followed by a more sluggish (sympathetic) response in heart failure vs. hypertensive patients and healthy controls.
In response to standing, BPV Lf power doubled (0.16 ± 0.10 vs. 0.30 ± 0.15 n.u., P = 0.022) in healthy controls but did not change in HF patients. Furthermore, the HRV Lf/Hf ratio increased in healthy controls and HT patients only (1.05 ± 0.80 vs. 2.15 ± 1.66 ms2/ms2 (P = 0.049) and 0.72 ± 0.47 vs. 1.34 ± 1.25 ms2/ms2 (P = 0.044), respectively).
New York Heart Association class I vs. II vs. III
The abnormal postural BP and HR responses were not different across NYHA classes (Figure 3), whereas the smallest reduction in SV was observed in class III vs. class II and class I HF patients throughout the full period of standing (P ≤ 0.015). The increase in TPR was absent in class III vs. class I HF patients at 240 s upright (0 ± 7% vs. 12 ± 15%, P = 0.048) with an intermediate response in class II HF patients (7 ± 19%). The magnitude of the haemodynamic postural response in HF patients was directly related to NYHA classification (Figure 4).
Figure 3.
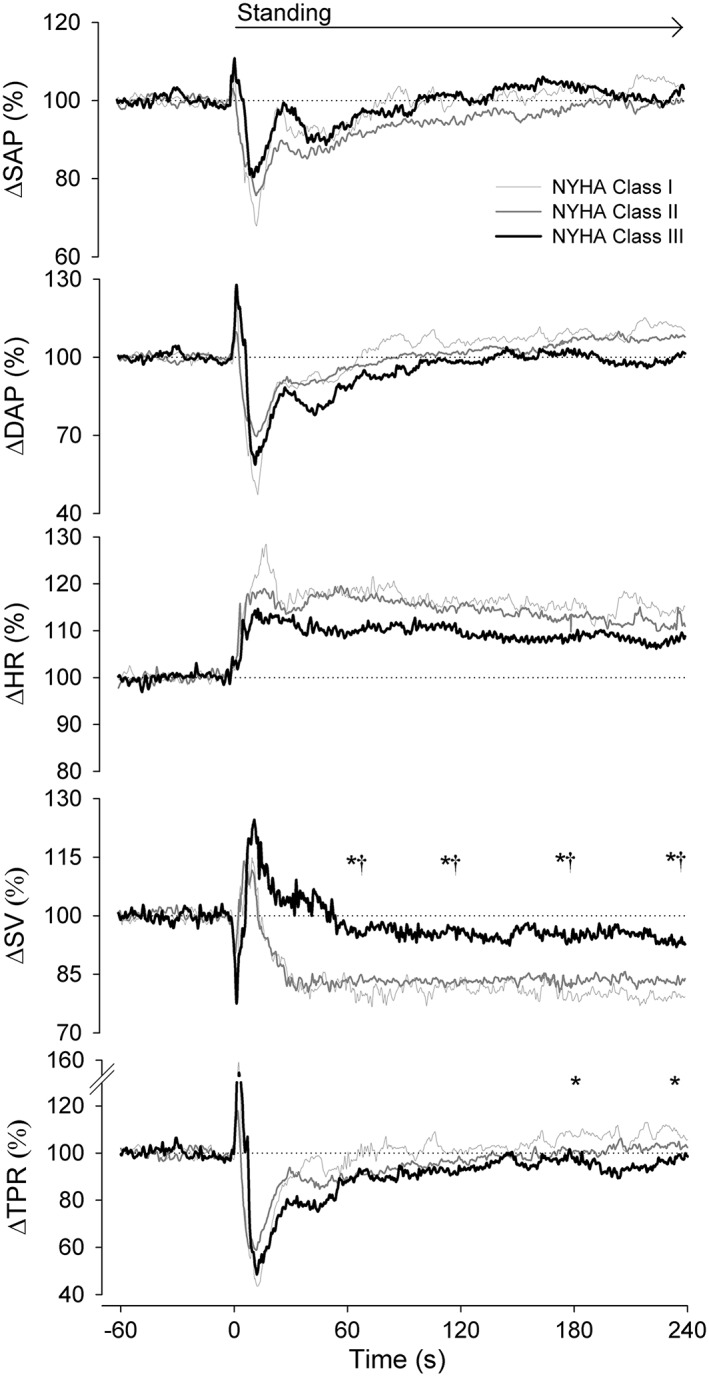
Postural response in NYHA class I (light grey lines), NYHA class II (dark grey lines), and NYHA class III (black lines) heart failure patients. Abbreviations: refer to Figure 1. *P < 0.05 NYHA class I vs. class III; †P < 0.05 NYHA class II vs. class III.
Figure 4.
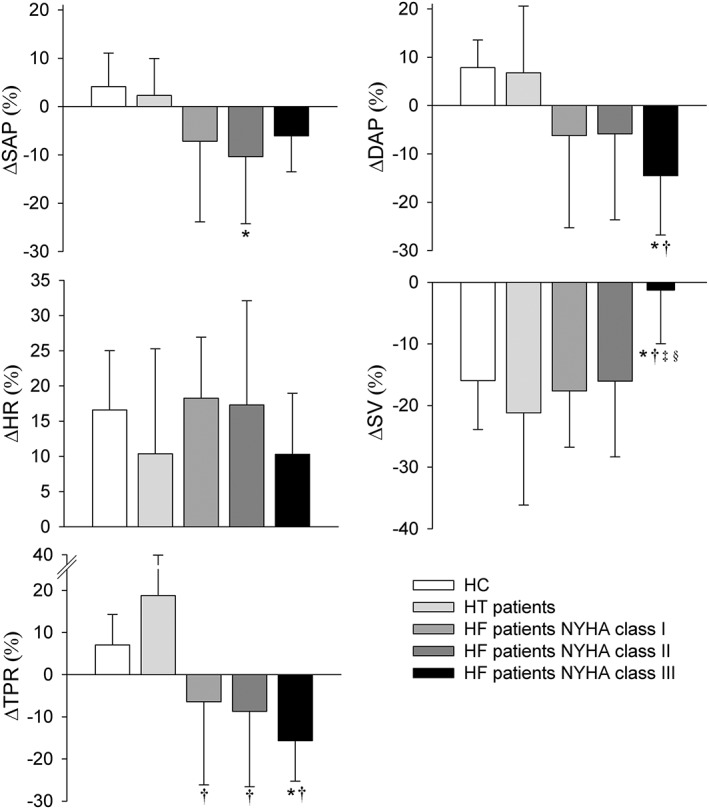
Postural response at 60 s of standing in HF patients (NYHA I, II, and III), HT patients, and healthy controls with respect to the supine position. Abbreviations: refer to Figure 1. *P < 0.01 vs. healthy controls; †P < 0.01 vs. HT patients; ‡P < 0.01 vs. HF patients NYHA class I; §P < 0.01 vs. HF patients NYHA class II.
Discussion
The findings of the present study are three‐fold. First, the postural haemodynamic response in treated HF patients is abnormal but not similar to the previously reported ‘heart failure response’. Second, HR and BP variability measures were attenuated in HF patients during rest and in response to standing. Finally, the magnitude of the postural HR, SV, and TPR responses was inversely related to NYHA class with a large incidence of orthostatic hypotension equally divided across groups.
Heart failure response
In the pre‐neurohormonal blockade era, the abnormal postural response in HF, designated as ‘heart failure response’, was characterized by maintained CO and blood pressure and absent reflex increases in HR and TPR, attributed to overfilling of the heart.5, 6 Accordingly, the majority of patients with HF tolerated the upright position remarkably well as the volume overload prevented the postural shift of blood towards the lower parts of the body, that is, venous pooling. In the minority of patients in whom venous pooling was present, some demonstrated a normal postural response while others suffered from postural hypotension related to absent reflex increase of vascular resistance.7 Treatment with predominantly venous and/or arteriolar dilators and ACE inhibition lowered resting ventricular filling pressures and TPR with enhanced cardiac index.19, 20 Also, the haemodynamic response to the upright posture shifted from no orthostatic changes to a postural fall in cardiac filling pressures and CO indicating re‐establishment of postural pooling.21, 22 Nevertheless, postural reflex increments in HR and in TPR remained absent with a high incidence of orthostatic hypotension.21 The paradigm shift towards multidrug HF therapy in the last two decades reduced mortality further23 (for a timeline in HF therapy, see Sacks et al.8). A new finding of the current study is the partial re‐establishment of the initial postural reflex increase in HR in the HF patients (Figure 2). Differences in latency and time course of the HR response can be applied to differentiate between the efferent parasympathetic and sympathetic limbs of the reflex arc: vagally mediated HR changes have a latency of a few hundred milliseconds and a time constant of a few seconds, whereas these values are 1–3 and 10 sec, respectively, for sympathetically mediated HR changes.24 Data of the present study demonstrated an early (first 4 s) increase in HR that was similar for HF patients and healthy controls suggesting recovery of parasympathetic HR control. Interestingly, resting HR in the HF patients vs. HT and healthy controls was comparable and in accordance with the effect of chronic beta‐adrenergic blockade treatment.25 In contrast, the secondary sympathetically mediated HR increase from 4 s on24 was sluggish compared with healthy controls and HT patients and therefore designated as subnormal. Together with the slacking TPR response, this exemplifies persistence of abnormal sympathetic circulatory control in HF patients. During the development of pacing‐induced HF in dogs, the fall in CO precedes the increase in TP that has been attributed to involve a reduction in intrinsic peripheral vascular tone despite neurohumoral activation.26 In the present study, the shift towards a more balanced preload and afterload with re‐establishment of postural pooling but with a still abnormal vasomotor response may render them prone to orthostatic hypotension. Also given the normal response in treated HT, we consider the abnormal postural haemodynamic response in HF patients attributable to cardiac dysfunction rather than treatment.
Blood pressure and heart rate variability
Postural stress is associated with parasympathetic withdrawal and sympathetic activation, and the increase in the Lf vs. Hf component of the BP/HR spectrum in the HT patients and healthy controls corresponded to that.27 The attenuated Lf BP/HR power at rest and its failure to increase during standing in the HF group may be interpreted as signs of central autonomic impairment. We acknowledge that the approach based on spectral analysis of HR and BP signals has limitations that inherently prevent us from substituting the frequency domain data for sympathetic cardiovascular drive.28 With these in mind, this data conforms to results of previous studies who revealed a decreased Lf component of HR and BP variability in HF,29, 30 which is closely coherent to simultaneous recordings of resting muscle sympathetic nerve activity.29 Collectively, these data suggest that in extremes of stress when sympathetic drive is high—as is the case in chronic HF—all physiological mechanisms are mobilized to the maximum to maintain homeostasis such that the system has no reserve to maintain its variability. Possible explanations for these findings are a saturatingly high level of sympathetic input attenuating the responsiveness of sinus nodal cells to neural modulation,17 beta adrenoreceptor downregulation,31 or dysfunctional baroreceptor reflexes.32
Severity of cardiac dysfunction
The haemodynamic abnormalities in response to postural stress ran parallel with disease severity, being more pronounced in NYHA class III HF patients with absent postural decrease in SV. In addition, the baroreflex mediated increase in TPR and HR were most slackened in NYHA class III HF patients with an intermediate to almost normal response in respectively NYHA class II and I HF patients. Also, the magnitude of HR and BP variability was inversely related to NYHA class. Kubo et al. demonstrated that neurohormonal responses to head‐up tilt were relatively normal in the early symptomatic stages of HF compared with healthy controls,33 and the present study showed this for haemodynamic responses as well. The finding of the inverse relationship between the postural response and severity of cardiac dysfunction provides further evidence that haemodynamic and neurovascular abnormalities to postural stress are restrained to severe rather than mild HF. An association between orthostatic hypotension and severity of HF according to NYHA functional class was suggested by Gorelik and colleagues.34 In contrast, Potocka‐Plazak and Plazak demonstrated that orthostatic hypotension was equally present across NYHA classes,35 which is in line with findings from the present study. This suggests that in NYHA class I vs. III patients the larger postural SV decline is balanced by the less sluggish vasomotor response and vice versa.
Limitations
For obvious reasons, it is not justified to discontinue pharmacological therapy in HF patients for research purposes, and all patients studied were on pharmacological treatment. HT patients with a similar constellation of therapies but without HF served as surrogate reference subjects to account for use of medication. Evidently hypertension and chronic HF are distinct clinical entities in many aspects. Their common bond is that pathological mechanisms are in part or in whole mediated by an elevated TPR with implications for pharmacologically counteracting the prevailing vasoconstriction. The HT patients did not receive AIIRI antagonist or nitrates, and we acknowledge that HT did not match HF patients for use of diuretics that may have induced differences in postural changes in central blood volume and CO. Our finding of a similar postural reduction in SV for HT vs. HF patients contests this assumption, leaving the view of high incidence of orthostatic hypotension among HF patients being explained by an abnormal vasomotor response rather than by a reduction in SV unchallenged.
In summary, the mechanism regarding the standing‐up response in treated HF patients is complex and includes abnormalities of autonomic cardiovascular function and neurohumoral volume control. The magnitude of the haemodynamic postural response in HF patients was directly related to NYHA classification. The abnormal vasomotor response in HF patients relates to cardiac condition rather than to pharmacological treatment rendering them prone to orthostatic hypotension.
Conflict of interest
None declared.
Funding
This work was supported by the Rembrandt Institute for Cardiovascular Science (project award 2011) and by the Dutch Heart Foundation (grant # 99182).
Acknowledgements
We would like to thank Dr Gert van Montfrans, Tonny Veelenturf, and Ronald Zwart for their assistance pertaining to the data collection and support of this research.
Bronzwaer, A.‐S. G. T. , Bogert, L. W. J. , Westerhof, B. E. , Piek, J. J. , Daemen, M. J. A. P. , and van Lieshout, J. J. (2017) Abnormal haemodynamic postural response in patients with chronic heart failure. ESC Heart Failure, 4: 146–153. doi: 10.1002/ehf2.12127.
This research was performed in the Academic Medical Centre, University of Amsterdam, Amsterdam, the Netherlands.
References
- 1. Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 1986; 73: 615–621. [DOI] [PubMed] [Google Scholar]
- 2. Francis GS, Goldsmith SR, Levine TB, Olivari MT, Cohn JN. The neurohumoral axis in congestive heart failure. Ann Intern Med 1984; 101: 370–377. [DOI] [PubMed] [Google Scholar]
- 3. Parwani P, Ryan J. Heart failure patients with low blood pressure: how should we manage neurohormonal blocking drugs? Circ Heart Fail 2012; 5: 819. [DOI] [PubMed] [Google Scholar]
- 4. Borst C, Van Brederode JFM, Wieling W, Van Montfrans GA, Dunning AJ. Mechanisms of initial blood pressure response to postural stress. Clin Sci 1984; 67: 321–327. [DOI] [PubMed] [Google Scholar]
- 5. Abelmann WH, Fareeduddin K. Increased tolerance of orthostatic stress in patients with heart disease. Am J Cardiol 1969; 23: 353–363. [DOI] [PubMed] [Google Scholar]
- 6. Jeffrey FE, Fareeduddin K, Abelmann WH. Increased tolerance of patients with circulatory congestion to circulatory stress. Am J Med Sci 1970; 259: 323–332. [DOI] [PubMed] [Google Scholar]
- 7. Kubo SH, Cody RJ. Circulatory autoregulation in chronic congestive heart failure: responses to head‐up tilt in 41 patients. Am J Cardiol 1983; 52: 512–518. [DOI] [PubMed] [Google Scholar]
- 8. Sacks CA, Jarcho JA, Curfman GD. Paradigm shifts in heart‐failure therapy–a timeline. N Engl J Med 2014; 371: 989–991. [DOI] [PubMed] [Google Scholar]
- 9. Bozkurt B. Response to Ryan and Parwani: heart failure patients with low blood pressure: how should we manage neurohormonal blocking drugs? Circ Heart Fail 2012; 5: 820–821. [DOI] [PubMed] [Google Scholar]
- 10. Ko DT, Hebert PR, Coffey CS, Curtis JP, Foody JM, Sedrakyan A, Krumholz HM. Adverse effects of beta‐blocker therapy for patients with heart failure: a quantitative overview of randomized trials. Arch Intern Med 2004; 164: 1389–1394. [DOI] [PubMed] [Google Scholar]
- 11. Ricci F, De Caterina R, Fedorowski A. Orthostatic hypotension: epidemiology, prognosis, and treatment. J Am Coll Cardiol 2015; 66: 848–860. [DOI] [PubMed] [Google Scholar]
- 12. Gorelik O, Feldman L, Cohen N. Heart failure and orthostatic hypotension. Heart Fail Rev 2016; 21: 529–538. [DOI] [PubMed] [Google Scholar]
- 13. Peñáz J. Photoelectric measurement of blood pressure, volume and flow in the finger. Digest of the 10th international conference on medical and biological engineering – Dresden 1973.
- 14. Bogert LW, Wesseling KH, Schraa O, Van Lieshout EJ, de Mol BA, van Goudoever J, Westerhof BE, van Lieshout JJ. Pulse contour cardiac output derived from non‐invasive arterial pressure in cardiovascular disease. Anaesthesia 2010; 65: 1119–1125. [DOI] [PubMed] [Google Scholar]
- 15. Shaffer F, McCraty R, Zerr CL. A healthy heart is not a metronome: an integrative review of the heart's anatomy and heart rate variability. Front Psychol 2014; 5: 1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Billman GE. The Lf/Hf ratio does not accurately measure cardiac sympatho‐vagal balance. Front Physiol 2013; 4: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Camm A, Malik M, Bigger J, Günter B, Cerutti S, Choen R. Task force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation 1996; 93: 1043–1065. [PubMed] [Google Scholar]
- 18. Freeman R, Wieling W, Axelrod FB, Benditt DG, Benarroch E, Biaggioni I, Cheshire WP, Chelimsky T, Cortelli P, Gibbons CH, Goldstein DS, Hainsworth R, Hilz MJ, Jacob G, Kaufmann H, Jordan J, Lipsitz LA, Levine BD, Low PA, Mathias C, Raj SR, Robertson D, Sandroni P, Schatz I, Schondorff R, Stewart JM and Van Dijk JG. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. ClinAutonRes 2011; 21: 69–72. [DOI] [PubMed] [Google Scholar]
- 19. Chatterjee K, Massie B, Rubin S, Gelberg H, Brundage BH, Ports TA. Long‐term outpatient vasodilator therapy of congestive heart failure. Consideration of agents at rest and during exercise. Am J Med 1978; 65: 134–145. [DOI] [PubMed] [Google Scholar]
- 20. DiCarlo L, Chatterjee K, Parmley WW, Swedberg K, Atherton B, Curran D, Cucci M. Enalapril: a new angiotensin‐converting enzyme inhibitor in chronic heart failure: acute and chronic hemodynamic evaluations. J Am Coll Cardiol 1983; 2: 865–871. [DOI] [PubMed] [Google Scholar]
- 21. Cody RJ, Franklin KW, Laragh JH. Postural hypotension during tilt with chronic captopril and diuretic therapy of severe congestive heart failure. Am Heart J 1982; 103: 480–484. [DOI] [PubMed] [Google Scholar]
- 22. Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long‐term converting‐enzyme inhibition. Circulation 1982; 66: 135–142. [DOI] [PubMed] [Google Scholar]
- 23. Levy WC, Mozaffarian D, Linker DT, Sutradhar SC, Anker SD, Cropp AB, Anand I, Maggioni A, Burton P, Sullivan MD, Pitt B, Poole‐Wilson PA, Mann DL, Packer M. The Seattle Heart Failure Model: prediction of survival in heart failure. Circulation 2006; 113: 1424–1433. [DOI] [PubMed] [Google Scholar]
- 24. Wieling W, Borst C, Van Lieshout JJ, Sprangers RL, Karemaker JM, Van Brederode JF, Van Montfrans GA, Dunning AJ. Assessment of methods to estimate impairment of vagal and sympathetic innervation of the heart in diabetic autonomic neuropathy. Neth J Med 1985; 28: 383–392. [PubMed] [Google Scholar]
- 25. Doughty RN, Whalley GA, Gamble G, MacMahon S, Sharpe N. Left ventricular remodeling with carvedilol in patients with congestive heart failure due to ischemic heart disease. Australia‐New Zealand Heart Failure Research Collaborative Group. J Am Coll Cardiol 1997; 29: 1060–1066. [DOI] [PubMed] [Google Scholar]
- 26. Kiuchi K, Shannon RP, Sato N, Bigaud M, Lajoie C, Morgan KG, Vatner SF. Factors involved in delaying the rise in peripheral resistance in developing heart failure. Am J Physiol 1994; 267: H211–H216. [DOI] [PubMed] [Google Scholar]
- 27. Vybiral T, Bryg RJ, Maddens ME, Boden WE. Effect of passive tilt on sympathetic and parasympathetic components of heart rate variability in normal subjects. Am J Cardiol 1989; 63: 1117–1120. [DOI] [PubMed] [Google Scholar]
- 28. Grassi G, Esler M. How to assess sympathetic activity in humans. J Hypertens 1999; 17: 719–734. [DOI] [PubMed] [Google Scholar]
- 29. van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low‐frequency variability of sympathetic nerve activity in severe heart failure. Circulation 1997; 95: 1449–1454. [DOI] [PubMed] [Google Scholar]
- 30. Radaelli A, Perlangeli S, Cerutti MC, Mircoli L, Mori I, Boselli L, Bonaita M, Terzoli L, Candotti G, Signorini G, Ferrari AU. Altered blood pressure variability in patients with congestive heart failure. J Hypertens 1999; 17: 1905–1910. [DOI] [PubMed] [Google Scholar]
- 31. Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta‐adrenergic‐receptor density in failing human hearts. N Engl J Med 1982; 307: 205–211. [DOI] [PubMed] [Google Scholar]
- 32. Ferguson DW, Berg WJ, Roach PJ, Oren RM, Mark AL. Effects of heart failure on baroreflex control of sympathetic neural activity. Am J Cardiol 1992; 69: 523–531. [DOI] [PubMed] [Google Scholar]
- 33. Kubo SH, Clark M, Laragh JH, Borer JS, Cody RJ. Identification of normal neurohormonal activity in mild congestive heart failure and stimulating effect of upright posture and diuretics. Am J Cardiol 1987; 60: 1322–1328. [DOI] [PubMed] [Google Scholar]
- 34. Gorelik O, Almoznino‐Sarafian D, Litvinov V, Alon I, Shteinshnaider M, Dotan E, Modai D, Cohen N. Seating‐induced postural hypotension is common in older patients with decompensated heart failure and may be prevented by lower limb compression bandaging. Gerontology 2009; 55: 138–144. [DOI] [PubMed] [Google Scholar]
- 35. Potocka‐Plazak K, Plazak W. Orthostatic hypotension in elderly women with congestive heart failure. Aging Clin Exp Res 2001; 13: 378–384. [DOI] [PubMed] [Google Scholar]