

Abstract
Spinal muscular atrophy (SMA) is a hereditary neurodegenerative disease with severity ranging from progressive infantile paralysis and premature death (type I) to limited motor neuron loss and normal life expectancy (type IV). Without disease‐modifying therapies, the impact is profound for patients and their families. Improved understanding of the molecular basis of SMA, disease pathogenesis, natural history, and recognition of the impact of standardized care on outcomes has yielded progress toward the development of novel therapeutic strategies and are summarized. Therapeutic strategies in the pipeline are appraised, ranging from SMN1 gene replacement to modulation of SMN2 encoded transcripts, to neuroprotection, to an expanding repertoire of peripheral targets, including muscle. With the advent of preliminary trial data, it can be reasonably anticipated that the SMA treatment landscape will transform significantly. Advancement in presymptomatic diagnosis and screening programs will be critical, with pilot newborn screening studies underway to facilitate preclinical diagnosis. The development of disease‐modifying therapies will necessitate monitoring programs to determine the long‐term impact, careful evaluation of combined treatments, and further acceleration of improvements in supportive care. In advance of upcoming clinical trial results, we consider the challenges and controversies related to the implementation of novel therapies for all patients and set the scene as the field prepares to enter an era of novel therapies. Ann Neurol 2017;81:355–368
Spinal muscular atrophy (SMA) is characterized by muscle weakness and severe physical disability attributed to motor neuron degeneration in the spinal cord and brainstem. It continues to represent the leading genetic cause of infant death attributed to respiratory insufficiency, with a pan‐ethnic incidence of approximately 1 in 11,000 live births and a carrier frequency of 1 in 40 to 67.1 The most common form of SMA is caused by mutations in the survival motor neuron 1 (SMN1) gene, resulting in SMN protein deficiency.2 The almost identical survival motor neuron 2 (SMN2) gene produces a small amount of functional SMN protein, and SMN2 copy number is recognized as a major modulator of the SMA phenotype.3 There have been significant advances in the understanding of the underlying pathogenic process in SMA. Concomitantly, there has been progress in defining disease progression and natural history, with a concerted effort in developing outcome measures and clinical trial readiness. Consequently, novel genetic therapies, aimed at modulating SMN protein expression, have resulted in significant clinical improvement in SMA patients for the first time, thereby providing much needed hope for the treatment of this devastating disease. As such, the present review will focus on recent advances made toward developing novel therapeutic strategies and future challenges as the field enters into a new treatment era.
Progress in Understanding the Natural History of SMA
SMA has a broad range of age of onset, severity, rate of progression, and variability between and within subtypes (Table 1).4, 5 In type I SMA, earlier age of onset is associated with worse prognosis and mortality6; the median age to death or ventilation (>16 hours per day) is 13.5 months and 10.5 months for patients with 2 copies of SMN2.5 Patients with SMA type II have a better prognosis than those with type I disease, with 93% surviving to 25 years.6 After age 15, a relative stability in function develops with subsequent gradual decline over time.7 Age of onset is also a predictor of functional ability, with patients classified as SMA IIIa having a 73% probability of walking 10 years after diagnosis, whereas SMA type IIIb patients have a 97% probability of walking 10 years after diagnosis.8
Table 1.
Classification and Subtypes of Spinal Muscular Atrophy
| Type | Age of Onset | Maximal Motor Milestone | Motor Ability and Additional Features | Prognosisc |
|---|---|---|---|---|
| SMA 0 | Before birth | None | Severe hypotonia; unable to sit or rolla | Respiratory insufficiency at birth; death within weeks |
| SMA I |
2 weeks (Ia) 3 months (Ib) 6 months (Ic) |
None | Severe hypotonia; unable to sit or rollb | Death/ventilation by 2 years |
| SMA II | 6 to 18 months | Sitting | Proximal weakness; unable to walk independently | Survival into adulthood |
| SMA III |
<3 years (IIIa) >3 years (IIIb) >12 years (IIIc) |
Walking | May lose ability to walk | Normal life span |
| SMA IV | >30 years or 10 to 30 years | Normal | Mild motor Impairment | Normal life span |
Need for respiratory support at birth; contractures at birth, reduced fetal movements.
Ia joint contractures present at birth; Ic may achieve head control.
Prognosis varies with phenotype and supportive care interventions.
A better understanding of the natural history of SMA has been crucial to the development of relevant outcome measures and implementation of clinical trials. Understanding variability in the rate of clinical progression in SMA related to age, SMA type, and ambulation status will assist in the development of appropriate motor function scales that are able to monitor subtle, but clinically meaningful changes (see Finkel et al9). The clinical heterogeneity in motor function is a challenge and connected with the range of possibilities for change in short time frames. The pattern of age‐related changes in motor function in SMA types II and III is nonlinear, and there are different patterns of progression between ambulant and nonambulant patients.7, 10 In nonambulant patients, variable improvement in motor function occurs up to 4 to 5 years of age, before functional ability (eg, in upper limbs) declines between 5 and 15 years. After age 15, a relative stability in function develops with subsequent gradual decline over time.7 Decline in motor and respiratory function within a 12‐month period was often minor, although progression was variable between individuals.7, 9 This slow rate of progression, particularly in milder phenotypes, poses a major challenge to clinical trials in SMA because most trials need to be completed within 1 to 2 years. Rather than SMA phenotype, ambulant status may be more relevant to the trajectory of disease progression and consequently in trial design and outcome measure development. For instance, different outcome measures are required to monitor clinically important differences among ambulant and nonambulant cohorts, yet it will also be important to better connect scales that measure different functional levels to be able to more accurately demonstrate improvements.10
In addition to motor function scales, neurophysiological studies have provided insights into clinical progression, timing of motor neuron loss, and compensation in SMA; however, these studies have predominantly focused on later stages of disease. Electrophysiological outcomes compound muscle action potential (CMAP) amplitude and motor unit number estimation (MUNE) correlate with age, SMA type, functional status, and SMN2 copy number.11 Gradual decline in motor and respiratory function may, in part, be related to physical growth in SMA types II and III, with the provision that CMAP amplitude remains stable.9 In addition, the CMAP amplitude may also remain constant despite reduction in MUNE values, a finding explained by the presence of motor unit loss with compensatory collateral sprouting and supported by axonal excitability studies.12 Transcranial magnetic stimulation techniques assessing central motor networks also suggest adaptive changes.13 Neurophysiological studies are rare in presymptomatic infants, but suggest that motor function is initially relatively preserved.11, 14 In SMA type I, this loss of motor function is followed by early and precipitous reductions in CMAP and MUNE responses. The onset, time course, and extent of motor neuron loss has not been established in SMA types II and III, yet is important in determining whether there is a specific therapeutic window. Preclinical studies of SMN restoring therapies, such as gene therapy and antisense oligonucleotides to correct SMN2 messenger RNA (mRNA) splicing, provide support for the utilization of electrophysiological biomarkers for treatment stratification, determining response and defining therapeutic windows.15
Clinical Care
In parallel with preclinical advances, continued improvements in multidisciplinary care and technological advances have altered the natural history of SMA since the Consensus Statement for standards of care in SMA was published in 2007 (Table 2; Appendix 1)16; however, even in areas of general consensus, marked variability in the implementation of the standards of care, particularly in the use of ventilation, nutritional support, and scoliosis surgery, have been observed.17, 18 For patients participating in clinical trials, it is crucial to standardize the management of modifiable factors, particularly nutrition and respiratory support, given that differences in care practices may impact outcome. However, at the most severe end of the SMA phenotypic spectrum, in the setting of uncertain therapeutic efficacy, this remains a challenge. Advances in drug development are likely to impact the standards of care for SMA, particularly given that successful disease modification will inevitably alter natural history and necessitate new standards of supportive care and interventions.
Table 2.
Current Management of Spinal Muscular Atrophy
| Assessments | Interventions | |
|---|---|---|
| Respiratory |
Cough effectiveness; respiratory muscle function tests; overnight oximetry; forced vital capacity (>6 years) Overnight polysomnography if disordered breathing suspected Acute respiratory infections |
Referral to respiratory specialist Routine immunizations Annual influenza vaccination Airway clearance techniques and cough assistance—chest physiotherapy, postural drainage, mechanical or manual cough assistance Noninvasive ventilation (nocturnal and/or daytime if indicated)a Antibiotics intensified airway clearance, increased ventilation supporta |
| Gastrointestinal and nutritional |
Feeding and swallowing assessment Assess caloric intake Assess for signs of reflux or aspiration Assess for constipation |
Nutritional supplementation, modifying food consistency, optimizing oral intake, positioning and seating alterations Nasogastric, nasojejunal, or percutaneous gastronomy—as soon as reduced oral intake is recognized Nissen fundoplication (if indicated) Hydration, regular oral aperients |
| Orthopedic and rehabilitation |
Posture, mobility, function Contractures Scoliosis Hip subluxation/dislocation |
Equipment to assist with mobility, self‐care, and function Physiotherapy, standing frames, orthoses Spinal surgeryb |
| Psychological | Assess for depression/anxiety | Counseling, pharmacotherapy |
The management of SMA incorporates a multidisciplinary and supportive approach, including neurologists (adult and pediatric), respiratory physicians, geneticists, gastroenterologists, palliative care physicians, rehabilitation specialists, orthopedic surgeons, and allied health.
The appropriate level of interventional support to prolong life, particularly in SMA type1, is controversial and the consensus statement16 recognizes the importance of discussions with the family to explore and define potential quality‐of‐life and palliative care issues. The philosophy and introduction of proactive respiratory support in patients with SMA type 1 varies considerably and practice varies internationally.
There is no consensus on management of scoliosis or hip subluxation/dislocation in nonambulant patients.
Respiratory Management
Respiratory complications are the major cause of morbidity and mortality in SMA. The onset of peripheral hypoventilation may be asymptomatic and initially occur during sleep, but with deterioration daytime respiratory dysfunction develops. Objective measures of respiratory function are not routinely performed in children younger than 4 to 6 years because of complexity of the required maneuvers; potential alternative measures of respiratory function, for example, sniff nasal inspiratory pressure and forced oscillation techniques, have been proposed in this population because they are noninvasive and require less patient cooperation.19 Attempts to identify nighttime hypoventilation using pulmonary testing in SMA patients have been largely unsuccessful,20 highlighting the continued benefit of overnight polysomnography. Assessment frequency needs to be individualized, based on current functional status and rate of disease progression, and should be supplemented with other clinical observations, such as assessment for paradoxical breathing and chest wall growth, among others.
The major respiratory complications faced by SMA patients include impaired cough—resulting in reduced clearance of lower airway secretions—hypoventilation, chest wall and lung underdevelopment, and recurrent infections that exacerbate muscle weakness. Although contentious, proactive management with noninvasive ventilation, even before the onset of paradoxical respirations, has led to improved survival, prevention, and improvement in chest wall deformity as well as improved quality of life.21, 22 In addition, optimizing airway clearance is important for acute and chronic management of SMA patients with secretion mobilization techniques, such as assisted coughing,23, 24, 25 physiotherapy, and postural drainage.26 The decision to progress to invasive ventilation with tracheostomy remains an ethical dilemma, and considerable variability exists between countries with no consensus in guidelines.17, 18 The goal of interventions should always be to improve quality of life with the provision of support and assistance to parents in making difficult decisions consonant with their values and beliefs.
Nutritional Support
Malnutrition is prevalent in SMA, with bulbar dysfunction and deterioration of nutritional status preceding and exacerbating respiratory failure with disease progression.27, 28 Appropriate nutritional management of SMA patients is critical for improving quality of life and optimizing survival,29 although no clear consensus exists on the timing of initiation of nutritional support.
Orthopedic Considerations
Scoliosis is a common complication of SMA, present in 60% to 95% of patients, secondary to progressive muscle weakness. In SMA types I and II, scoliosis occurs earlier and a more‐severe, progressive curvature is evident compared to SMA type III.30 Progression of scoliosis may exacerbate respiratory dysfunction, gastrointestinal reflux, and increase postural discomfort.30 Management of scoliosis includes nonsurgical options, such as physical therapy, bracing, and seating modification, and depending on the strategy used, may slow, but not necessarily prevent, curve progression. Additionally, surgical approaches are utilized in progressive scoliosis, most frequently in type II and III SMA. Posterior spinal fusion is typically implemented after skeletal maturity during adolescence in SMA, with iliac fixation used to assist correction of pelvic obliquity. Innovative surgical techniques utilizing growing rods (eg, vertical prosthetic titanium rib or magnetic rods) enable spinal growth while avoiding repeated invasive surgeries; however, medium‐ to longer‐term implications remain unclear. Although surgery does not reverse the respiratory reserve lost because of scoliosis, it leads to improved life quality31 and can slow deterioration of respiratory function.32 Finally, with an intrathecally administered therapy showing promise in phase 3 clinical trials, construction of bony windows may be considered to facilitate drug administration.
Genetic and Environmental Insights Into Pathogenesis
Whereas mutations in SMN1 characterize SMA, disease severity is also linked to a number of genetic modifiers. These modifiers are of relevance in enabling patient stratification in clinical trials, better prediction of an individual's prognosis, and establishing newborn screening. Patients have variable copy numbers of the SMN2 gene, a related gene that differs from SMN1 by only five nucleotides, altering splicing and leading to transcription of a nonfunctional SMN protein lacking exon 7 in the majority of transcripts (Fig 1).33 SMN2 copy number is the main determinant of phenotype, although not solely sufficient to predict severity.34 Sequence variations within the SMN2 gene and upregulation of modifier proteins, such as plastin 3, may also positively modify phenotype.35, 36, 37 In addition, nutritional deficiency, oxidative stress, and hypoxia, partly attributed to gastrointestinal dysfunction, may cause widespread splicing alterations, including SMN2, and accelerate SMA progression.38, 39, 40
Figure 1.
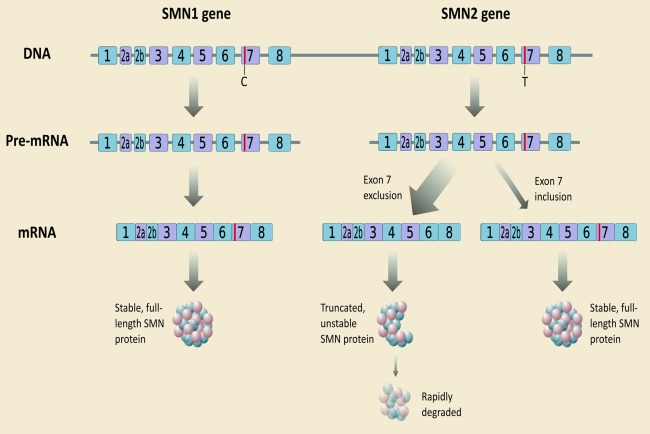
Genetics of Spinal Muscular Atrophy. In humans, the SMN protein is encoded by the SMN1 and SMN2 genes. The C to T substitution in exon 7 of SMN2 is translationally silent, but alters splicing such that the majority of SMN2 transcripts lack exon 7 and the truncated protein is unstable. Normally, SMN1 produces abundant SMN protein. In SMA, homozygous mutation of SMN1 results in only a small amount of functional SMN protein contributed by the varying copy numbers of SMN2. mRNA = messenger RNA; SMN = survival motor neuron [Color figure can be viewed at wileyonlinelibrary.com].
How Low Levels of SMN Cause SMA
Recent insights into the role of SMN within motor neurons have furthered our understanding of the implications of SMN deficiency.41 The best characterized role of the SMN complex is in the assembly of Sm proteins (a distinctive family of RNA associated small proteins) onto small nuclear RNAs (snRNAs), forming small nuclear ribonucleoproteins (snRNPs), which are essential components of pre‐mRNA splicing machinery in cells.42 SMN deficiency and therefore reduced snRNP assembly capacity are proposed to cause aberrant splicing or transport of RNPs to the detriment of motor neurons.43 A recent study showed transcriptional dysregulation in motor neurons isolated from very young presymptomatic SMA mice that preferentially affected a small subset of genes involved in synaptogenesis and maintenance of neuromuscular junctions (NMJs).43 Furthermore, some of these dysregulated motor neuron relevant genes showed underlying splicing changes, strengthening a potential link between aberrant splicing and motor neuron vulnerability.43
A second view of SMA pathogenesis contends that SMN has axonal function independent of splicing that may be disrupted in SMA. Consequently, SMN deficiency may impair targeting and local translation of axonal mRNAs essential for motor neuron development and maintenance.44, 45 Furthermore, SMN regulates several other fundamental cellular processes in the neuronal cytoplasm that are critical for maintaining axonal and synaptic health, including endocytic pathways, local translation, mitochondrial transport, and targeting to axons and ubiquitin homeostasis.46, 47, 48, 49, 50, 51
Animal Models of SMA
Current understanding of SMA pathogenesis has been generated largely as a result of the availability of mouse models of SMA. These have been largely generated by targeting the endogenous mouse Smn gene, while using transgenic strategies to add variable copy numbers of the human SMN2 transgene.52 These animal models tend to phenocopy severe forms of the human disease, resulting in the majority of animal‐based work focusing on early postnatal phenotypes. Recently, alternative strategies have been used to generate mice modeling less‐severe forms of SMA, allowing investigation of disease pathogenesis and preclinical drug testing more relevant to type II and III SMA.53, 54 Additional animal models of SMA are also beginning to play an important role in SMA research, including Drosophila and zebrafish,55, 56 and recent developments suggesting that large animal models (eg, pigs) may be forthcoming.57
Although alpha motor neurons in the spinal cord remain the primary pathological target in SMA,58 there is now accumulating evidence suggesting that other cells, tissues, and organs contribute to disease symptoms (Fig 2).59, 60 For example, there is now experimental evidence suggesting a non‐cell‐autonomous contribution to motor neuron degeneration from astrocytes and Schwann cells.61, 62 Likewise, low levels of SMN in skeletal muscle have been implicated in SMA pathogenesis with significant disruption of the molecular composition of skeletal muscle evident in presymptomatic severe SMA mice in the absence of detectable changes in lower motor neurons.63 One potential unifying factor may be a deficiency in the development of vasculature in SMA; the resulting hypoxia would likely impact motor neurons as well as skeletal muscle and possibly contribute to the gastrointestinal defects (gastroesophageal reflux, constipation, and delayed gastric emptying) commonly observed in SMA patients.64Although the mechanisms mediating the effects of vascular depletion have not been fully elucidated, hypoxia has been identified as a modifier of SMN2 splicing, potentially explaining some of the splicing alterations observed in SMA.38, 39, 65
Figure 2.
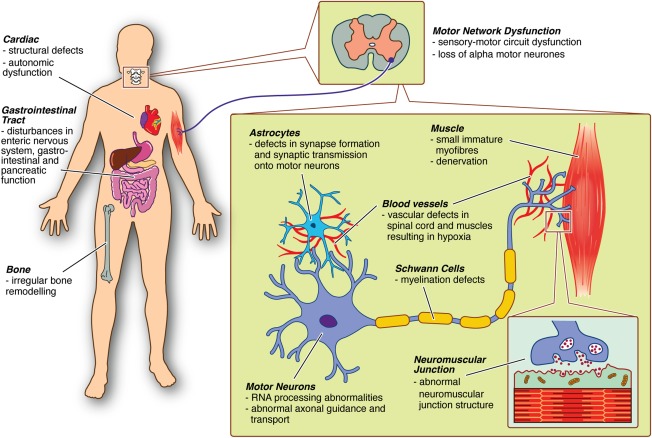
Pathophysiological findings in SMA. Multiple functional abnormalities in motor networks have been identified in SMA mice and humans, including defects in astrocytes, Schwann cells, motor neurons, and skeletal muscle. Disease‐associated phenotypes have also been reported across a range of other organs in SMA mice (in some cases supported by data from human patients), including cardiac structural and functional abnormalities, gastrointestinal tract dysfunction, and irregular bone remodeling. One potential unifying factor may be a deficiency in the development of vasculature in SMA, with the resulting hypoxia likely impacting a range of cell types. SMA = spinal muscular atrophy. [Color figure can be viewed at wileyonlinelibrary.com]
Disease‐associated phenotypes have been reported across a range of other organs in SMA mice (in some cases supported by data from human patients). These include functional and structural cardiac defects,66 abnormal development of the gastrointestinal tract, liver, and spleen,64, 67, 68 and irregular bone remodeling and skeletal pathology.69 These findings suggest that successful treatment of SMA may require systemic targeting of a range of affected tissues. However, how these findings will translate to humans is uncertain, for example, heart defects are rare in humans.
Defining the Therapeutic Window in Animal Models
In severe SMA mice, induction of SMN expression at gestation or in the early postnatal period substantially improves survival, whereas later induction is less effective.70, 71 Mice are resistant to SMN depletion after early postnatal stages, suggesting that there is a period of sensitivity to low SMN levels and that high SMN levels are required during this early postnatal stage.70, 72 Notably, in mouse models, the time period when SMN function is required coincides with the neonatal period of NMJ establishment, development, and maturation, suggesting that the mechanistic underpinnings of the therapeutic window are based on the pathways driving normal NMJ maturation. These observations imply that early correction of SMN levels in SMA types II and III is likely to be necessary and sufficient to protect the neuromuscular system, and lifelong expression of SMN may not be required.
Whereas most preclinical studies investigating SMN restoration in animals were limited to evaluating presymptomatic administration of gene therapy with viral vectors or antisense oligonucleotides to correct SMN2 splicing and enhance SMN expression, which, in most cases, robustly improves the health of SMA mice, some studies have tested the impact of pre‐ and postsymptomatic SMN restoration. Systemic delivery of these approaches largely rescued SMA mice's motor function, neuromuscular physiology, and life span when delivered within the first 3 postnatal days (P0–P3), but were less effective beyond P5 and gene therapy was not effective at P10, confirming the presence of a narrow therapeutic window.73, 74 However, SMN restoration using intravenous injection of self‐complementary adeno‐associated virus (scAAV9)‐SMN vectors given at symptom onset had a marked effect with amelioration of severe proximal weakness and electrophysiological indices, in a porcine model of SMA57 which may represent a more‐relevant model for predicting efficacy in humans. Even so, presymptomatic delivery prevented the development of symptoms, suggesting that therapeutic windows are still critical in this model. Whereas it is accepted that animal models cannot recapitulate human SMA precisely, the translation of concepts of motor neuron degeneration to humans suggests that presymptomatic or early‐symptomatic restoration of SMN (during NMJ maturation) will likely produce the best response to therapy. An unresolved issue remains as to whether commencing therapy in older patients will be effective. The time course and extent of motor neuron loss in type III or IV has never properly been mapped, largely attributed to a paucity of robust animal models of less‐severe forms of SMA. Encouragingly, recent results from models of milder SMA phenotypes suggest that some therapeutic efficacy may be possible even at late disease stages.75 Although the therapeutic window for SMA types III and IV has not been defined, the normal early motor development may suggest that it is linked to age of presentation and broader than types I and II. Preliminary clinical trial data are emerging and indicating that with SMN repletion motor neurons may not be irreversibly doomed. However, animal models recapitulating severe SMA show rapid postnatal motor neuron attrition and reduced efficacy with delayed treatments, such that the optimal success may ultimately arise from presymptomatic provision of therapy.73, 76
Therapeutic Developments
The pipeline of therapies for SMA encompasses four different strategies, including SMN1 gene replacement, modulation of SMN2 encoded full‐length protein levels, neuroprotection, and targeted improvements of muscle strength and function (Fig 3). Translational research continues to progress and clinical trials have recently reporting positive preliminary results related to safety and efficacy of the newest approaches (Table 3). This follows a number of negative clinical trials of repurposed drugs, including valproic acid and acetyl‐L‐carnitine, phenylbutyrate hydroxyurea, riluzole, and somatotropin,77, 78, 79 despite promising preclinical data. Importantly, these negative studies have informed clinical trial design, validated the reliability and feasibility of specific outcome measures, and highlighted the importance of patient stratification.
Figure 3.
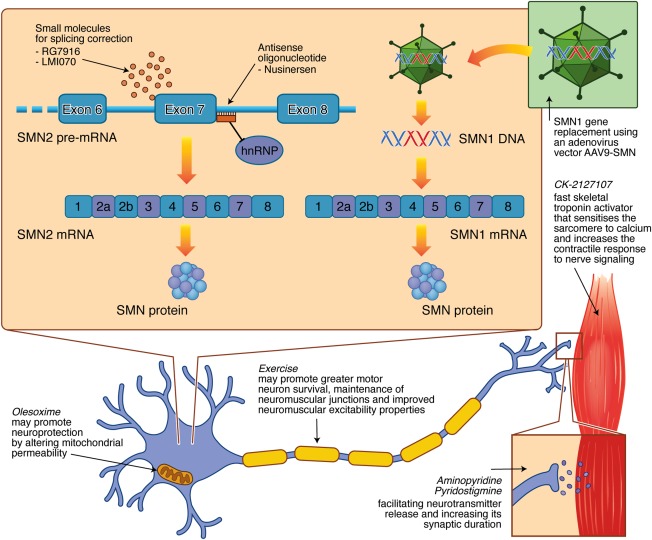
Therapeutic targets for SMA being investigated in clinical trials. SMN1 gene replacement therapy utilizes a self‐complementary adeno‐associated viral vector (AAV9‐SMN) that crosses the blood–brain barrier following intravenous administration. Compounds that increase the production of fully functional SMN protein by modifying the splicing of SMN2 include the orally available small molecules, RG7916 and LMI070, and the intrathecally administered antisense oligonucleotide, nusinersen, which acts by displacing heterogenous nuclear ribonucleoprotein (hnRP) proteins from the intronic splicing silencer site on the SMN2 pre‐mRNA. The neuroprotective effects of olesoxime, through altered mitochondrial permeability, and exercise, through greater motor neuron survival, maintenance of neuromuscular junctions, and improved neuromuscular excitability properties, are being investigated. Additional strategies focused on improving neuromuscular function and physical performance include CK‐2127107, a fast skeletal troponin activator that sensitizes the sarcomere to calcium and increases the contractile response to nerve signaling, and 4‐aminopyridine and pyridostigmine that may facilitate neurotransmitter release and increase its synaptic duration. mRNA = messenger RNA; SMN = survival motor neuron. [Color figure can be viewed at wileyonlinelibrary.com]
Table 3.
New Therapeutic Approaches in Spinal Muscular Atrophy: Current Clinical Trials
| Approach | Study Description | Preliminary Results |
|---|---|---|
| SMN1 gene replacement | ||
| AVXS‐101a, b |
Phase 1/2a gene transfer of SMN1 in SMA type I infants Two cohorts treated with a single dose of AVXS‐101 delivered intravenously |
Safety, survival, and motor function have been promising with all patients event free (death or continuous noninvasive ventilation greater than 16 hours per day) and stabilization of pulmonary outcomes.80 |
| Modulation of SMN2 full‐length protein | ||
| Nusinersen (IONIS–SMNRX)a |
10 phase 1 to phase 3 studies Nusinersen administered by intrathecal injection in SMA type I infants (0–6 months) and later‐onset type II/III participants (age range, 2–14 years) |
Favorable safety, tolerability, and encouraging clinical efficacy83, 84; intrathecal administration tolerated, no drug‐related adverse events82
Phase 3 ENDEAR study in SMA type I and phase 3 CHERISH study in childen aged 2 to 12 years with SMA type II; primary endpoint met in each study at interim analysis with statistically significant improvement in motor milestones |
| RG7916 (RO7034067) | Phase 2 in adult and paediatric patients with type II and III SMA with oral delivery | |
| LMI070 | Phase 1/2 study in infants with type I SMA (1–7 months) of oral LMI070 | |
| Neuroprotection: promote survival of motor neurons | ||
| Olesoxime (TRO19622) | Phase 2 studies in 3‐ to 25‐year‐olds with type II or nonambulant type III SMA | A greater percentage of patients were stable or improved compared with placebo; however, the primary endpoint was not met (p = 0.07).89 |
| Exercise | Pilot study of a physiotherapeutic approach tailored to type II and III SMA patients aged 5 to 10 years | |
| Exercise | Muscle‐strengthening program using hand weights and resistance bands in combination with a home‐based cycle ergometry in type III patients aged 8 to 50 years | |
| Enhancing Nerve or Muscle Function | ||
| CK‐2127107 | A phase 2 oral compound in SMA type II to IV (aged 12 years+). | |
| Pyridostigmine | Phase 2 study in SMA type III (aged 6 years+) | |
| 4‐aminopyridine | Phase 2/3 study assessing changes in walking ability and endurance in 18‐ to 50‐year‐olds with SMA type III | |
AVXS‐101 and Nusinersen have been granted US Food and Drug (FDA) and European Medicines Agency orphan drug status and FDA fast‐track approval.
AVXS‐101 has been granted FDA Breakthrough Therapy Designation. Following a FDA Type B meeting on September 30, 2016, a single‐arm pivotal trial has been announced.
In a mouse model of severe SMA, postnatal intravenous gene therapy using a viral vector rescued motor function and neurophysiology and extended survival from 2 weeks to beyond 250 days.70 A phase 1/2a clinical trial for AVXS‐101(a self‐complementary AAV9 carrying the SMN gene under the control of a hybrid cytomegalovirus enhancer/chicken‐β‐actin promoter) in SMA type I infants has completed enrollment and initial observations in safety, survival, and motor function have been promising with all patients event free (death or continuous noninvasive ventilation greater than 16 hours per day) and stabilization of pulmonary outcomes reported.80 Modest improvements in motor function were observed in patients receiving a low dose of the study drug and greater improvements were shown in patients receiving the proposed therapeutic dose; 2 patients achieved normal motor function 4.9 and 10.3 months following treatment as measured by the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP‐INTEND), a marked change from the natural history of SMA type I.81
The most advanced compound known to increase production of fully functional SMN protein is nusinersen (IONIS‐SMNRX), an antisense oligonucleotide administered intrathecally that modifies the splicing of SMN2. A phase 1 open label study of nusinersen in 28 SMA II and III patients aged 2 to 14 years reported that the drug was well tolerated with no safety concerns identified.82 Transient back pain and postlumbar puncture headache were of a similar frequency to previous reports in infants and children undergoing lumbar puncture. Favorable time‐ and dose‐dependent increases in muscle function were reported in patients 9 to 14 months postdosing.83 Observations from a phase 2 open‐label study of nusinersen in 20 infants with SMA type I show increases in motor function, ranging from stable independent sitting to walking (the latter patients having 3 copies of SMN2, not the standard type I SMA copy number of 2), with no evidence of a therapeutic plateau in motor skills yet.84 Interim analyses of phase 3 clinical trials evaluating nusinersen, including infants with SMA type I (ENDEAR) and children with SMA type II (CHERISH), have reached primary endpoints with improvement in motor milestones and favorable safety profiles. Regulatory filings have recently been submitted and an expanded access program initiated for SMA type I. The effect of presymptomatic administration of nusinersen is currently being evaluated and will provide pivotal insights into therapeutic windows. Further advances include the development of peptide‐mediated oligonucleotides to enable systemic therapy and overcome difficulties with central nervous system delivery by repeated intrathecal injections, with recent efficacy demonstrated in rodent models.85
Orally bioavailable small molecules are being developed for selective SMN2 splicing correction, and several (RG 7916 and LM1070) are entering early phase clinical trials. Administration of these compounds to mice with severe SMA increased SMN protein levels, motor function, and survival (from 18 to beyond 150 days).76, 86 Further clinical trials are also needed to define the efficacy of salbutamol, a β agonist that promotes exon 7 inclusion in SMN2 transcripts, following encouraging early results from several pilot studies in SMA patients.87 Increasing the expression of SMN2 with small molecules, such as quinazoline‐derived compounds, moderately increased SMN mRNA and protein levels as well as survival in severe SMA mice.88 However, plans to progress beyond phase 1 clinical trials have been terminated, reflecting challenges in translating disease‐modifying benefits from mouse to human.
Olesoxime has entered clinical trials in SMA patients following demonstration of its neuroprotective properties motor neurons in cell culture and SMA mice. Phase 2 trials have been completed and though the primary endpoint was not statistically significant, a greater percentage of patients were stable or improved compared to placebo, suggesting that olesoxime may slow decline in motor function over 2 years in already symptomatic patients with SMA types II and III.89 However, further data are needed to determine whether this is a clinically meaningful effect.
Additional strategies focused on improving neuromuscular function and physical performances in SMA patients are also being assessed in clinical trials. Among these is CK‐2127107, which slows calcium release from fast skeletal muscle troponin and sensitizes the sarcomere to calcium thus increasing contractile response to nerve signalling; studies have demonstrated its efficacy in mouse models of motor neuron disease and it appears safe in healthy human volunteers.90 Additionally, exercise‐induced neuroprotection has recently been demonstrated in SMA‐like mice with greater motor neuron survival, maintenance of neuromuscular junctions, and improved neuromuscular excitability properties, accompanied by positive metabolic and behavioral changes.91 The benefits and risks of different types of exercise are being evaluated in SMA patients, and initial studies have demonstrated that resistance training is feasible, safe, and well tolerated and aerobic training increases oxidative capacity.92, 93 Further outcomes will be important in planning patient therapy and rehabilitation.
A number of potential SMN independent therapeutic targets have been identified in preclinical studies. These include the compounds, Fasudil and Y‐27632, that regulate actin cytoskeleton integrity through Rho‐associated protein kinase inhibition,94, 95 the antioxidant flavonoid, quercetin, that suppresses beta‐catenin signaling,96 BAY 55‐9837 that indirectly stabilizes SMN mRNA,97 and compounds that activate the mammalian target of rapamycin pathway.98 In addition, RNA sequencing of motor neurons may identify novel downstream targets of splicing alterations. Stabilization of endogenous SMN protein provides a further therapeutic strategy, with STL‐182 showing promising preclinical efficacy.99
Conclusions and Future Directions
There have been tremendous advances in therapeutic development in SMA, with treatment options rapidly evolving and preliminary results of clinical trials in patients producing new hope. In parallel, there has been substantial progress in understanding clinical disease progression and natural history to accelerate the implementation of clinical trials. Rodent models suggest that requirements for normal SMN levels are paramount during development of the motor unit, with SMN restoring therapies most effective early. The translation of these concepts to humans is needed to determine whether therapy in later stages of disease is beneficial. Critical for timely access to novel disease‐modifying treatments is the rapid recognition of clinical manifestations and diagnosis, with presymptomatic diagnosis to guard against disease onset and progression the ultimate aim. Population‐based newborn screening pilot programs are determining the feasibility and reliability of presymptomatic diagnosis, and effective molecular methods have been validated on dried blood spots, including real‐time polymerase chain reaction and high‐resolution melting analysis.100, 101, 102, 103
Improvements in multidisciplinary clinical care, together with advances in technology, have changed the natural history for patients with SMA. With new therapeutics emerging, it is likely that profound shifts in management approaches will transpire in severe SMA. These will also necessitate additional validation of nonmotor standardized and reliable outcome measures, particularly respiratory assessments, given that these are functionally meaningful and contribute substantially to morbidity. Secondary complications, such as scoliosis and contractures, may further limit the value of existing motor outcome measures. Furthermore, individual motor function scales are relevant to specific levels of SMA severity, and it will be important to better connect scales that measure different functional levels to be able to more accurately demonstrate improvements. Whereas motor function scales are a major focus and most relevant to SMA, strength testing, electrophysiological assessments, and metabolomic and proteomic outcomes measures are also being integrated into natural history studies and clinical trials in SMA.104
With the tantalizing prospect of novel therapies moving closer to clinical reality, more questions arise, compelling the formation of collaborative and linked future monitoring programs to determine the impact of these therapies. Longer‐term monitoring programs should also include assessments of cognition, growth, autonomic function, and adverse events and enable a comparison and evaluation of combined treatments. These will serve to understand how novel therapies may affect phenotype over the longer term and the duration of effect—will they reduce progression, as well as stabilize or improve function? How will they affect requirements for permanent ventilation or age of death? What is the potential variability of responsiveness of different motor neuron subpopulations attributed to drug distribution or inherent differences in reversibility? Furthermore, the possibility that reinnervation may stress remaining motor neurons, resulting in a post‐polio‐like condition with late deterioration, must be considered. It is likely that combined therapies increasing SMN levels while also enhancing and preserving neuromuscular function and preventing additional systemic pathology will provide the best approach. In the setting of a first‐in‐class approved therapy, continued progress in developing second‐generation and combination therapies will require innovative approaches in trial design. In addition, new challenges are arising with emerging therapies, including difficulties with access to treatment associated with the complexities, costs, and expertise required with intrathecal administration. Further efforts to ascertain optimal routes of drug delivery and distribution and defining the therapeutic window will be essential.
As the field looks toward a new treatment era, it is necessary to focus on timely access to novel, disease‐modifying therapy and endeavoring to develop therapies for patients with SMA of all ages and severities.
Author Contributions
The authors contributed equally to the literature search and the writing and formatting of the review, and to critically reviewing the manuscript.
Potential Conflicts of Interest
Dr Farrar has received an honoraria from Biogen. Professor Gillingwater is named on a patent application filed by the University of Edinburgh covering the use of beta‐catenin inhibitors for the treatment of SMA (DD/P206869GB, filed October 7, 2013).
Supporting information
Additional supporting information can be found in the online version of this article.
Appendix 1: Additional evidence base for management of spinal muscular atrophy subsequent to 2007 standards of care
Acknowledgment
M.F. received support from the Motor Neurone Diseases Research Institute of Australia Beryl Bayley MND Postdoctoral Fellowship. T.G. received support from the UK SMA Research Consortium (SMA Trust) and Muscular Dystrophy UK. B.T. received support from the National Health and Medical Research Council project grant (#1104299), Stafford Fox Medical Research Foundation, and Cure for MND Foundation. K.J.S. received support from the National Institutes of Child Health and Development grant R01‐ HD69045 from the National Institutes of Health.
References
- 1. Sugarman EA, Nagan N, Zhu H, et al. Pan‐ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet 2012;20:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 1995;80:155–165. [DOI] [PubMed] [Google Scholar]
- 3. Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999;8:1177–1183. [DOI] [PubMed] [Google Scholar]
- 4. Dunaway S, Montes J, Ryan PA, et al. Spinal muscular atrophy type III: trying to understand subtle functional change over time: a case report. J Child Neurol 2012;27:779–785. [DOI] [PubMed] [Google Scholar]
- 5. Finkel RS, McDermott MP, Kaufmann P, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 2014;83:810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Farrar MA, Vucic S, Johnston HM, et al. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr 2013;162:155–159. [DOI] [PubMed] [Google Scholar]
- 7. Mercuri E, Finkel R, Montes J, et al. Patterns of disease progression in type 2 and 3 SMA: implications for clinical trials. Neuromuscul Disord 2016;26:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zerres K, Rudnik‐Schoneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol 1995;52:518–523. [DOI] [PubMed] [Google Scholar]
- 9. Finkel R, Bertini E, Muntoni F, et al. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7–9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord 2015;25:593–602. [DOI] [PubMed] [Google Scholar]
- 10. Kaufmann P, McDermott MP, Darras BT, et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012;79:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol 2005;57:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farrar MA, Vucic S, Lin CS, et al. Dysfunction of axonal membrane conductances in adolescents and young adults with spinal muscular atrophy. Brain 2011;134:3185–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farrar MA, Vucic S, Johnston HM, et al. Corticomotoneuronal integrity and adaptation in spinal muscular atrophy. Arch Neurol 2011;69:467–473. [DOI] [PubMed] [Google Scholar]
- 14. Finkel RS. Electrophysiological and motor function scale association in a pre‐symptomatic infant with spinal muscular atrophy type I. Neuromuscul Disord 2013;23:112–115. [DOI] [PubMed] [Google Scholar]
- 15. Arnold W, McGovern VL, Sanchez B, et al. The neuromuscular impact of symptomatic SMN restoration in a mouse model of spinal muscular atrophy. Neurobiol Dis 2016;87:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 2007;22:1027–1049. [DOI] [PubMed] [Google Scholar]
- 17. Bladen CL, Thompson R, Jackson JM, et al. Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J Neurol 2014;261:152–163. [DOI] [PubMed] [Google Scholar]
- 18. Oskoui M, Ng P, Liben S, et al. Physician driven variation in the care of children with spinal muscular atrophy type 1. Pediatr Pulmonol 2016. Sep 29. doi: 10.1002/ppul.23616. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 19. Khirani S, Colella M, Caldarelli V, et al. Longitudinal course of lung function and respiratory muscle strength in spinal muscular atrophy type 2 and 3. Eur J Paediatr Neurol 2013;17:552–560. [DOI] [PubMed] [Google Scholar]
- 20. Bersanini C, Khirani S, Ramirez A, et al. Nocturnal hypoxaemia and hypercapnia in children with neuromuscular disorders. Eur Respir J 2012;39:1206–1212. [DOI] [PubMed] [Google Scholar]
- 21. Han YJ, Park JD, Lee B, et al. Home mechanical ventilation in childhood‐onset hereditary neuromuscular diseases: 13 years' experience at a single center in Korea. PLoS One 2015;10:e0122346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lemoine TJ, Swoboda KJ, Bratton SL, et al. Spinal muscular atrophy type 1: are proactive respiratory interventions associated with longer survival? Pediatr Crit Care Med;13:e161–e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chatwin M, Bush A, Simonds AK. Outcome of goal‐directed non‐invasive ventilation and mechanical insufflation/exsufflation in spinal muscular atrophy type I. Arch Dis Child 2011;96:426–432. [DOI] [PubMed] [Google Scholar]
- 24. Lacombe M, Del Amo Castrillo L, Bore A, et al. Comparison of three cough‐augmentation techniques in neuromuscular patients: mechanical insufflation combined with manually assisted cough, insufflation‐exsufflation alone and insufflation‐exsufflation combined with manually assisted cough. Respiration 2014;88:215–222. [DOI] [PubMed] [Google Scholar]
- 25. Stehling F, Bouikidis A, Schara U, et al. Mechanical insufflation/exsufflation improves vital capacity in neuromuscular disorders. Chron Respir Dis 2015;12:31–35. [DOI] [PubMed] [Google Scholar]
- 26. Dunaway S, Montes J, McDermott MP, et al. Physical therapy services received by individuals with spinal muscular atrophy (SMA). J Pediatr Rehabil Med 2016;9:35–44. [DOI] [PubMed] [Google Scholar]
- 27. Mehta NM, Newman H, Tarrant S, et al. Nutritional status and nutrient intake challenges in children with spinal muscular atrophy. Pediatr Neurol 2016;57:80–83. [DOI] [PubMed] [Google Scholar]
- 28. Davis RH, Godshall BJ, Seffrood E, et al. Nutritional practices at a glance: spinal muscular atrophy type I nutrition survey findings. J Child Neurol 2014;29:1467–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mannaa MM, Kalra M, Wong B, et al. Survival probabilities of patients with childhood spinal muscle atrophy. J Clin Neuromuscul Dis 2009;10:85–89. [DOI] [PubMed] [Google Scholar]
- 30. Fujak A, Raab W, Schuh A, et al. Natural course of scoliosis in proximal spinal muscular atrophy type II and IIIa: descriptive clinical study with retrospective data collection of 126 patients. BMC Musculoskelet Disord 2013;14:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suk KS, Baek JH, Park JO, et al. Postoperative quality of life in patients with progressive neuromuscular scoliosis and their parents. Spine J 2015;15:446–453. [DOI] [PubMed] [Google Scholar]
- 32. Chua K, Tan CY, Chen Z, et al. Long‐term follow‐up of pulmonary function and scoliosis in patients with duchenne's muscular dystrophy and spinal muscular atrophy. J Pediatr Orthop 2016;36:63–69. [DOI] [PubMed] [Google Scholar]
- 33. Lorson CL, Hahnen E, Androphy EJ, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A 1999;96:6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feldkotter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real‐time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002;70:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008;320:524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet 2009;85:408–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bernal S, Alias L, Barcelo MJ, et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet 2010;47:640–642. [DOI] [PubMed] [Google Scholar]
- 38. Bebee TW, Dominguez CE, Samadzadeh‐Tarighat S, et al. Hypoxia is a modifier of SMN2 splicing and disease severity in a severe SMA mouse model. Hum Mol Genet 2012;21:4301–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sahashi K, Hua Y, Ling KK, et al. TSUNAMI: an antisense method to phenocopy splicing‐associated diseases in animals. Genes Dev 2012;26:1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seo J, Singh NN, Ottesen EW, et al. Oxidative stress triggers body‐wide skipping of multiple exons of the spinal muscular atrophy gene. PLoS One 2016;11:e0154390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 2009;10:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Q, Fischer U, Wang F, et al. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 1997;90:1013–1021. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Z, Pinto AM, Wan L, et al. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc Natl Acad Sci U S A 2013;110:19348–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Akten B, Kye MJ, Hao le T, et al. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci U S A 2011;108:10337–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fallini C, Donlin‐Asp PG, Rouanet JP, et al. Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J Neurosci 2016;36:3811–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Powis R, Karyka E, Boyd P, et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 2016;1:e87908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sanchez G, Dury AY, Murray LM, et al. A novel function for the survival motoneuron protein as a translational regulator. Hum Mol Genet 2013;22:668–684. [DOI] [PubMed] [Google Scholar]
- 48. Wishart TM, Mutsaers CA, Riessland M, et al. Dysregulation of ubiquitin homeostasis and beta‐catenin signaling promote spinal muscular atrophy. J Clin Invest 2014;124:1821–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu CC, Denton KR, Wang ZB, et al. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis Model Mech 2016;9:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hosseinibarkooie S, Peters M, Torres‐Benito L, et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet 2016;99:647–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dimitriadi M, Derdowski A, Kalloo G, et al. Decreased function of survival motor neuron protein impairs endocytic pathways. Proc Natl Acad Sci U S A 2016;113:E4377–E4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Awano T, Kim JK, Monani UR. Spinal muscular atrophy: journeying from bench to bedside. Neurotherapeutics 2014;11:786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bogdanik LP, Osborne MA, Davis C, et al. Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc Natl Acad Sci U S A 2015;112:E5863–E5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bowerman M, Murray LM, Beauvais A, et al. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul Disord 2012;22:263–276. [DOI] [PubMed] [Google Scholar]
- 55. Praveen K, Wen Y, Gray KM, et al. SMA‐causing missense mutations in survival motor neuron (Smn) display a wide range of phenotypes when modeled in Drosophila. PLoS Genet 2014;10:e1004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Giacomotto J, Rinkwitz S, Becker TS. Effective heritable gene knockdown in zebrafish using synthetic microRNAs. Nat Commun 2015;6:7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Duque SI, Arnold WD, Odermatt P, et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann Neurol 2015;77:399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Powis RA, Gillingwater TH. Selective loss of alpha motor neurons with sparing of gamma motor neurons and spinal cord cholinergic neurons in a mouse model of spinal muscular atrophy. J Anat 2016;228:443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 2013;19:40–50. [DOI] [PubMed] [Google Scholar]
- 60. Shababi M, Lorson CL, Rudnik‐Schoneborn SS. Spinal muscular atrophy: a motor neuron disorder or a multi‐organ disease? J Anat 2014;224:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rindt H, Feng Z, Mazzasette C, et al. Astrocytes influence the severity of spinal muscular atrophy. Hum Mol Genet 2015;24:4094–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hunter G, Aghamaleky Sarvestany A, Roche SL, et al. SMN‐dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum Mol Genet 2014;23:2235–2250. [DOI] [PubMed] [Google Scholar]
- 63. Mutsaers CA, Wishart TM, Lamont DJ, et al. Reversible molecular pathology of skeletal muscle in spinal muscular atrophy. Hum Mol Genet 2011;20:4334–4344. [DOI] [PubMed] [Google Scholar]
- 64. Sintusek P, Catapano F, Angkathunkayul N, et al. Histopathological defects in intestine in severe spinal muscular atrophy mice are improved by systemic antisense oligonucleotide treatment. PLoS One 2016;11:e0155032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Somers E, Lees RD, Hoban K, et al. Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann Neurol 2016;79:217–230. [DOI] [PubMed] [Google Scholar]
- 66. Shababi M, Habibi J, Yang HT, et al. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet 2010;19:4059–4071. [DOI] [PubMed] [Google Scholar]
- 67. Szunyogova E, Zhou H, Maxwell GK, et al. Survival motor neuron (SMN) protein is required for normal mouse liver development. Sci Rep 2016;6:34635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Thomson AK, Somers E, Powis RA, et al. Survival of motor neurone protein is required for normal postnatal development of the spleen. J Anat 2016;230:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shanmugarajan S, Tsuruga E, Swoboda KJ, et al. Bone loss in survival motor neuron (Smn(‐/‐) SMN2) genetic mouse model of spinal muscular atrophy. J Pathol 2009;219:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Le TT, McGovern VL, Alwine IE, et al. Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet 2011;20:3578–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lutz CM, Kariya S, Patruni S, et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest 2011;121:3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kariya S, Obis T, Garone C, et al. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest 2014;124:785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Foust KD, Wang X, McGovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol 2010;28:271–274. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74. Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long‐term rescue of a severe spinal muscular atrophy mouse model. Nature 2011;478:123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feng Z, Ling KK, Zhao X, et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum Mol Genet 2016;25:964–975. [DOI] [PubMed] [Google Scholar]
- 76. Naryshkin NA, Weetall M, Dakka A, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014;345:688–693. [DOI] [PubMed] [Google Scholar]
- 77. Kirschner J, Schorling D, Hauschke D, et al. Somatropin treatment of spinal muscular atrophy: a placebo‐controlled, double‐blind crossover pilot study. Neuromuscul Disord 2014;24:134–142. [DOI] [PubMed] [Google Scholar]
- 78. Wadman RI, Bosboom WM, van der Pol WL, et al. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev 2012:Cd006282. [DOI] [PubMed] [Google Scholar]
- 79. Wadman RI, Bosboom WM, van der Pol WL, et al. Drug treatment for spinal muscular atrophy type I. Cochrane Database Syst Rev 2012:Cd006281. [DOI] [PubMed] [Google Scholar]
- 80. Shell RD, Kotha K, Al‐zaidy S, et al. Gene therapy for spinal muscular atrophy type 1 improves survival and stabilizes pulmonary outcomes in a phase I/IIa safety study. Am J Respir Crit Care Med 2016;193:A1040. [Google Scholar]
- 81. Mendell JR, Al‐Zaidy S, Shell R, et al. Gene therapy for spinal muscular atrophy type 1 shows potential to improve survival and motor functional outcomes. Mol Ther 2016;24:s190. [Google Scholar]
- 82. Hache M, Swoboda KJ, Sethna N, et al. Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J Child Neurol 2016;31:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS‐SMNRx) in children with spinal muscular atrophy. Neurology 2016;86:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile‐onset spinal muscular atrophy with nusinersen: a phase 2, open‐label, dose‐escalation study. Lancet 2017;388:3017–3026. [DOI] [PubMed] [Google Scholar]
- 85. Hammond SM, Hazell G, Shabanpoor F, et al. Systemic peptide‐mediated oligonucleotide therapy improves long‐term survival in spinal muscular atrophy. Proc Natl Acad Sci U S A 2016;113:10962–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Palacino J, Swalley SE, Song C, et al. SMN2 splice modulators enhance U1‐pre‐mRNA association and rescue SMA mice. Nat Chem Biol 2015;11:511–517. [DOI] [PubMed] [Google Scholar]
- 87. Giovannetti AM, Pasanisi MB, Černiauskaitė M, et al. Perceived efficacy of salbutamol by persons with SMA: a mixed methods study. Muscle Nerve 2016;54:843–849. [DOI] [PubMed] [Google Scholar]
- 88. Butchbach ME, Singh J, Thorsteinsdottir M, et al. Effects of 2,4‐diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet 2010;19:454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dessaud E, Andre C, Scherrer B, et al. Results of a phase II study to assess safety and efficacy of olesoxime (TRO19622) in 3–25 years old spinal muscular atrophy patients. Neuromuscul Disord 2014;24:791–924. [Google Scholar]
- 90. Hwee DT, Kennedy A, Ryans J, et al. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL‐SOD1G93A ALS mouse model. PLoS One 2014;9:e96921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chali F, Desseille C, Houdebine L, et al. Long‐term exercise‐specific neuroprotection in spinal muscular atrophy‐like mice. J Physiol 2016;594:1931–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Madsen KL, Hansen RS, Preisler N, et al. Training improves oxidative capacity, but not function, in spinal muscular atrophy type III. Muscle Nerve 2015;52:240–244. [DOI] [PubMed] [Google Scholar]
- 93. Lewelt A, Krosschell KJ, Stoddard GJ, et al. Resistance strength training exercise in children with spinal muscular atrophy. Muscle Nerve 2015;52:559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bowerman M, Beauvais A, Anderson CL, et al. Rho‐kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum Mol Genet 2010;19:1468–1478. [DOI] [PubMed] [Google Scholar]
- 95. Bowerman M, Murray LM, Boyer JG, et al. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med 2012;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Uzunalli G, Bora‐Tatar G, Dayangac‐Erden D, et al. Effects of flavonoid quercetin on survival of motor neuron gene expression. Cell Biol Int 2015;39:350–354. [DOI] [PubMed] [Google Scholar]
- 97. Hadwen J, MacKenzie D, Shamim F, et al. VPAC2 receptor agonist BAY 55‐9837 increases SMN protein levels and moderates disease phenotype in severe spinal muscular atrophy mouse models. Orphanet J Rare Dis 2014;9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Little D, Valori CF, Mutsaers CA, et al. PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Mol Ther 2015;23:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Calder AN, Androphy EJ, Hodgetts KJ. Small molecules in development for the treatment of spinal muscular atrophy. J Med Chem 2016;59:10067–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dobrowolski SF, Pham HT, Downes FP, et al. Newborn screening for spinal muscular atrophy by calibrated short‐amplicon melt profiling. Clin Chem 2012;58:1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A 2010;152a:1608–1616. [DOI] [PubMed] [Google Scholar]
- 102. Pyatt RE, Prior TW. A feasibility study for the newborn screening of spinal muscular atrophy. Genet Med 2006;8:428–437. [DOI] [PubMed] [Google Scholar]
- 103. Taylor JL, Lee FK, Yazdanpanah GK, et al. Newborn blood spot screening test using multiplexed real‐time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin Chem 2015;61:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kolb SJ, Coffey CS, Yankey JW, et al. Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study. Ann Clin Transl Neurol 2016;3:132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.
Appendix 1: Additional evidence base for management of spinal muscular atrophy subsequent to 2007 standards of care