

Abstract
Heart failure with reduced ejection fraction (HFrEF) is a progressive disorder whereby cardiac structure and function continue to deteriorate, often despite the absence of clinically apparent signs and symptoms of a worsening disease state. This silent yet progressive nature of HFrEF can contribute to the increased risk of death—even in patients who are ‘clinically stable’, or who are asymptomatic or only mildly symptomatic—because it often goes undetected and/or undertreated. Current therapies are aimed at improving clinical symptoms, and several agents more directly target the underlying causes of disease; however, new therapies are needed that can more fully address factors responsible for underlying progressive cardiac dysfunction. In this review, mechanisms that drive HFrEF, including ongoing cardiomyocyte loss, mitochondrial abnormalities, impaired calcium cycling, elevated LV wall stress, reactive interstitial fibrosis, and cardiomyocyte hypertrophy, are discussed. Additionally, limitations of current HF therapies are reviewed, with a focus on how these therapies are designed to counteract the deleterious effects of compensatory neurohumoral activation but do not fully prevent disease progression. Finally, new investigational therapies that may improve the underlying molecular, cellular, and structural abnormalities associated with HF progression are reviewed.
Keywords: Heart failure, Progressive deterioration, Treatment, Mechanism, Stable heart failure
Introduction
Patients with heart failure (HF) are often considered clinically stable if they are receiving treatment and show no physical signs and symptoms suggestive of worsening cardiac function. However, a lack of clinically observable signs and symptoms of worsening HF may not always be indicative of a patient's long‐term prognosis. Although currently available therapies for HF with reduced ejection fraction (HFrEF) can be used to minimize clinical symptoms, most do little to correct the underlying pathology responsible for progressive cardiac dysfunction. Recent studies have shown that most patients with clinically stable HFrEF have chronic low‐level elevations in plasma troponin, a marker for myocardial injury and cardiomyocyte loss that is associated with neurohormonal activation, inflammation, oxidative stress, and cell wall stretch.1, 2, 3 HF is thus a progressive disorder, and ongoing cardiac structural and functional deterioration is present in many patients who are asymptomatic or mildly symptomatic.4 Once LV dysfunction is established, it can worsen over the course of months or years without any clinically apparent symptoms;5 thus, progressive yet silent worsening of the HF state often remains unrecognized, and, consequently, may be left undertreated.4 Despite their apparent clinical stability, these patients remain at high risk of adverse outcome. Sudden cardiac death, for example, is most common in patients with mild or moderate HF symptoms who could be considered ‘clinically stable’.6 Furthermore, in patients with HFrEF, gradual functional and structural deterioration can lead to progressive lowering of the LVEF and progressive LV dilation, both of which are surrogate indicators of poor long‐term outcome.7, 8
Recognition of the underlying processes in HFrEF that drive the progression of cardiac functional and structural deterioration in the absence of clinically apparent signs and symptoms is essential for the development of novel therapies needed to counteract and prevent this ongoing deterioration. However, HF is extremely complex, and a number of cellular and subcellular maladaptations may exist in the failing heart that can have a direct or indirect impact on driving the slow but ongoing state of deterioration that leads to worsening disease.9 Many of these processes can also occur as part of ageing and, therefore, are not always identified as mediators of the gradually deteriorating HF state.10 A principal challenge is to identify which derangements can be reversed to impact a patient's clinical trajectory (i.e. to discern underlying risk factors from clinical risk markers).
In this review, we will examine the nature of these adverse, biologically active processes; evaluate the impact of existing therapies on them; and discuss novel treatment strategies that can ameliorate or limit their impact. In doing so, physicians can aim to prevent or, at the very least, slow the pace of ongoing cardiac structural and functional deterioration toward intractable HFrEF.
Underlying intrinsic mechanisms that drive left ventricular dysfunction
Cardiomyocyte apoptosis
Early studies of the pathophysiology of HFrEF identified cardiomyocyte apoptosis as a potentially important factor in the progression of LV dysfunction, and cardiomyocyte loss is now recognized as a defining feature of HFrEF.5, 11, 12, 13 Adult cardiomyocytes have a limited capacity for self‐renewal, and the gradual loss of functional cardiac units through apoptosis contributes to disease progression.12, 13, 14 The increase in cardiomyocyte apoptosis is associated with increased levels of cytotoxic pro‐inflammatory cytokines and reactive oxygen species (ROS).14 Additionally, members of the tumour necrosis factor (TNF)/TNF‐receptor superfamily have been linked to numerous adverse processes that include cell death, hypertrophy, and fibrosis.15 Low levels of soluble TNF‐like weak inducer of apoptosis (sTWEAK) are associated with increased risk of functional impairment and mortality, and sTWEAK levels are decreased in patients with stable HFrEF.16 Cardiomyocyte apoptosis and autophagy can also occur as a result of increased wall stress.3, 17 Recent evidence has suggested that TNF may also initiate necroptosis, a tightly controlled form of necrosis that also results in cardiomyocyte death.18
Mitochondrial abnormalities
In addition to cardiomyocyte loss through apoptosis and necrosis, the function of residual viable cardiomyocytes of the failing heart can be impaired as a result of deficiencies in availability of energy related primarily to abnormalities in mitochondria.19 Abnormalities of mitochondrial structure and function exist in the failing heart of both humans and experimental animals, as evidenced by hyperplasia, reduced organelle size, diminished rate of ATP synthesis, and increased formation of ROS.19, 20, 21, 22, 23 Mitochondrial functional abnormalities in the failing heart include poor respiration, reduced membrane potential (Δψm), and opening of the permeability transition pore.19, 21, 22 Examination of these mitochondrial abnormalities, along with other identified metabolic defects in HF, led, in part, to the emergence of the concept that the failing heart is ‘energy starved’, a condition synonymous with ‘an engine out of fuel’.24 Although this is perhaps an overly simplified explanation of HF, since energy starvation is probably a downstream consequence of profound disruption in myocardial architecture, it highlights the importance of considering modulations of both extracellular and cellular components as key contributors to worsening LV function. Further research is needed to understand how different processes may be inter‐related to drive the cascade of events that lead to progression of HF.
In addition to abnormalities of ATP synthesis, impairments in mitochondrial function are associated with derangements in the electron transport chain and excessive production of ROS. Excessive production of ROS by mitochondria can exert widespread adverse effects on the failing heart through damage to cellular and subcellular components that include components of key signalling pathways, membrane lipid sublayers, and components of the extracellular matrix.25, 26, 27
Impaired intracellular calcium cycling
Calcium signalling plays an important role in modulating systolic and diastolic function, and in regulating excitation–contraction coupling.28, 29 Abnormalities of intracellular calcium handling are known to develop in failing cardiomyocytes.30, 31, 32 These abnormalities are characterized by reduced sarcoplasmic reticulum activity and expression of calcium ATPase (SERCA2a), reduced phosphorylation of phospholamban, and increased phosphorylation of the ryanodine channel with subsequent development of a ryanodine calcium leak.28, 33, 34 The net result of these abnormalities is the development of intracellular calcium irregularities, which can lead to calcium overload, arrhythmogenesis, cardiomyocyte dysfunction, and often death of the cardiomyocyte.28, 33, 34
Wall stress
During the course of evolving HFrEF, the left ventricle progressively enlarges and its shape transforms from elliptical to one that more closely approximates a sphere.35, 36 These structural changes promote worsening HF symptoms and predict poor long‐term outcome.37 An important contributor to progressive LV systolic dysfunction in HFrEF, and supported by Laplace's Law, is the elevation of LV wall stress that results from progressive LV enlargement and wall thinning. Wall stress or tension is a major determinant of myocardial oxygen consumption (MVO2). Structural LV changes that develop during disease progression can, therefore, lead to increased wall stress and consequently increased MVO2 with increased energy demands; a condition the failing heart can ill afford.38
Fibrosis and cardiomyocyte hypertrophy
Global remodelling of the failing left ventricle is invariably accompanied by structural changes at the cellular level characterized by cardiomyocyte hypertrophy, secondary accumulation of collagen in the interstitium termed ‘reactive interstitial fibrosis’, reduced capillary density, and increased oxygen diffusion distance.39, 40, 41 These abnormalities favour increased LV stiffness and the development of hypoxia that can lead to progressive worsening of LV function.40
Cardiac fibroblast activation may also play a primary role in myocardial disease as a result of microRNA‐21 stimulation of mitogen‐activated protein kinase signalling in response to cardiac stress. This increase in cardiac fibroblast survival leads to fibrosis, hypertrophy, and cardiac dysfunction.42 It is also noteworthy that clinical evaluation of myocardial fibrosis using cardiovascular magnetic resonance extracellular volume fraction measures may predict HF prognosis. In a recent study, Schelbert and colleagues showed that such measures of myocardial fibrosis were associated with risk of HF hospitalization, death, or both.43 However, available techniques for measuring fibrotic changes associated with myocardial interstitial disease may not be practical for clinical use, making it difficult to detect and easy to overlook such changes.44
Hypertrophy of the residual viable myocardium develops early after myocardial injury in response to increased workload. As such, it is always present in the failing heart even under clinically stable conditions. Therefore, therapies that reduce the burden of interstitial fibrosis and attenuate cardiomyocyte hypertrophy should improve tissue oxygenation, improve capillary density, and reduce LV stiffness, and, in doing so, improve global LV systolic and diastolic function as well as ‘passive’ LV filling.
Extrinsic mechanisms that drive left ventricular dysfunction
Heart failure with reduced ejection fraction is characterized by a reduction of cardiac output that leads to a reduction in perfusion pressure to vital organs.45 The body's natural response is to compensate for these changes via neurohormonal activation—namely activation of the sympathetic nervous system (SNS), the renin–angiotensin–aldosterone system (RAAS), and the natriuretic peptide system (NPS), all designed to maintain homeostasis.45 Activation of the SNS results in the release of norepinephrine and epinephrine, which lead to increased heart rate and cardiac contractility, thereby increasing cardiac output and perfusion pressure to vital organs.28, 45 Activation of the RAAS results in the formation and release of a host of potent vasoconstrictors, namely angiotensin‐II, aldosterone, and vasopressin, all of which are intended to restore homeostasis.45 NPS activation counteracts the vasoconstrictive effects of the RAAS and SNS in blood vessels by stimulating vasodilation via adjustments to salt and water excretion.45 However, as HF advances, there is inadequate activation of, or a diminished response to, natriuretic peptides (NPs). This subdued response when coupled with enhanced and sustained activation of the RAAS fails to elicit the necessary balance between vasoconstriction and vasodilation.46 Ultimately, vasoconstriction prevails, thus setting the stage for a sustained elevation of systemic vascular resistance that is characteristic of HFrEF.
Enhanced and sustained activation of the SNS and RAAS results in undesirable consequences that include cardiomyocyte injury and loss, development of cellular hypertrophy and reactive interstitial fibrosis, water retention, increased sodium reabsorption, and increased vascular resistance—all of which contribute to the deteriorative state of HFrEF.45, 47 Further, enhanced and sustained neurohormonal activation can also lead to the development of malignant ventricular arrhythmias with increased incidence of sudden cardiac death.45 The adverse consequences of sustained neurohumoral activation include a sustained increase of heart rate, LV filling pressure, and contractility, along with structural LV remodelling manifested by progressive LV dilation and increased LV chamber sphericity with attendant increase of LV wall stress and tension.
Elevated heart rate, LV wall stress and tension, contractility, and elevated preload and afterload are all major determinants of MVO2. The resultant increase of MVO2, an energy demand state that the failing heart can ill afford and indeed cannot supply, can itself drive progressive dysfunction and loss of functional cardiac units, and thus worsening of HF. Injury to tissue secondary to the toxic effects of sustained neurohumoral activation can also trigger an inflammatory response with elaboration of proinflammatory cytokines that themselves become part of the deleterious arsenal targeting the heart and other organs.
Limitations of current heart failure treatments
Current treatments for HF are aimed at counteracting the sustained enhancement of neurohumoral compensation and its adverse pathological consequences.45 Digoxin reduces SNS and RAAS activation, and diuretics relieve fluid retention that can cause pulmonary congestion and peripheral oedema.45 ACE inhibitors and ARBs reduce RAAS activation and reverse the maladaptive effects of angiotensin‐II and, as such, act as vasodilators, evidenced by reduction of systemic pressure and lowering of systemic vascular resistance.45 Certain ACE inhibitors and ARBs have demonstrated the ability to decrease levels of proinflammatory cytokines and other markers of inflammation.48 However, the efficacy of ACE inhibitors may decrease over time due to aldosterone escape.49
Beta‐blockers protect the heart and vasculature from the adverse effects of SNS overactivity and reduce the heart rate, allowing for a reduction of MVO2 through at least a heart rate reduction pathway.45 Beta‐blockers can also improve calcium handling within the sarcoplasmic reticulum through restoration of physiological calcium release via improvement of ryanodine receptor function and restoration of SERCA2a activity and expression.31 Some beta‐blockers have anti‐inflammatory properties and can reduce circulating levels of inflammatory cytokines.48 Aldosterone antagonists (mineralocorticoid receptor blockers) directly inhibit the actions of aldosterone and have been shown to attenuate the burden of reactive interstitial fibrosis.50 Positive cAMP‐stimulating inotropic agents such as beta‐1 agonists and phosphodiesterase‐III inhibitors are used in HF to increase cardiac output through a direct increase of intrinsic myocardial contractility. However, these agents are indicated for short‐term use only, and not recommended for chronic use.
Ivabradine is a specific and selective inhibitor of the If current that reduces heart rate and has been reported to reverse some structural, biochemical, and molecular maladaptations found in HF.51 In the Phase III Systolic Heart Failure Treatment With the If Inhibitor Ivabradine Trial (SHIFT) of patients with chronic HFrEF, ivabradine reduced the rates of hospital admissions for worsening HF and deaths due to HF compared with placebo, although the primary endpoint was driven by a reduction in hospitalizations.52 The fundamental reduction of heart rate elicited by ivabradine and the expected consequential decrease in MVO2 is the key to its effectiveness in this patient population. Ivabradine is currently approved in Europe and the USA.
When viewed in aggregate, current pharmacological therapies for HF elicit their benefits by reducing cardiac workload through partial mitigation of the adverse consequences of sustained neurohumoral activation. The effective reduction of cardiac workload through lowering of heart rate and systemic vascular resistance elicits a highly desirable reduction of MVO2, thus lowering myocardial energy demands to levels in line with reduced myocardial energy supply. However, it has become clear over the last decade that, despite improvement in the haemodynamic status of the affected patient afforded by current pharmacological therapies, HF remains a progressive disease of significant mortality and morbidity. It is true that the past three decades have seen enormous success in the management of patients with HF with the use of current medical therapies. However, if a patient with HF is to achieve greater survival and a better quality of life, then other mechanisms responsible for the progressive deterioration of heart, kidney, and skeletal muscle function must be addressed.
Novel therapies that target extrinsic and intrinsic factors of heart failure
Considerable advances have been made in treatment strategies targeting extrinsic factors through modulation of the NPS. The approach holds promise in that these peptides promote vasodilation and directly or indirectly counteract pathological growth, fibrosis, and cardiac dysfunction.49 The BNP, nesiritide, is approved in the USA and Canada for the treatment of acute decompensated HF. In the pivotal VMAC trial, nesiritide reduced pulmonary capillary wedge pressure compared with nitroglycerin and placebo, and was associated with improvements in some self‐reported symptoms (e.g. dyspnoea) compared with placebo.53 However, conflicting findings have been observed on the efficacy and safety of nesiritide in subsequent studies. Some studies (including ADHERE) showed a similar or decreased risk of short‐term mortality or worsening renal function with nesiritide vs. comparators,54, 55, 56, 57, 58 while findings from meta‐analyses have shown an increased risk of short‐term death59 and worsening renal function.60 In the ASCEND‐HF trial, nesiritide was associated with a modestly reduced symptom burden compared with conventional therapy in European but not American patients with acute decompensated HF, and did not affect rates of death or rehospitalization. Based on these results, the authors of ASCEND‐HF recommended against routine use of nesiritide in patients with acute HF.61
The atrial natriuretic peptide (ANP)‐like peptide urodilatin regulates sodium and water handling and protects against hypertension, and in Phase II studies of synthetic urodilatin (ularitide), favourable haemodynamic, neurohumoral, and symptomatic effects have been observed, without adverse effects on renal function.62, 63 The TRUE‐AHF randomized, double‐blind, placebo‐controlled, Phase III study of ularitide in patients with acute HF (NCT01661634) was recently completed, but, to date, results have not been published.64 Another ANP‐like peptide, carperitide, is approved in Japan for the treatment of HF. In the randomized, controlled PROTECT study (n = 49), carperitide was associated with significantly reduced rates of death and rehospitalzation during 18 months of follow‐up compared with controls.65 However, a recent retrospective propensity score‐matched analysis found that patients treated with carperitide (n = 402) had an increased rate of in‐hospital mortality.66
Currently available ANPs and BNPs are limited by short bioavailability and the risk of hypotension.49 To minimize the undesirable effects of recombinant NPs, several ‘designer NPs’ have been developed.49 Many of these drugs have been designed to mimic the antifibrotic, antiproliferative, and antihypertrophic effects of C‐type natriuretic peptide (CNP).49 Another novel approach to augmenting the NPS involves inhibition of neutral endopeptidase (neprilysin), which hydrolyses CNP, ANP, and BNP, as well as many other vasoactive peptides such as angiotensin‐I, bradykinin, and adrenomedullin.49 However, possibly due to its broad affinity for many peptides beyond the NP, monotherapy with neprilysin inhibitors has demonstrated limited clinical efficacy.67 Given the opposing roles of the RAAS and NPS in HF, it was hypothesized that the addition of a neprilysin inhibitor could enhance the beneficial effects of RAAS inhibition.49 These beneficial effects were confirmed in clinical studies, but combining neprilysin inhibition with ACE inhibition was associated with serious angioedema.68, 69 Because ARBs are less likely to cause angioedema than ACE inhibitors, the combination of neprilysin inhibition with ARB therapy was explored as a treatment option for HF.49 Sacubitril/valsartan (LCZ696)—a salt complex comprising the prodrug sacubitril (a neprilysin inhibitor) with the ARB valsartan in a 1:1 ratio49, 70—is a novel drug that delivers simultaneous neprilysin inhibition and angiotensin‐II type 1 (AT1) receptor blockade, thus capitalizing on the beneficial effects of the NPS, while blocking the detrimental effects of an overactive RAAS (Figure 1).
Figure 1.
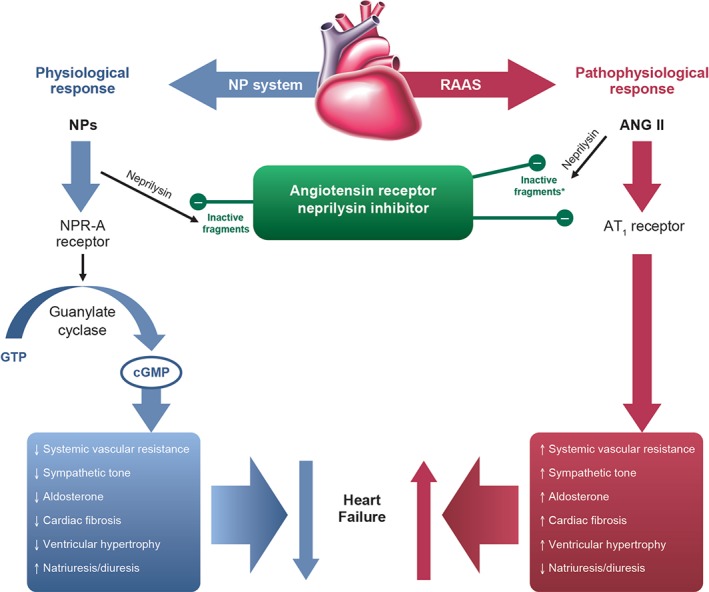
Mechanism of action for sacubitril/valsartan.93 Reprinted from Langenickel TH, Dole WP. Angiotensin receptor–neprilysin inhibition with LCZ696: a novel approach for the treatment of heart failure. Drug Discov Today Ther Strateg 2013; 9:e131–e139. ANG, angiotensin; AT1, angiotensin‐II type 1; cGMP, cyclic guanosine monophosphate; GTP, guanosine‐5'‐triphosphate; NP, natriuretic peptide (e.g. atrial natriuretic peptide, BNP); NPR‐A, NP receptor‐A; RAAS, renin–angiotensin–aldosterone system. *In vitro evidence.
Because the neprilysin enzyme degrades NPs as well as other vasoactive peptides, inhibition of neprilysin preserves the NPs and, therefore, enhances their effects on vasorelaxation, natriuresis, diuresis, and inhibition of cardiac fibrosis and cardiomyocyte hypertrophy. Through simultaneous blockade of the AT1 receptor, sacubitril/valsartan also suppresses the long‐term harmful effects of RAAS overactivation, such as vasoconstriction and sodium and water retention, and, in doing so, ameliorates the congestive HF state. Further, the combined natriuretic and vasodilator properties of sacubitril/valsartan may provide renal protection by reducing intraglomerular pressure and proteinuria.71, 72, 73
In pre‐clinical studies, sacubitril/valsartan reduced cardiomyocyte hypertrophy and cardiomyocyte apoptosis, improved endothelial function, prevented adverse cardiac remodelling, and attenuated cardiac dysfunction.74 Pre‐clinical studies of dual RAAS–neprilysin inhibitors have also shown increased synthesis of nitric oxide, probably from endothelial nitric oxide synthase (eNOS), which can lead to improved mitochondrial respiration and biogenesis.75 The Phase II PARAMOUNT and Phase III PARADIGM‐HF studies compared sacubitril/valsartan with valsartan and enalapril, respectively.70, 76, 77 In both trials, sacubitril/valsartan significantly reduced levels of NT‐proBNP, a marker for LV wall stretch. Results from the PARAMOUNT study also showed that sacubitril/valsartan significantly reduced high‐sensitivity troponin‐T levels compared with valsartan in patients with HF with preserved ejection fraction (HFpEF), suggesting amelioration of ongoing cardiomyocyte degeneration and loss.78 When compared with enalapril in the PARADIGM‐HF trial of patients with HFrEF, sacubitril/valsartan treatment resulted in decreased mortality and hospitalizations for HF along with sustained reductions in circulating troponin‐T levels.70, 77
In addition to the recently approved sacubitril/valsartan, other agents in development that target improvement in cardiomyocyte function or survival include: phosphoinositide‐3 kinases; selective type 5 phosphodiesterase inhibitors (e.g. sildenafil); β3 adrenergic receptor agonists (e.g. nebivolol); gene therapies to increase SERCA2a expression; microRNAs that regulate cardiomyocyte hypertrophy, contractility, and function; myosin activators (e.g. omecamtiv mecarbil); and agents that may prevent cardiomyocyte death (e.g. cyclosporine A, necrostatin‐1, neuregulin 1, and heat shock proteins that act as cardioprotective molecular chaperones).79 Agents in development that may regulate interstitial remodelling processes include thrombospondins, secreted protein acid rich in cysteine (SPARC), syndecans, torasemide, and microRNAs that inhibit fibroblast proliferation and alter extracellular matrix properties.79
Other novel HF therapies that are currently under development may improve cardiac unloading and target the structural and functional abnormalities that exist in the failing heart. It is well known that vascular endothelial growth factor (VEGF), acting through its receptor, can promote cell proliferation and possibly cell migration and regeneration. Additionally, growth factors such as VEGF, basic fibroblast growth factor, and hepatocyte growth factor can promote angiogenesis through increased myocardial capillary density.80, 81, 82 Therapy with VEGF‐A, VEGF‐B, and other agents that boost cardiac angiogenesis is being explored in pre‐clinical and early‐phase clinical studies.79
New therapies are needed that target intrinsic structural and functional abnormalities of the failing heart rather than easing the work burden on the heart by targeting treatment to the periphery. To this end, considerable research was conducted in recent years to target and reverse abnormalities of sarcoplasmic reticulum calcium handling in the failing heart. These abnormalities include dysfunction of ryanodine receptor 2 (RyR2), evidenced by hyperphosphorylation of RyR2 and the presence of a pathological calcium leak, down‐regulation of SERCA2a along with reduced activity and reduced calcium uptake, and dysfunction of the sodium–calcium exchanger, which is evidenced by hyperphosphorylation and operation in ‘reverse mode’.28 Despite these efforts, no new drugs have currently been identified that directly target these signalling pathways. Recently published results from the randomized, double‐blind, multicentre CUPID 2 clinical study of SERCA2a gene therapy in patients with HFrEF were disappointing because this treatment failed to improve the clinical course of worsening HFrEF.83
Substantial research has been, and continues to be, expended in understanding the adverse ramifications of the excessive proinflammatory state that exists in HF, which is characterized by excess elaboration of proinflammatory cytokines including TNF‐α and interleukin‐6. Drugs approved for indications other than HF that have anti‐inflammatory properties or that target certain cytokines involved in the pathogenesis of HF (e.g. statins, etanercept, infliximab, methotrexate, and colchicine) have been tested in HF, but results of these studies to date have been disappointing or inconclusive.48
Serelaxin, a recombinant form of human relaxin‐2, has been reported to increase arterial compliance, cardiac output, and renal blood flow.84 Relaxin ligand–receptor binding has been shown to increase the expression of VEGF.85 Furthermore, in the failing heart, capillary density is reduced and oxygen diffusion distance is increased, leading to regional hypoxia, and relaxin can therefore relieve myocardial hypoxia, leading to improved myocardial oxygenation and improved LV function. Relaxin ligand–receptor binding can also stimulate the activity of eNOS and release of nitric oxide.86 In the Phase III RELAX‐AHF trial, serelaxin was associated with dyspnoea relief and reduced 180‐day cardiovascular mortality rates in patients with acute HF, but failed to reduce rates of all‐cause death or hospital readmission for HF or renal failure before day 60 compared with placebo.87
The soluble guanylate cyclase (sGC)–cGMP pathway has been identified as a potential target in patients with HF and sGC stimulators, and sGC activators may provide haemodynamic improvements and organ‐protective properties independently of haemodynamic changes.88 The sGC stimulator, vericiguat, is currently being evaluated in patients with worsening chronic HF in the Phase II SOCRATES programme, consisting of a trial for patients with HF and preserved LVEF ≥45% (SOCRATES‐PRESERVED) and another for patients with HF and reduced LVEF <45% (SOCRATES‐REDUCED).89
Other novel therapies that have been tested in pre‐clinical studies and are currently in Phase II clinical trials in patients with HF include (i) partial adenosine A1 receptor (pA1R) agonists; (ii) mitochondria‐targeting peptides; and (iii) myocardial augmentation with hydrogels. In dogs with chronic systolic HF, 12 weeks of therapy with a pA1R agonist resulted in improved LV systolic function and prevention of progressive LV enlargement, as evidenced by a significant improvement in LVEF and reduced LV end‐systolic volume (Figure 2).90 Unlike full adenosine A1 receptor agonists, pA1R agonists are not associated with bradycardia, heart block, sedation, or antidiuretic effects. The benefits of pA1R agonists for the treatment of HF lie in their ability to afford protection to the failing myocardium by limiting triggers of cell injury and death and potentially by providing the necessary energy to the working myocardium.90
Figure 2.
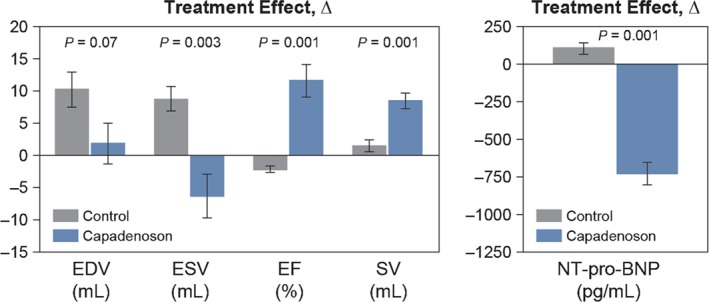
Treatment effect of a partial adenosine A1‐receptor agonist on LV function.90 Left: change Δ (treatment effect) between pre‐treatment and 12 weeks post‐treatment of LV end‐diastolic volume (EDV), end‐systolic volume (ESV), EF, and stroke volume (SV) in untreated control dogs and dogs treated for 3 months with capadenoson. Right: change Δ (treatment effect) between pre‐treatment and 12 weeks post‐treatment in plasma levels of NT‐proBNP. Data are shown as mean ± SEM. Probability values are comparisons between untreated control and capadenoson. Reprinted from Sabbah HN et al. Chronic therapy with a partial adenosine A1‐receptor agonist improves left ventricular function and remodelling in dogs with advanced heart failure. Circ Heart Fail 2013;6:563–571.
Elamipretide (MTP131), a novel mitochondria‐targeting peptide that enhances ATP synthesis and limits excessive ROS production, improved LV systolic function without increasing heart rate or decreasing mean arterial pressure compared with placebo in a canine model of chronic HF (Figure 3).91, 92 Additionally, elamipretide has been demonstrated to reverse abnormalities of mitochondrial function and normalize the rate of ATP synthesis in cardiomyocytes isolated from canines with experimentally induced advanced HF.91, 92 The benefits of this targeted approach to the treatment of HF lie in the ability of this peptide to improve mitochondrial function in HF, thus providing the necessary energy to the working myocardium through improved mitochondrial function, while at the same time significantly reducing the formation of tissue‐destructive ROS originating from the mitochondria.
Figure 3.
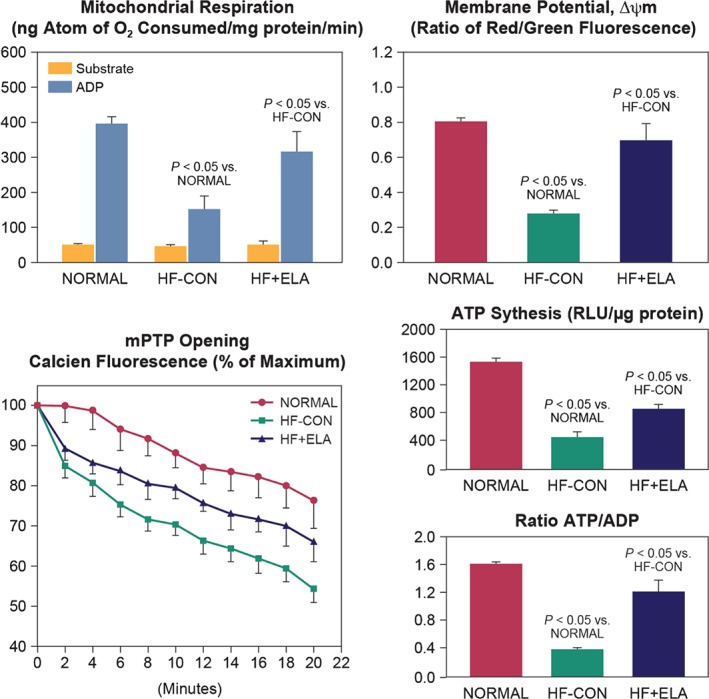
Mitochondrial function in cardiomyocytes of LV myocardium of normal dogs, untreated heart failure control dogs (HF‐CON), and dogs with heart failure treated with elamipretide (HF + ELA).92 Top left: mitochondrial state 3 and 4 respiration. Top right: mitochondrial membrane potential. Bottom left: mitochondrial permeability transition pore (mPTP). Bottom right: maximum rate of adenosine triphosphate (ATP) synthesis and ratio of ATP to adenosine diphosphate (ADP). Probability values are comparisons between normal dogs, HF‐CON, and HF + ELA dogs. Statistical significance based on one‐way analysis of variance (ANOVA) followed by the Student–Newman–Keuls test. All bar graphs are depicted as the mean ± SEM. Reprinted from Sabbah HN et al. Chronic therapy with elamipretide (MTP‐131), a novel mitochondria‐targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ Heart Fail 2016;9:e002206.
In dogs with advanced HF, augmentation of LV wall thickness with intramyocardial alginate hydrogel implants (AHIs) significantly improved LV systolic and diastolic function and was associated with preload‐independent improvement of LV performance.36 Therapy with AHIs that directly targeted the heart also reduced LV size, partially restored physiological LV shape and reduced LV wall stress, and had a favourable impact at the cellular level consistent with reverse LV remodelling.36
Conclusions
Despite current optimal medical therapy in patients with stable HF, the underlying structural and molecular abnormalities that drive disease progression persist in many, if not all, patients. This progression is often silent and frequently remains undetected, and is only manifested with the emergence of a serious life‐threatening event. Understanding the mechanisms contributing to progression of cardiac dysfunction in HF has led to the identification of therapeutic targets that may correct some of the key underlying cardiac abnormalities. Novel approaches to therapy, such as a neprilysin inhibitor plus ARB therapy, relaxin, pA1R agonists, sGC activators and stimulators, mitochondria‐targeting peptides, and direct cardiac wall thickness augmentation, have the potential to correct some of these abnormalities. Although promising new treatments for stable yet progressive HF may be on the horizon, continued efforts directed at the development of treatments that target the multitude of complex molecular mechanisms associated with HF progression are needed. More effort should be expended in the development of therapies that target structural and functional abnormalities of the heart rather than the periphery. These therapies should strive toward ‘haemodynamic neutrality’. New drugs that further lower heart rate and blood pressure beyond those that exist today are less likely to succeed and more likely to cause harm in this patient population. Finally, while we shift attention to the heart as the target of therapy and move away from targeting the periphery, we should not lose sight of the need to address abnormalities of skeletal muscle and the kidney in the setting of chronic HF. Abnormalities of skeletal muscle are the key to improving exercise tolerance in HF and, hence, quality of life of the affected patient. By the same token, renal dysfunction in HF, if left unattended, will undoubtedly contribute to poor survival. The ideal therapy for HF is one that reverses or prevents the progression of intrinsic cardiac structural and functional abnormalities, improves skeletal muscle function, and affords the necessary protection to the kidneys.
Acknowledgements
The author was fully responsible for all content and editorial decisions and received no financial support or other form of compensation related to the development of this manuscript. Technical assistance with editing and styling of the manuscript for submission was provided by Oxford PharmaGenesis Inc. This assistance was funded by Novartis Pharmaceuticals Corporation.
Conflicts of interest: H.N.S has received research grants from Bayer AG, Stealth Biotherapeutics, Cardioxyl Pharmaceuticals, Lonestar Heart, Amgen, Merck, Novartis, Mast Therapeutics, and Johnson and Johnson; is a consultant to Bayer AG, Stealth Biotherapeutics, and Mast Therapeutics; and has served as a member of the Scientific and/or Medical Boards for BioControl Medical, Inc., Cardioxyl Pharmaceuticals, Mast Therapeutics, Bayer, Novartis, Merck, and Takeda.
References
- 1. Lyons KS, McKeeman G, McVeigh GE, Harbinson MT. High‐sensitivity troponin T is detectable in most patients with clinically stable heart failure. Br J Cardiol 2014;21:33–36. [Google Scholar]
- 2. Alehagen U, Dahlström U, Rehfeld JF, Goetze JP. Prognostic assessment of elderly patients with symptoms of heart failure by combining high‐sensitivity troponin T and N‐terminal pro‐B‐type natriuretic peptide measurements. Clin Chem 2010;56:1718–1724. [DOI] [PubMed] [Google Scholar]
- 3. Januzzi JL Jr, Filippatos G, Nieminen M, Gheorghiade M. Troponin elevation in patients with heart failure: on behalf of the third Universal Definition of Myocardial Infarction Global Task Force: Heart Failure Section. Eur Heart J 2012;33:2265–2271. [DOI] [PubMed] [Google Scholar]
- 4. Goldberg LR, Jessup M. Stage B heart failure: management of asymptomatic left ventricular systolic dysfunction. Circulation 2006;113:2851–2860. [DOI] [PubMed] [Google Scholar]
- 5. Sabbah HN, Sharov VG, Goldstein S. Programmed cell death in the progression of heart failure. Ann Med 1998;30 Suppl 1:33–38. [PubMed] [Google Scholar]
- 6. Carson P, Anand I, O'Connor C, Jaski B, Steinberg J, Lwin A, Lindenfeld J, Ghali J, Barnet JH, Feldman AM, Bristow MR. Mode of death in advanced heart failure: the Comparison of Medical, Pacing, and Defibrillation Therapies in Heart Failure (COMPANION) trial. J Am Coll Cardiol 2005;46:2329–2334. [DOI] [PubMed] [Google Scholar]
- 7. Yeboah J, Bluemke DA, Hundley WG, Rodriguez CJ, Lima JA, Herrington DM. Left ventricular dilation and incident congestive heart failure in asymptomatic adults without cardiovascular disease: multi‐ethnic study of atherosclerosis (MESA). J Card Fail 2014;20:905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoon HJ, Jeong MH, Jeong Y, Kim KH, Song JE, Cho JY, Jang SY, Jeong HC, Lee KH, Park KH, Sim DS, Yoon NS, Hong YJ, Park HW, Kim JH, Ahn Y, Cho JG, Park JC, Kang JC. Progressive dilation of the left atrium and ventricle after acute myocardial infarction is associated with high mortality. Korean Circ J 2013;43:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mill JG, Stefanon I, dos Santos L, Baldo MP. Remodeling in the ischemic heart: the stepwise progression for heart failure. Braz J Med Biol Res 2011;44:890–898. [DOI] [PubMed] [Google Scholar]
- 10. Strait JB, Lakatta EG. Aging‐associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 2012;8:143–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sabbah HN, Sharov VG, Goldstein S. Cell death, tissue hypoxia and the progression of heart failure. Heart Fail Rev 2000;5:131–138. [DOI] [PubMed] [Google Scholar]
- 12. Sabbah HN. Apoptosis in heart failure: a real problem? Cardiovasc Drugs Ther 2001;15:525–528. [DOI] [PubMed] [Google Scholar]
- 13. Fiedler LR, Maifoshie E, Schneider MD. Mouse models of heart failure: cell signaling and cell survival. Curr Top Dev Biol 2014;109:171–247. [DOI] [PubMed] [Google Scholar]
- 14. Johnson FL. Pathophysiology and etiology of heart failure. Cardiol Clin 2014;32:9–19, vii. [DOI] [PubMed] [Google Scholar]
- 15. Novoyatleva T, Sajjad A, Engel FB. TWEAK–Fn14 cytokine–receptor axis: a new player of myocardial remodeling and cardiac failure. Front Immunol 2014;5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chorianopoulos E, Rosenberg M, Zugck C, Wolf J, Katus HA, Frey N. Decreased soluble TWEAK levels predict an adverse prognosis in patients with chronic stable heart failure. Eur J Heart Fail 2009;11:1050–1056. [DOI] [PubMed] [Google Scholar]
- 17. Jiang L, Huang Y, Hunyor S, dos Remedios CG. Cardiomyocyte apoptosis is associated with increased wall stress in chronic failing left ventricle. Eur Heart J 2003;24:742–751. [DOI] [PubMed] [Google Scholar]
- 18. Orogo AM, Gustafsson AB. Cell death in the myocardium: my heart won't go on. IUBMB Life 2013;65:651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 2000;32:2361–2367. [DOI] [PubMed] [Google Scholar]
- 20. Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol 1992;24:1333–1347. [DOI] [PubMed] [Google Scholar]
- 21. Sharov VG, Goussev A, Lesch M, Goldstein S, Sabbah HN. Abnormal mitochondrial function in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol 1998;30:1757–1762. [DOI] [PubMed] [Google Scholar]
- 22. Sharov VG, Todor A, Khanal S, Imai M, Sabbah HN. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J Mol Cell Cardiol 2007;42:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugamura K, Keaney JF Jr. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med 2011;51:978–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neubauer S. The failing heart—an engine out of fuel. N Engl J Med 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 25. Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H2O2 regulates cardiac myocyte phenotype via concentration‐dependent activation of distinct kinase pathways. J Mol Cell Cardiol 2003;35:615–621. [DOI] [PubMed] [Google Scholar]
- 26. Siwik DA, Colucci WS. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev 2004;9:43–51. [DOI] [PubMed] [Google Scholar]
- 27. Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 2013;61:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lou Q, Janardhan A, Efimov IR. Remodeling of calcium handling in human heart failure. Adv Exp Med Biol 2012;740:1145–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res 2013;113:690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end‐stage heart failure. Circ Res 1987;61:70–76. [DOI] [PubMed] [Google Scholar]
- 31. Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D, Burkhoff D, Marks AR. β‐Blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation 2003;107:2459–2466. [DOI] [PubMed] [Google Scholar]
- 32. George I, Sabbah HN, Xu K, Wang N, Wang J. β‐Adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in a coronary embolization model of heart failure in canines. Cardiovasc Res 2011;91:447–455. [DOI] [PubMed] [Google Scholar]
- 33. Braunwald E. Heart failure. JACC Heart Fail 2013;1:1–20. [DOI] [PubMed] [Google Scholar]
- 34. Mishra S, Sabbah HN, Rastogi S, Imai M, Gupta RC. Reduced sarcoplasmic reticulum Ca2+ uptake and increased Na+–Ca2+ exchanger expression in left ventricle myocardium of dogs with progression of heart failure. Heart Vessels 2005;20:23–32. [DOI] [PubMed] [Google Scholar]
- 35. Sabbah HN, Kono T, Stein PD, Mancini GB, Goldstein S. Left ventricular shape changes during the course of evolving heart failure. Am J Physiol 1992;263:H266–H270. [DOI] [PubMed] [Google Scholar]
- 36. Sabbah HN, Wang M, Gupta RC, Rastogi S, Ilsar I, Sabbah MS, Kohli S, Helgerson S, Lee RJ. Augmentation of left ventricular wall thickness with alginate hydrogel implants improves left ventricular function and prevents progressive remodeling in dogs with chronic heart failure. JACC Heart Fail 2013;1:252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Douglas PS, Morrow R, Ioli A, Reichek N. Left ventricular shape, afterload and survival in idiopathic dilated cardiomyopathy. J Am Coll Cardiol 1989;13:311–315. [DOI] [PubMed] [Google Scholar]
- 38. Kronenberg MW, Cohen GI, Leonen MF, Mladsi TA, Di Carli MF. Myocardial oxidative metabolic supply–demand relationships in patients with nonischemic dilated cardiomyopathy. J Nucl Cardiol 2006;13:544–553. [DOI] [PubMed] [Google Scholar]
- 39. Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin‐converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest 1997;99:1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sabbah HN, Sharov VG, Lesch M, Goldstein S. Progression of heart failure: a role for interstitial fibrosis. Mol Cell Biochem 1995;147:29–34. [DOI] [PubMed] [Google Scholar]
- 41. Sabbah HN, Stanley WC, Sharov VG, Mishima T, Tanimura M, Benedict CR, Hegde S, Goldstein S. Effects of dopamine β‐hydroxylase inhibition with nepicastat on the progression of left ventricular dysfunction and remodeling in dogs with chronic heart failure. Circulation 2000;102:1990–1995. [DOI] [PubMed] [Google Scholar]
- 42. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA‐21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008;456:980–984. [DOI] [PubMed] [Google Scholar]
- 43. Schelbert EB, Piehler KM, Zareba KM, Moon JC, Ugander M, Messroghli DR, Valeti US, Chang CC, Shroff SG, Diez J, Miller CA, Schmitt M, Kellman P, Butler J, Gheorghiade M, Wong TC. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc 2015;4:e002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Collier P, Ledwidge M, McDonald K. Diagnostics and therapeutic interventions in myocardial interstitial disease, a previously neglected pathology. QJM 2012;105:721–724. [DOI] [PubMed] [Google Scholar]
- 45. Kemp CD, Conte JV. The pathophysiology of heart failure. Cardiovasc Pathol 2012;21:365–371. [DOI] [PubMed] [Google Scholar]
- 46. Tsutamoto T, Kanamori T, Morigami N, Sugimoto Y, Yamaoka O, Kinoshita M. Possibility of downregulation of atrial natriuretic peptide receptor coupled to guanylate cyclase in peripheral vascular beds of patients with chronic severe heart failure. Circulation 1993;87:70–75. [DOI] [PubMed] [Google Scholar]
- 47. Chatterjee K. Pathophysiology of systolic and diastolic heart failure. Med Clin North Am 2012;96:891–899. [DOI] [PubMed] [Google Scholar]
- 48. Bouras G, Giannopoulos G, Hatzis G, Alexopoulos D, Leventopoulos G, Deftereos S. Inflammation and chronic heart failure: from biomarkers to novel anti‐inflammatory therapeutic strategies. Med Chem 2014;10:682–699. [DOI] [PubMed] [Google Scholar]
- 49. von Lueder TG, Sangaralingham SJ, Wang BH, Kompa AR, Atar D, Burnett JC, Jr. , Krum H. Renin–angiotensin blockade combined with natriuretic peptide system augmentation: novel therapeutic concepts to combat heart failure. Circ Heart Fail 2013;6:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suzuki G, Morita H, Mishima T, Sharov VG, Todor A, Tanhehco EJ, Rudolph AE, McMahon EG, Goldstein S, Sabbah HN. Effects of long‐term monotherapy with eplerenone, a novel aldosterone blocker, on progression of left ventricular dysfunction and remodeling in dogs with heart failure. Circulation 2002;106:2967–2972. [DOI] [PubMed] [Google Scholar]
- 51. Sabbah HN, Gupta RC, Kohli S, Wang M, Zhang K, Rastogi S. Heart rate reduction with ivabradine improves left ventricular function and reverses multiple pathological maladaptations in dogs with chronic heart failure. ESC Heart Fail 2014;1:94–102. [DOI] [PubMed] [Google Scholar]
- 52. Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost‐Brama A, Lerebours G, Tavazzi L, Investigators SHIFT. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo‐controlled study. Lancet 2010;376:875–885. [DOI] [PubMed] [Google Scholar]
- 53. Publication Committee for the VMAC Investigators . Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 2002;287:1531–1540. [DOI] [PubMed] [Google Scholar]
- 54. Abraham WT, Adams KF, Fonarow GC, Costanzo MR, Berkowitz RL, LeJemtel TH, Cheng ML, Wynne J, ADHERE Scientific Advisory Committee and Investigators, ADHERE Study Group . In‐hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the Acute Decompensated Heart Failure National Registry (ADHERE). J Am Coll Cardiol 2005;46:57–64. [DOI] [PubMed] [Google Scholar]
- 55. Arora RR, Venkatesh PK, Molnar J. Short and long‐term mortality with nesiritide. Am Heart J 2006;152:1084–1090. [DOI] [PubMed] [Google Scholar]
- 56. Kurien S, Warfield KT, Wood CM, Miller WL. Effects of standard heart failure therapy and concomitant treatment with intravenous furosemide or inotropes (dobutamine, dopamine, and/or milrinone) on renal function and mortality in patients treated with nesiritide. Am J Cardiol 2006;98:1627–1630. [DOI] [PubMed] [Google Scholar]
- 57. Kelesidis I, Mazurek J, Khullar P, Saeed W, Vittorio T, Zolty R. The effect of nesiritide on renal function and other clinical parameters in patients with decompensated heart failure and preserved ejection fraction. Congest Heart Fail 2012;18:158–164. [DOI] [PubMed] [Google Scholar]
- 58. Witteles RM, Kao D, Christopherson D, Matsuda K, Vagelos RH, Schreiber D, Fowler MB. Impact of nesiritide on renal function in patients with acute decompensated heart failure and pre‐existing renal dysfunction: a randomized, double‐blind, placebo‐controlled clinical trial. J Am Coll Cardiol 2007;50:1835–1840. [DOI] [PubMed] [Google Scholar]
- 59. Sackner‐Bernstein JD, Kowalski M, Fox M, Aaronson K. Short‐term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA 2005;293:1900–1905. [DOI] [PubMed] [Google Scholar]
- 60. Sackner‐Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation 2005;111:1487–1491. [DOI] [PubMed] [Google Scholar]
- 61. O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, Nieminen MS, Reist CJ, Rouleau JL, Swedberg K, Adams KF Jr, Anker SD, Atar D, Battler A, Botero R, Bohidar NR, Butler J, Clausell N, Corbalán R, Costanzo MR, Dahlstrom U, Deckelbaum LI, Diaz R, Dunlap ME, Ezekowitz JA, Feldman D, Felker GM, Fonarow GC, Gennevois D, Gottlieb SS, Hill JA, Hollander JE, Howlett JG, Hudson MP, Kociol RD, Krum H, Laucevicius A, Levy WC, Méndez GF, Metra M, Mittal S, Oh BH, Pereira NL, Ponikowski P, Tang WH, Tanomsup S, Teerlink JR, Triposkiadis F, Troughton RW, Voors AA, Whellan DJ, Zannad F, Califf RM. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med 2011;365:32–43. [DOI] [PubMed] [Google Scholar]
- 62. Mitrovic V, Lüss H, Nitsche K, Forssmann K, Maronde E, Fricke K, Forssmann WG, Meyer M. Effects of the renal natriuretic peptide urodilatin (ularitide) in patients with decompensated chronic heart failure: a double‐blind, placebo‐controlled, ascending‐dose trial. Am Heart J 2005;150:1239.e1231–e1238. [DOI] [PubMed] [Google Scholar]
- 63. Mitrovic V, Seferovic PM, Simeunovic D, Ristic AD, Miric M, Moiseyev VS, Kobalava Z, Nitsche K, Forssmann WG, Lüss H, Meyer M. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J 2006;27:2823–2832. [DOI] [PubMed] [Google Scholar]
- 64. Packer M, Holcomb R, Abraham WT, Anker S, Dickstein K, Filippatos G, Krum H, Maggioni AP, McMurray JJ, Mebazaa A, O'Connor C, Peacock F, Ponikowski P, Ruschitzka F, van Veldhuisen DJ, Holzmeister J, TRUE‐AHF Investigators and Committees . Rationale for and design of the TRUE‐AHF trial: the effects of ularitide on the short‐term clinical course and long‐term mortality of patients with acute heart failure. Eur J Heart Fail 2016. Nov 13. doi: 10.1002/ejhf.698 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 65. Hata N, Seino Y, Tsutamoto T, Hiramitsu S, Kaneko N, Yoshikawa T, Yokoyama H, Tanaka K, Mizuno K, Nejima J, Kinoshita M. Effects of carperitide on the long‐term prognosis of patients with acute decompensated chronic heart failure: the PROTECT multicenter randomized controlled study. Circ J 2008;72:1787–1793. [DOI] [PubMed] [Google Scholar]
- 66. Matsue Y, Kagiyama N, Yoshida K, Kume T, Okura H, Suzuki M, Matsumura A, Yoshida K, Hashimoto Y. Carperitide is associated with increased in‐hospital mortality in acute heart failure: a propensity score‐matched analysis. J Card Fail 2015;21:859–864. [DOI] [PubMed] [Google Scholar]
- 67. von Lueder TG, Atar D, Krum H. Current role of neprilysin inhibitors in hypertension and heart failure. Pharmacol Ther 2014;144:41–49. [DOI] [PubMed] [Google Scholar]
- 68. Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens 2004;17:103–111. [DOI] [PubMed] [Google Scholar]
- 69. Packer M, Califf RM, Konstam MA, Krum H, McMurray JJ, Rouleau JL, Swedberg K, OVERTURE Study Group . Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation 2002;106:920–926. [DOI] [PubMed] [Google Scholar]
- 70. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR, PARADIGM‐HF Investigators and Committees . Angiotensin–neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014;371:993–1004. [DOI] [PubMed] [Google Scholar]
- 71. Segura J, Salazar J, Ruilope LM. Dual neurohormonal intervention in CV disease: angiotensin receptor and neprilysin inhibition. Expert Opin Investig Drugs 2013;22:915–925. [DOI] [PubMed] [Google Scholar]
- 72. Vardeny O, Miller R, Solomon SD. Combined neprilysin and renin–angiotensin system inhibition for the treatment of heart failure. JACC Heart Fail 2014;2:663–670. [DOI] [PubMed] [Google Scholar]
- 73. Judge P, Haynes R, Landray MJ, Baigent C. Neprilysin inhibition in chronic kidney disease. Nephrol Dial Transplant 2015;30:738–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. von Lueder TG, Wang BH, Kompa AR, Huang L, Webb R, Jordaan P, Atar D, Krum H. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail 2015;8:71–78. [DOI] [PubMed] [Google Scholar]
- 75. Benigni A, Zoja C, Zatelli C, Corna D, Longaretti L, Rottoli D, Maggioni P, Todeschini M, Noris M, Remuzzi G. Vasopeptidase inhibitor restores the balance of vasoactive hormones in progressive nephropathy. Kidney Int 2004;66:1959–1965. [DOI] [PubMed] [Google Scholar]
- 76. Solomon SD, Zile M, Pieske B, Voors A, Shah A, Kraigher‐Krainer E, Shi V, Bransford T, Takeuchi M, Gong J, Lefkowitz M, Packer M, McMurray JJ, Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fracTion (PARAMOUNT) Investigators . The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double‐blind randomised controlled trial. Lancet 2012;380:1387–1395. [DOI] [PubMed] [Google Scholar]
- 77. Packer M, McMurray JJ, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile M, Andersen K, Arango JL, Arnold JM, Bĕlohlávek J, Böhm M, Boytsov S, Burgess LJ, Cabrera W, Calvo C, Chen CH, Dukat A, Duarte YC, Erglis A, Fu M, Gomez E, Gonzàlez‐Medina A, Hagège AA, Huang J, Katova T, Kiatchoosakun S, Kim KS, Kozan O, Llamas EB, Martinez F, Merkely B, Mendoza I, Mosterd A, Negrusz‐Kawecka M, Peuhkurinen K, Ramires FJ, Refsgaard J, Rosenthal A, Senni M, Sibulo AS Jr, Silva‐Cardoso J, Squire IB, Starling RC, Teerlink JR, Vanhaecke J, Vinereanu D, Wong RC, PARADIGM‐HF Investigators and Coordinators . Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015;131:54–61. [DOI] [PubMed] [Google Scholar]
- 78. Jhund PS, Claggett BL, Voors AA, Zile MR, Packer M, Pieske BM, Kraigher‐Krainer E, Shah AM, Prescott MF, Shi V, Lefkowitz M, McMurray JJ, Solomon SD, PARAMOUNT Investigators . Elevation in high‐sensitivity troponin T in heart failure and preserved ejection fraction and influence of treatment with the angiotensin receptor neprilysin inhibitor LCZ696. Circ Heart Fail 2014;7:953–959. [DOI] [PubMed] [Google Scholar]
- 79. Tarone G, Balligand JL, Bauersachs J, Clerk A, De Windt L, Heymans S, Hilfiker‐Kleiner D, Hirsch E, Iaccarino G, Knöll R, Leite‐Moreira AF, Lourenco AP, Mayr M, Thum T, Tocchetti CG. Targeting myocardial remodelling to develop novel therapies for heart failure: a position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur J Heart Fail 2014;16:494–508. [DOI] [PubMed] [Google Scholar]
- 80. Yoon YS, Uchida S, Masuo O, Cejna M, Park JS, Gwon HC, Kirchmair R, Bahlman F, Walter D, Curry C, Hanley A, Isner JM, Losordo DW. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 2005;111:2073–2085. [DOI] [PubMed] [Google Scholar]
- 81. Kawasuji M, Nagamine H, Ikeda M, Sakakibara N, Takemura H, Fujii S, Watanabe Y. Therapeutic angiogenesis with intramyocardial administration of basic fibroblast growth factor. Ann Thorac Surg 2000;69:1155–1161. [DOI] [PubMed] [Google Scholar]
- 82. Azuma J, Taniyama Y, Takeya Y, Iekushi K, Aoki M, Dosaka N, Matsumoto K, Nakamura T, Ogihara T, Morishita R. Angiogenic and antifibrotic actions of hepatocyte growth factor improve cardiac dysfunction in porcine ischemic cardiomyopathy. Gene Ther 2006;13:1206–1213. [DOI] [PubMed] [Google Scholar]
- 83. Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double‐blind, placebo‐controlled, phase 2b trial. Lancet 2016;387:1178–1186. [DOI] [PubMed] [Google Scholar]
- 84. Teichman SL, Unemori E, Teerlink JR, Cotter G, Metra M. Relaxin: review of biology and potential role in treating heart failure. Curr Heart Fail Rep 2010;7:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Unemori EN, Lewis M, Constant J, Arnold G, Grove BH, Normand J, Deshpande U, Salles A, Pickford LB, Erikson ME, Hunt TK, Huang X. Relaxin induces vascular endothelial growth factor expression and angiogenesis selectively at wound sites. Wound Repair Regen 2000;8:361–370. [DOI] [PubMed] [Google Scholar]
- 86. Bani‐Sacchi T, Bigazzi M, Bani D, Mannaioni PF, Masini E. Relaxin‐induced increased coronary flow through stimulation of nitric oxide production. Br J Pharmacol 1995;116:1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, Dorobantu MI, Grinfeld LR, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin TM, Metra M, for the RELAXin in Acute Heart Failure (RELAX‐AHF) Investigators . Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised, placebo‐controlled trial. Lancet 2013;381:29–39. [DOI] [PubMed] [Google Scholar]
- 88. Gheorghiade M, Marti CN, Sabbah HN, Roessig L, Greene SJ, Böhm M, Burnett JC, Campia U, Cleland JG, Collins SP, Fonarow GC, Levy PD, Metra M, Pitt B, Ponikowski P, Sato N, Voors AA, Stasch JP, Butler J, Academic Research Team in Heart Failure . Soluble guanylate cyclase: a potential therapeutic target for heart failure. Heart Fail Rev 2013;18:123–134. [DOI] [PubMed] [Google Scholar]
- 89. Pieske B, Butler J, Filippatos G, Lam C, Maggioni AP, Ponikowski P, Shah S, Solomon S, Kraigher‐Krainer E, Samano ET, Scalise AV, Muller K, Roessig L, Gheorghiade M, SOCRATES Investigators and Coordinators . Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES). Eur J Heart Fail 2014;16:1026–1038. [DOI] [PubMed] [Google Scholar]
- 90. Sabbah HN, Gupta RC, Kohli S, Wang M, Rastogi S, Zhang K, Zimmermann K, Diedrichs N, Albrecht‐Küpper BE. Chronic therapy with a partial adenosine A1‐receptor agonist improves left ventricular function and remodeling in dogs with advanced heart failure. Circ Heart Fail 2013;6:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sabbah HN, Gupta RC, Wang M, Zhang K, Rastogi S. Long‐term therapy with bendavia (MTP‐131), a novel mitochondria‐targeting peptide, reverses mitochondrial functional abnormalities in left ventricular myocardium of dogs with advanced heart failure [abstract]. J Am Coll Cardiol 2013;61:E709. Presentation number: 1223‐1310. [Google Scholar]
- 92. Sabbah HN, Gupta RC, Kohli S, Wang M, Hachem S, Zhang K. Chronic therapy with elamipretide (MTP‐131), a novel mitochondria‐targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ Heart Fail 2016;9:e002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Langenickel TH, Dole WP. Angiotensin receptor–neprilysin inhibition with LCZ696: a novel approach for the treatment of heart failure. Drug Discov Today Ther Strateg 2013;9:e131–e139. [Google Scholar]