

Summary
Epoxyeicosatrienoic acids (EETs) are endogenous ligands that undergo hydrolysis by soluble epoxide hydrolase (sEH). The responses of 11, 12‐EET in comparison with other vasodilator agonists including carbachol and sodium nitroprusside (SNP) were investigated. The effect of 1‐cyclohexyl‐3‐dodecyl urea (CDU), a sEH, was tested on the vasodilator effect induced by 11, 12‐EET in the perfused mesenteric beds isolated from normo‐glycaemic and type‐1 STZ‐diabetic rats.
In the perfused mesenteric beds of control and diabetic animals, 11, 12‐EET produced vasodilation in a dose‐dependent manner. The vasodilator response induced by 11, 12‐EET was significantly decreased in tissues obtained from diabetic animals, but this was significantly corrected through inhibition of sEH.
The effects of nitric oxide synthase inhibitor, cyclo‐oxygenase inhibitor, specific potassium channel inhibitors, soluble guanylyl cyclase inhibitor and transient receptor potential channel V4 inhibitor, on vasodilator response to 11, 12‐EET were investigated.
In tissues isolated from control animals, vasodilator responses to 11, 12‐EET were not inhibited by acute incubation with l‐NAME, l‐NAME with indomethacin, glibenclamide, iberiotoxin, charybdotoxin, apamin or ODQ.
Incubation with the transient receptor potential channel V4 inhibitor ruthenium red caused significantly reduced vasodilator responses induced by 11, 12‐EET.
In conclusion, results from this study indicate that 11, 12‐EET has a vasodilator effect in the perfused mesenteric bed, partly through activation of vanilloid receptor. A strategy to elevate the levels of EETs may have a significant impact in correcting microvascular abnormality associated with diabetes.
Keywords: mesenteric bed, CDU, diabetes, vascular dysfunction, ruthenium red
1. Introduction
Epoxyeicosatrienoic acids (EETs) are epoxide derivatives of AA, which are synthesized through cytochrome P450 epoxygenases. EETs are formed in the endothelial layer and operate by activating the large‐conductance Ca2+‐activated K+ channels (BKCa). The hyperpolarizing effect that results due to the activation of BKCa channels will lead to relaxation of the smooth muscle in the vasculature, causing hyperpolarization of the vascular smooth muscle and consequently vasorelaxation. Therefore, EETs are recognized to act as ‘endothelium‐derived hyperpolarizing factor’ (EDHF) in several components of the vasculature such as the renal and coronary blood vessels, resulting in reduction of the blood pressure.1, 2 Several effects of EET have been shown to occur due to its binding to a membrane receptor which leads to the activation of ion channels and signalling transduction pathways.2 It is also reported that cells can take up EETs and integrate them into phospholipids, binding to cytosolic proteins and nuclear receptors.1 It is possible that they contribute to the pathogenesis of several disorders.3 The conversion of EETs to dihydroxyeicosatrienoic acids (DHET) is attained by soluble epoxide hydrolase (sEH).1 The main EET catabolic pathway is conversion to the corresponding DHET by sEH. Moreover, it is common that EETs may have one action in the periphery and an opposite effect in the pulmonary system. Therefore, such observations raise attention about the role of EETs in pulmonary hypertension, especially in cases of hypoxia.4
According to the definition of Hesselink, an autacoid is a modulating factor that is produced locally; it influences the function of cells and/or tissues in a limited region or position. An autacoid is produced in response to demand and is subsequently broken down in the same cells and/or tissues. Therefore, 11, 12‐EET can be considered an autacoid.5 The effect of 11, 12‐EET (C20H32O3) on intestinal vasculature is not well established. The mesenteric bed comprises mainly of arterioles that significantly contribute to the total peripheral resistance. Accordingly, the objective of this investigation was to examine the effect of 11, 12‐EET in the rat perfused mesenteric bed. The contribution of nitric oxide and cyclo‐oxygenase has been examined using inhibitors of nitric oxide synthase and cyclo‐oxygenase. In addition, the participation of potassium channels in the modulation of the vasodilation response to 11, 12‐EET was studied using specific potassium channel inhibitors: K+ATP channel blocker (glibenclamide), large‐conductance calcium‐activated potassium channel blocker (iberiotoxin), intermediate‐conductance calcium‐activated potassium channel blocker (charybdotoxin) and small‐conductance calcium‐activated potassium channel blocker (apamin). The role of guanylyl cyclase and receptor potential channel V4 (TRPV4) in mediating vasodilator responses induced by 11, 12‐EET was investigated using ODQ and ruthenium red, respectively. A model of type 1 diabetic animals, induced by an injection of streptozotocin (STZ), were used to examine the vasodilator responses of 11, 12‐EET in tissues from hyperglycaemic animals in comparison with control.4, 5 Abnormal vascular responsiveness of the mesenteric beds from diabetic animals to 11, 12‐EET would reflect vascular dysfunction induced by diabetes.
2. Materials and Methods
2.1. Animal groups
Two groups of animals were used in this project (N=12 per group). Group 1 included control male Sprague‐Dawley (SD) rats, while Group 2 included diabetic male SD rats. Diabetes was induced by one intraperitoneal injection (55 mg/kg, body weight) of streptozotocin (STZ).6, 7, 8 Rats were confirmed to be diabetic and approved for this investigation when fasting blood glucose levels were measured to be over 250 mg/dL. The animals involved in this project retained open entry to food and water during the investigation. All studies involving animals were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication number 85‐23, Revised in 1985) as approved by Kuwait University Research Administration.
2.2. Experimental procedure
Rats were anesthetized with ketamine (50 mg/mL) 4 weeks after inducing diabetes and then euthanized. Mesenteric beds were carefully isolated, cannulated and placed in a warm water‐jacketed chamber and perfused with Krebs‐Henseleit solution (KH) at 37°C. The isolated tissues received oxygenation with 95% oxygen and 5% carbon dioxide mixture in KH. The constituents of KH solution included the following (mmol/L): NaCl (118.3), KCl (4.7), CaCl2 (2.5), MgSO4 (1.2), NaHCO3 (25), KH2PO4 (1.2) and glucose (11.2). The mesenteric bed was perfused at a regular flow rate of 6 mL/min, employing a multichannel masterflex peristaltic pump.6, 7, 8 A pressure transducer linked with a Lectromed graph recorder measured changes in perfusion pressure. The perfused tissue was left to stabilize for 45 minutes.
2.3. Effect of 11, 12‐EET in the mesenteric vasculature
Tissues were obtained from normo‐glycaemic and hyperglycaemic animals as described earlier. Perfusion pressure was raised using phenylephrine (PE) (3×10−6 mol/L) added to KH solution. Subsequent to setting up a constant raising in perfusion pressure, ensuing dosages of 11, 12‐EET ranging from 10−13 to 10−8 moles were given as bolus injections at regular intervals to establish a dose‐response curve. The vasorelaxant effect due to 11, 12‐EET in control as well as diabetic tissues was measured as a percentage reduction in the level of perfusion pressure induced by PE. E max (maximum response) was calculated from the dose‐response curves of 11, 12‐EET using GraphPad Prism software version 5 for windows (GraphPad Software, Inc., La Jolla, California, USA). In a different part of the investigation, the vasorelaxant reaction to carbachol (10−13‐10−7 moles) and sodium nitroprusside (SNP) (10−13‐10−7 moles) was tested as explained for 11, 12‐EET.8, 9, 10
2.4. Influence of CDU on the reaction of the mesenteric vasculature to vasorelaxant agonists
Vasodilation due to 11, 12‐EET, carbachol or SNP was established in isolated perfused mesenteric vasculature obtained from normo‐glycaemic and hyperglycaemic animals. Thereafter, the tissues have been perfused with a soluble epoxide hydrolase (sEH) inhibitor, CDU (10−6 mol/L) for 30 minutes. After the incubation period, the effect of 11, 12‐EET, carbachol or SNP was examined in the presence of CDU. Separate tissues were used to test the effect of different agonists.
2.5. Effects of NO synthase and cyclo‐oxygenase inhibitors on vasorelaxant responses induced by 11, 12‐EET in control animals
In this experimental protocol, the effect of NG‐nitro‐l‐arginine methyl ester (l‐NAME) and indomethacin, inhibitors of nitric oxide synthase (NOS) and cyclo‐oxygenase, respectively, was investigated on 11, 12‐EET‐induced vasodilation. l‐NAME was used either single or added together along with indomethacin. Dose‐response curves for 11, 12‐EET were carried out as reported earlier. Then, mesenteric beds obtained from control rats were perfused with KH solution that contained l‐NAME (10−4 mol/L) alone or in combination with indomethacin (10−6 mol/L) for 30 minutes. Dose‐response curves for 11, 12‐EET were reconstructed in the presence of l‐NAME and indomethacin. After treatment with inhibitors for 30 minutes, tissues were precontracted with PE (10−6 mol/L) to test the vasodilator responses to 11, 12‐EET. Successive doses of 11, 12‐EET were given to establish a dose‐response curve. Different tissues were used to examine the effects of the inhibitors.
2.6. Effect of potassium channel inhibitors on vasorelaxant responses induced by 11, 12‐EET
In this group of investigations, the influence of inhibition of specific potassium channel on the vasorelaxant responses to 11, 12‐EET in was investigated. The tested inhibitors included glibenclamide (a K+ (ATP) channel blocker), iberiotoxin (an inhibitor of large‐conductance calcium‐activated potassium BK channel), charybdotoxin (an inhibitor of intermediate‐conductance calcium‐activated potassium SK4 channel) and apamin (an inhibitor of small‐conductance calcium‐activated potassium SK3 channel). Mesenteric vasculature isolated from normo‐glycaemic rats was perfused, and vasorelaxant responses to 11, 12‐EET were recorded. The mesenteric beds were then perfused with KH solution containing glibenclamide (10−5 mol/L), iberiotoxin (5×10−8 mol/L), charybdotoxin (5×10−8 mol/L) or apamin (10−7 mol/L) for 30 minutes. Following, the incubation period, tissues were precontracted with PE (10−6 mol/L) and a stable level of rising in perfusion pressure was accomplished. Thereafter, the vascular reaction to 11, 12‐EET was examined by giving successive doses of 11, 12‐EET (0.1, 1.0 and 10 nmol) to establish a dose‐response curve. Different animal tissues were used to examine the effects of the various inhibitors.
2.7. Effect of soluble guanylyl cyclase inhibitor
Contribution of soluble guanylyl cyclase to the vasorelaxant response induced by 11, 12‐EET was examined. 1H‐[1, 2, 4] oxadiazolo [4, 3‐α] guinoxalin‐1‐one (ODQ) causes inhibition of soluble guanylyl cyclase. The impact of ODQ on the vasorelaxation generated by 11, 12‐EET was investigated. Dose‐response curve for 11, 12‐EET has been constructed using perfused mesenteric bed of control animals. Thereafter, vascular mesenteric bed was perfused with KH solution including ODQ (10−5 mol/L) for 30 minutes. After the incubation period, the effect of 11, 12‐EET was examined by establishing a dose‐response curve as described earlier.
2.8. Influence of transient receptor potential channel V4 (TRPV4) inhibitor
This part of the study involved examining the impact of the transient receptor potential channel V4 (TRPV4) inhibitor, ruthenium red (RuR) on vasorelaxant responses induced by 11, 12‐EET in the mesenteric vasculature. After constructing a dose‐response curve to 11, 12‐EET, the mesenteric beds obtained from control animals were perfused for 30 minutes with the inhibitor of TRPV4 channel, RuR (10−6 mol/L), added to the KH solution. Thereafter, vasorelaxant responses to 11, 12‐EET were examined while the inhibitor was present in the perfusing solution, as described previously.
2.9. Drugs and chemicals
Carbachol, sodium nitroprusside (SNP), phenylephrine, l‐NAME, indomethacin, glibenclamide, iberiotoxin, charybdotoxin, apamin, ODQ and ruthenium red were obtained from Sigma Biochemical (St. Louis, Missouri, USA). 11, 12‐EET was purchased from Chayman chemical (Ann Arbor, Michigan, USA). CDU was obtained from CalBiochem (San Diego, California, United States).
2.10. Statistical analysis
Results obtained from the experiments were evaluated by applying GraphPad Prism software version 5 for windows. Data presented as ‘mean±SEM of number of experiments (n)’. Comparison of mean values was accomplished by adopting student t test, one‐way analysis of variance (ANOVA). Thereafter, a post hoc test (Bonferroni) was performed. A significant difference between the mean values was recognized if P‐value was less than .05 (P<.05).
3. Results
3.1. Hyperglycaemia along with changes in body weight
Diabetes was induced by a single intraperitoneal injection of STZ that caused a considerable augmentation in the concentration of glucose. Hyperglycaemia persisted with the diabetic animals and was 562.68±9.64 mg/dL after 4 weeks of diabetes induction compared with 91.0±0.55 mg/dL in the normo‐glycaemic rats (P<.05). Diabetes persistent for 4 weeks caused a considerable decrease in STZ‐diabetic rats body weight (257.40±1.30 g) compared to control animals (310±1.87 g) (P<.05).
3.2. 11, 12‐EET‐induced responses in mesenteric vasculature from normo‐glycaemic and hyperglycaemic animals
11, 12‐EET, carbachol and SNP resulted in vasodilation of mesenteric beds of normo‐glycaemic rats (Figures 1 and 2). In tissues isolated from diabetic animals, the vasodilator response induced by 11, 12‐EET or carbachol has shown to be attenuated in comparison with control rats (P<.05) (Figures 1 and 2). Results indicating reduction in carbachol‐induced vasodilator response in the mesenteric vasculature isolated from diabetic rats agree with our previous findings.11 SNP‐induced vasodilation was not found to be different in tissues from STZ rats compared to normo‐glycaemic animals (Figure 2).
Figure 1.
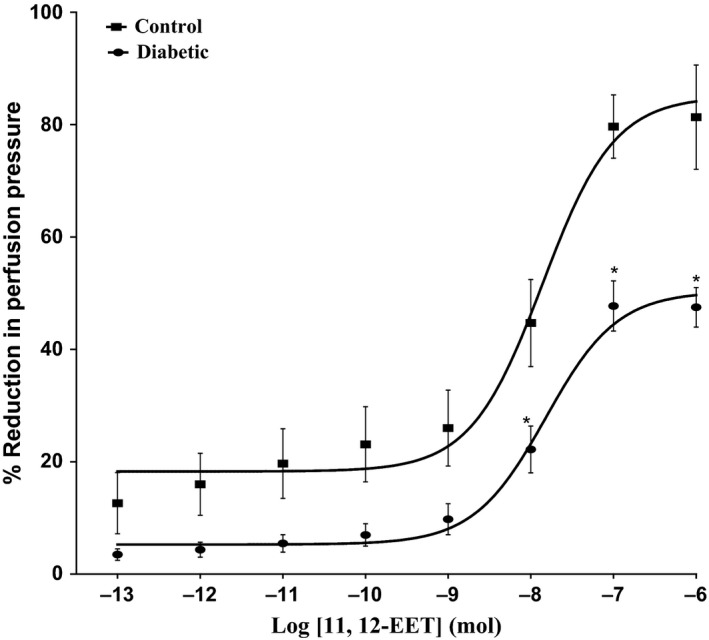
Effect of 11, 12‐epoxyeicosatrienoic acids in the perfused mesenteric beds isolated from control and diabetic Sprague‐Dawley male rats. Values are shown as mean±SEM, N=10 (*) Significantly different compared to control (P<.05)
Figure 2.
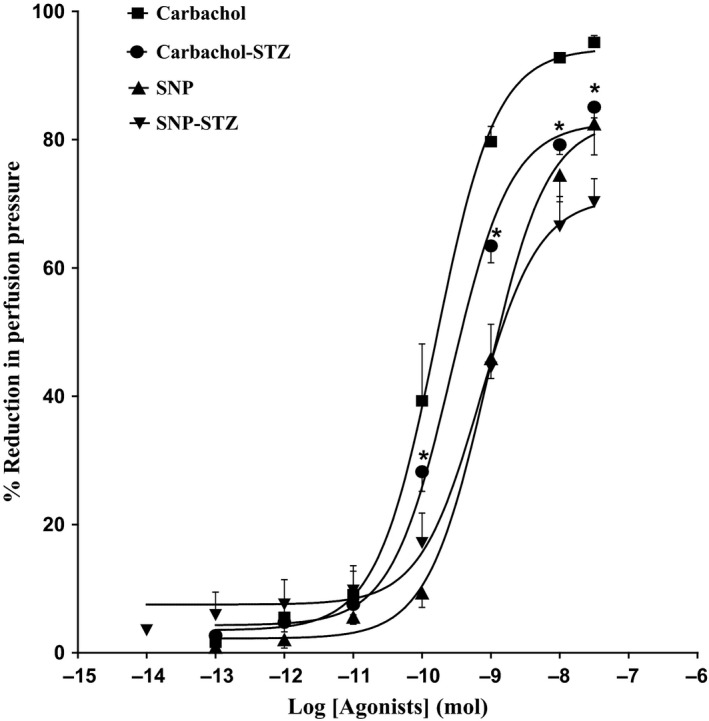
Effect of carbachol and SNP in the perfused mesenteric beds isolated from control and diabetic Sprague‐Dawley male rats. Values are shown as mean±SEM, N=4‐6 (*) Significantly different compared to control (P<.05)
3.3. Effect of soluble epoxide hydrolase inhibitor on vasodilator response to vasoactive agonists
Acute incubation of the mesenteric vasculature isolated from STZ‐diabetic rats with CDU caused a significant potentiation in the responses to 11, 12‐EET (Figure 3) or carbachol (Figure 4) compared with responses in diabetic tissues not incubated with CDU (Figures 3 and 4). Vasodilation induced by 11, 12‐EET or carbachol in tissues obtained from control rats has been maintained along with the existence of CDU (Figures 3 and 4). Incubation with CDU did cause any significant changes in the level of perfusion pressure raised by PE. Vasodilator responses to SNP were not changed in tissues isolated from normal or diabetic animals following incubation with CDU (Figure 5).
Figure 3.
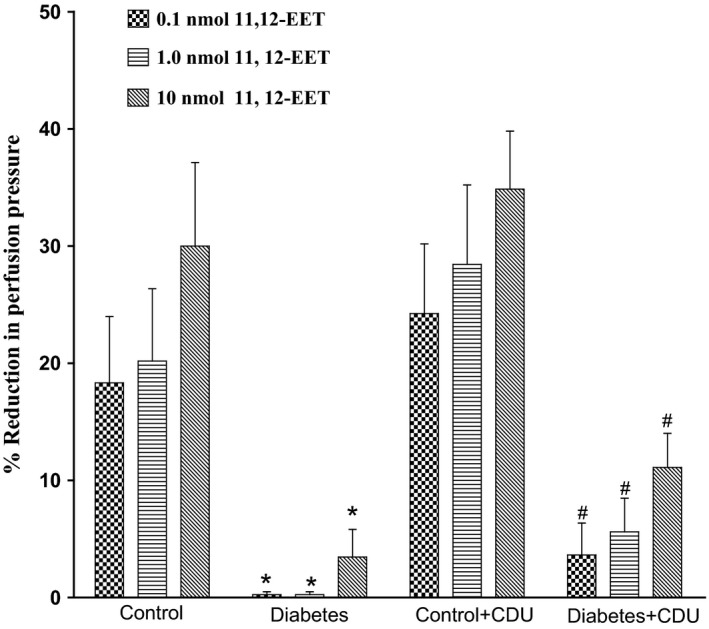
Effect of 11, 12‐epoxyeicosatrienoic acids in the perfused mesenteric beds isolated from control and diabetic Sprague‐Dawley male rats before and after incubation with CDU (10−6 mol/L). Values are shown as mean±SEM, N=4 (*) Significantly different compared to control (P<.05). (#) Significantly different compared to diabetes (P<0.05).
Figure 4.
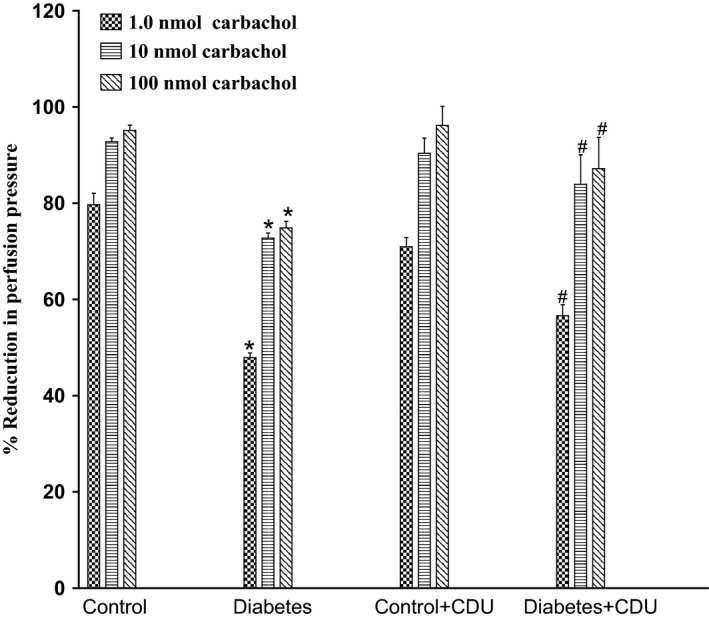
Effect of carbachol in the perfused mesenteric beds isolated from control and diabetic Sprague‐Dawley male rats before and after incubation with CDU (10−6 mol/L). Values are shown as mean±SEM, N=4 (*) Significantly different compared to control (P<.05). (#) Significantly different compared to diabetes (P<0.05).
Figure 5.
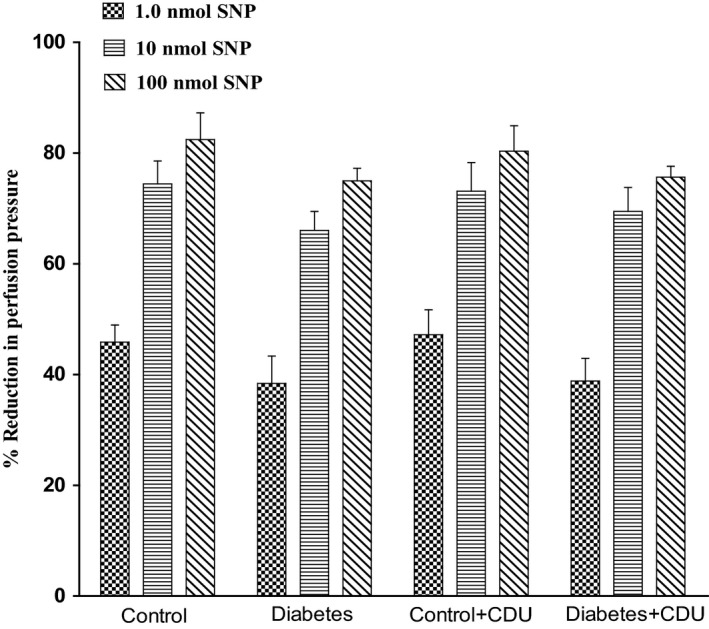
Effect of SNP in the perfused mesenteric beds isolated from control and diabetic Sprague‐Dawley male rats before and after incubation with CDU (10−6 mol/L). Values are shown as mean±SEM, N=6
3.4. Influence of l‐NAME and indomethacin on responses to 11, 12‐EET
11, 12‐EET induced vasodilator response at (0.1, 1.0, 10 nmol) in the mesenteric beds isolated from SD control rat. Following the perfusion with l‐NAME (10−4 mol/L), the vasodilator reaction to 11, 12‐EET was not attenuated (Figure 6A). Treatment with l‐NAME (10−4 mol/L) and indomethacin (10−6 mol/L) caused a noticeable increase in the vasodilator response to 11, 12‐EET (P<.05) (Figure 6B).
Figure 6.
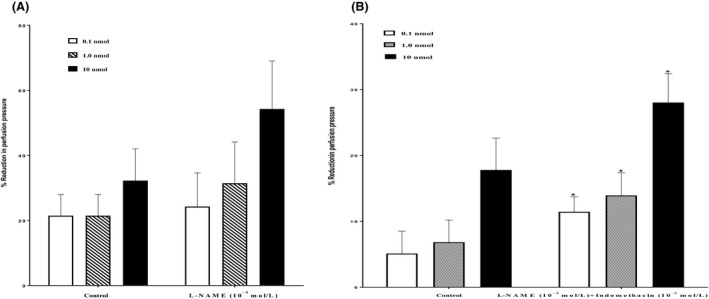
(A) Effect of nitric oxide synthase inhibitor (l‐NAME, 10−4 mol/L) on the response of 11, 12‐epoxyeicosatrienoic acid (EET) in the perfused mesenteric beds isolated from Sprague‐Dawley (SD) control male rats. Values are shown as mean±SEM, N=6 (N=number of animals per group). (B) Effect of nitric oxide synthase and cyclo‐oxygenase inhibitors (l‐NAME, 10−4 mol/L +Indomethacin, 10−6 mol/L) on the response of 11, 12‐EET in the perfused mesenteric beds isolated from SD control male rats. Values are shown as mean±SEM, N=6 (N=number of animals per group).
3.5. Vasodilator responses to11, 12‐EET in the presence of potassium channel inhibitors
In the mesenteric beds isolated from SD control male rats, 11, 12‐EET induced vasodilation at the doses of 0.1, 1.0 and 10 nmol. Following incubation with glibenclamide (10−5 mol/L), the vascular reactivity to 11, 12‐EET was not diminished. However, perfusion in the presence of glibenclamide caused an augmentation in vasodilator reaction to 11, 12‐EET (Figure 7A). In another group of experiments, incubation with iberiotoxin (5×10−8mol/L) resulted also in an increased vasodilator response to 11, 12‐EET at (1.0 and 10 nmol)in the mesenteric beds isolated from SD control male rats (Figure 7B). Incubation of the perfused mesenteric bed with charybdotoxin (5×10−8mol/L) caused a significant potentiation in the vasodilation induced by 11, 12‐EET (Figure 7C). Vasodilator responses induced by 11, 12‐EET were significantly enhanced after incubation with apamin for 30 minutes in the perfusate (Figure 7D).
Figure 7.
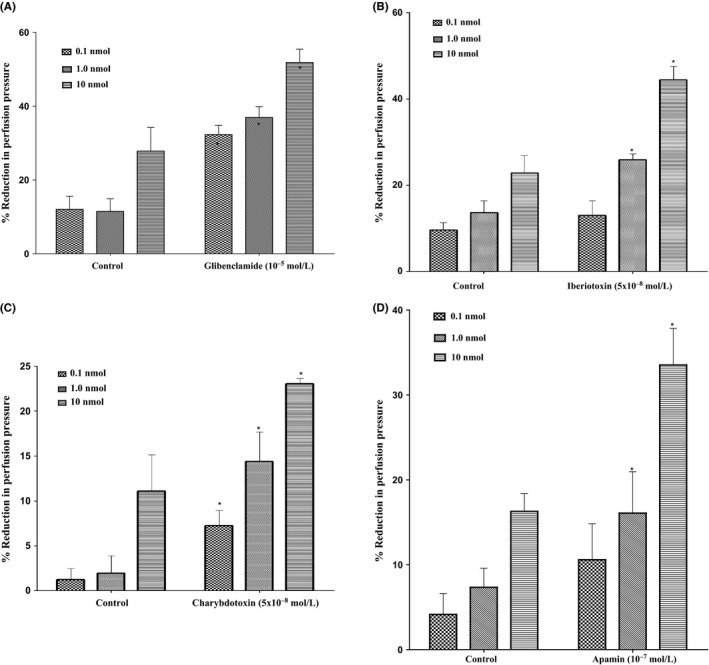
(A) Effect of 11, 12‐epoxyeicosatrienoic acid (EET) in the perfused mesenteric beds isolated from Sprague‐Dawley (SD) control male rats following incubation with K+ ATP channel blocker (glibenclamide, 10−5 mol/L). Values are shown as mean±SEM, N=4 (N=number of animals per group). (B) Effect of 11, 12‐EET in the perfused mesenteric beds isolated from SD control male rats following incubation with large‐conductance calcium‐activated potassium channel blocker (iberiotoxin, 5×10−8 mol/L). Values are shown as mean±SEM, N=4 (N=number of animals per group). (C) Effect of 11, 12‐EETin the perfused mesenteric beds isolated from SD control male rats following incubation with intermediate‐conductance calcium‐activated potassium channel blocker (charybdotoxin, 5×10−8 mol/L). Values are shown as mean±SEM, N=4 (N=number of animals per group). (D) Effect of 11, 12‐EET in the perfused mesenteric beds isolated from SD control male rats following incubation with small‐conductance calcium‐activated potassium channel blocker (Apamin, 10−7 mol/L). Values are shown as mean±SEM, N=4 (N=number of animals per group). (*) Significantly different compared to the treatment with inhibitors (P < 0.05).
3.6. Effect of inhibition of soluble guanylyl cyclase on 11, 12‐EET responses
Incubation of the mesenteric bed with ODQ (10−5mol/L) (a soluble guanylyl cyclase inhibitor) could not inhibit vasodilator responses caused by 11, 12‐EET. However, incubation with ODQ accounted for a considerable potentiation in responses induced by 11, 12‐EET (P<.05) (Figure 8).
Figure 8.
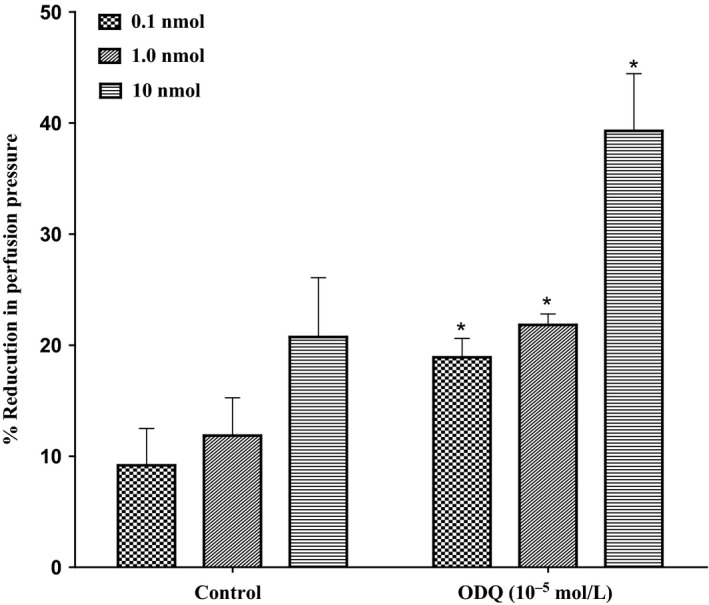
Effect of 11, 12‐epoxyeicosatrienoic acids response in the perfused mesenteric beds isolated from Sprague‐Dawley control male rats after incubation with soluble guanylyl cyclase inhibitor (ODQ, 10−5 mol/L). Values are shown as mean±SEM, N=6 (N=number of animals per group). (#) Significantly different compared to diabetes (P<0.05).
3.7. Influence of transient receptor potential channel V4 (TRPV4) inhibitor on 11, 12‐EET‐induced responses
In the perfused mesenteric beds isolated from control SD rats, 11, 12‐EET‐induced vasodilator responses at (0.1, 1.0, 10 nmol) were significantly decreased following incubation with ruthenium red (RuR) (10−6 mol/L) (P<.05) (Figure 9). Incubation with RuR for 30 minutes leads to a significant decrease in perfusion pressure induced by 11, 12‐EET. Vasodilator responses (%) decreased from 17.9%±0.9% and 22%±0.8% to 2.3%±1% and 11%±4.5% in response to 0.1 and 1.0 nmol of 11, 12‐EET before and after incubation with ruthenium red (RuR) (10−6 mol/L).
Figure 9.
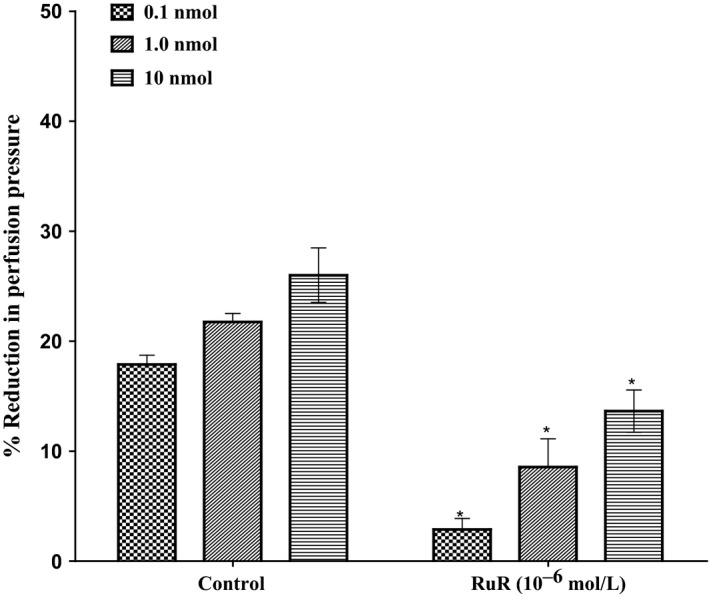
Effect of 11, 12‐epoxyeicosatrienoic acids response in the perfused mesenteric beds isolated from Sprague‐Dawley control male rats following incubation with (ruthenium red, RuR, 10−6 mol/L). Values are shown as mean±SEM, N=4 (N=number of animals per group). (*) Significantly different compared to the treatment with inhibitors (P < 0.05)
4. Discussion
Findings from this study revealed that 11, 12‐EET resulted in a dose‐depending vasodilation response in perfused mesenteric vascular beds of SD male rats. The vasodilation response of 11, 12‐EET was independent from NO/COX pathways. Neither K+ATP channel, large, intermediate, small‐conductance calcium‐activated potassium channels nor c‐GMP pathway had a role in mediating the vasodilator responses to 11, 12‐EET. Activation of vanilloid receptors may have an essential role in mediating 11, 12‐EET‐generated vasodilator responses. In mesenteric beds obtained from diabetic animals, the vasodilator responsiveness to 11, 12‐EET was noticeably reduced in comparison with control. Soluble epoxide hydrolase inhibitor (CDU) can effectively inhibit the hydrolysis of EETs; thus, vasodilation due to 11, 12‐EET as well as other vasodilator agonists such as carbachol was increased in mesenteric beds isolated from hyperglycaemic animals.
The responses to 11, 12‐EET in the perfused mesenteric vasculature collected from diabetic male rats have been investigated. We found that the vasodilation produced by 11, 12‐EET was considerably decreased in the perfused mesenteric beds isolated from diabetic animals in comparison with normo‐glycaemic. This finding was in accordance with the results obtained by Yousif and Benter7 who tested the responses of 11, 12‐EET in the corpus cavernosum of diabetic rats. EETs are released by the endothelial cells in response to different stimulants like the agonist acetylcholine.2 In this study, carbachol, a cholinergic agonist with similar properties to acetylcholine, was tested instead of acetylcholine because it is more stable for experimental procedures. Carbachol, SNP (an endothelium‐independent nitric oxide donor) and 11, 12‐EET resulted in a dose‐depending vasodilation in the mesenteric vascular beds of normal and hyperglycaemic animals. Vasodilation in response to 11, 12‐EET and carbachol was noticeably attenuated in mesenteric beds of diabetic animals.
Inflammation contributes to cardiovascular disease progression; therefore, the interactions between inflammation and the epoxygenase pathway can affect cardiovascular function in disease conditions.12, 13, 14, 15 Despite the fact that more studies are needed to resolve the precise cellular signalling mechanisms involved in the anti‐inflammatory actions of EETs, there is great evidence that EETs reduce inflammation. It is suggested that EETs inhibit the stimulation of the transcription factor nuclear factor to produce their vascular anti‐inflammatory responses.15, 16 Studies using inhibitors of soluble epoxide hydrolase (sEH) also support the idea that EETs have anti‐inflammatory actions.17, 18 Based on these findings, inhibitors of sEH could be protective against the detrimental effects of inflammation that occur in cardiovascular diseases and that they could afford cure for other inflammatory diseases.19 The effect of the sEH inhibitor, CDU, on the vascular responses to vasodilator agonists in the mesenteric vasculature isolated from normal and hyperglycaemic rats was investigated. Soluble epoxide hydrolase converts EETs to DHETS, and this metabolism limits many of the biological actions of EETs. Vasodilatation in response to carbachol or 11, 12‐EET has been increased after incubation of mesenteric vasculature from hyperglycaemic animals in CDU‐containing KH solution. However, the responsiveness to SNP was not affected among diabetic subjects. SNP is an NO (nitric oxide) donor, and its action is not influenced by possible endothelium dysfunction due to diabetes. Our observation was similar to those demonstrated in the corpus cavernosum.10 Further, as diabetes is associated with attenuated response to dilator agonists and exaggerated response to constrictor agonists,6 accordingly, it is suggested that inhibition of sEH by acute treatment with CDU could enhance the dilation of agonists by reducing the inactivation of EETs in diabetes and such approach may reverse the vascular dysfunction associated with diabetes.
This study showed that 11, 12‐EET produced dose‐dependent vasodilator response that was consistent with previous studies.20, 21 We have found that specific inhibitors of nitric oxide synthase and cyclo‐oxygenase inhibitors did not inhibit the vasodilatation due to 11, 12‐EET. Results from these experiments demonstrated that 11, 12‐EET is a vasodilator agent in the perfused rat mesenteric vasculature. In addition, the dilator response was independent from NO and COX, suggesting that NO/COX vasodilator pathways are not directly contributing in mediating the vasodilator responses of 11, 12‐EET in this vascular bed. Moreover, Wong and Vanhoutte22 reported that the endothelium produces several materials to regulate the basal force in the vascular smooth muscle. Endothelium‐derived relaxing factor is characterized to be NO.23 The basal force can be enhanced by producing endothelium‐derived contracting factors (EDCF) from the endothelial layer.24 It is reported that an increase in the release of EDCF can lead to abnormal function of the endothelium and that occurs in different various vascular disorders. A variety of stimuli can cause EDCF production, including agonists like acetylcholine, adenosine nucleotides or by stretch.25 Inflow of calcium ions into the cells of the endothelium would occur in response to the different triggering elements. Release of EDCF follows in response to the enhancement in the concentration of calcium ions intracellular. Activation of thromboxane‐prostanoids (TP) receptors that are present on the vascular smooth muscle cells will result in the contraction.22 This activation is initiated by several mechanisms including activation of phospholipase A2 (PLA2), cyclo‐oxygenases (COX) as well as release of reactive oxygen species (ROS) and vasoconstrictor prostanoids.22 Similarly, 11, 12‐EET may trigger the release of EDCF. It has been reported that EDCF is able to deeply influence the basal tone and thus can negate the relaxant effects of the substances produced by the endothelial layer. Thromboxane A2, PGH2 and superoxide anions may be formed through the endothelial cyclo‐oxygenase pathway. It is reported that the synthesis of cyclo‐oxygenase‐dependent endothelial‐derived contracting factors may be affected in some disease conditions.26 Therefore, when we blocked COX enzyme with indomethacin, there was a significant potentiation in the vasodilatation due to 11, 12‐EET in the perfused vascular bed.
Endothelium‐derived hyperpolarizing factor was defined pharmacologically by Campbell et al.27 EDHF is therefore known to provoke an endothelium‐dependent vasorelaxation that cannot be abolished by inhibitors of NO synthase and cyclo‐oxygenase.27 Others also reported that EDHF shows distinctive properties compared to NO and prostacyclin. For example, agonists may cause hyperpolarization while inhibitors of both cyclo‐oxygenase and NO synthase are present.28, 29 Thus, the above studies are consistent with our observations that 11, 12‐EET may act as EDHF in the mesenteric vasculature obtained from control animals. As additional support for our observations, Prattet al.30 indicated that EETs might display similar behaviour to EDHFs.
The impact of blocking potassium channels on the vasodilator response produced by 11, 12‐EET in the perfused mesenteric vascular bed was investigated. Incubation with a K+ ATP channel blocker (glibenclamide) did not change the vasodilator effect due to 11, 12‐EET in the mesenteric vasculature. Yousif and Benter7 reported that incubation with glibenclamide caused a considerable decrease in the relaxant effect to 11, 12‐EET in the corpus cavernosum isolated from normal Wister male. The results of this present study indicated that ATP‐sensitive K+ channels did not contribute in producing the vasodilator effect of 11, 12‐EET in the mesenteric vasculature obtained from normal rats.
Activation of large‐conductance calcium‐activated K+ channels (BKca) has been suggested to play a role in vasodilation induced by EETs.31, 32 Iberiotoxin, a selective and reversible inhibitor of high‐conductance calcium‐activated potassium channel (BKca inhibitor), could not block the vasodilator effect produced by 11, 12‐EET in the mesenteric vasculature. This result is found to be consistent with other reporters who indicated that EETs did not influence the BKca channel inside‐out detached membrane patches excised from VSM cells. Therefore, it was proposed that stimulation of BKca channel in vascular smooth muscle cells are not linked to the direct effects of EETs.33 Hercule et al.34 also reported that EDHF‐dependent relaxation in mouse mesenteric arteries was not prevented by iberiotoxin. This finding indicated that BKca channel in arterial smooth muscle cells had no contribution towards the effects of EDHF. Campbell et al.35 and Larsen et al.31 demonstrated that 20%‐30% of the 14, 15‐EET‐induced relaxation of coronary artery is resistant to the specific BKca channel blocker iberiotoxin.
Furthermore, to address whether the vasodilation induced by 11, 12‐EET was secondary to the stimulation of intermediate (IKca) and/or small (SKca) conductance calcium‐activated potassium channels, the effect of IKca inhibitor charybdotoxin (ChTx) and SKca inhibitor (apamin) was investigated. Incubation with specific calcium‐activated potassium channel inhibitors failed to attenuate the vasodilator effect of 11, 12‐EET in. This finding was similar to the results demonstrated by Hercule et al.34 in which the dilator responses to 5, 6‐EET, 11, 12‐EET and 14, 15‐EET were not changed due to apamin/ChTx in mouse mesenteric arteries. Therefore, our findings indicate that 11, 12‐EET may exhibit its vasodilator action in the mesenteric vasculature from SD rats without any involvement of SKca/IKca channels.
The possible contribution of c‐GMP to the vasodilator response induced by 11, 12‐EET in the perfused mesenteric beds was investigated. Incubation with ODQ, an inhibitor of soluble guanylyl cyclase, did not block the vasorelaxant response to 11, 12‐EET. This finding was in agreement with results of Sacerdoti et al.36 which reported that ODQ did not alter the reaction to 11, 12‐EET in the mesenteric microvessels isolated from Wister male rat. From our results, we suggest that the vasodilator effect induced by 11, 12‐EET in the perfused mesenteric vascular beds isolated from SD rats may not involve the activation of c‐GMP pathway. So far, in this study, the vasodilator response to 11, 12‐EET was resistant to inhibition by potassium channel blockers, nitric oxide synthase inhibitor, ODQ or l‐NAME‐indomethacin combination. This is an interesting observation that may be further investigated in future studies. It is reported that the particularity in relaxations induced by EET would differ in different species and when tested in various vasculature. More studies may be needed to elucidate the mechanism involved. Determination of EET receptor(s) continues to be an important topic for investigation that will have great therapeutic significance.
The possible involvement of vanilloid receptors in mediating the vasodilator action of 11, 12‐EET in the perfused mesenteric vascular beds we investigated.37, 38 The vasodilator response to 11, 12‐EET was considerably attenuated following acute treatment with ruthenium red (RUR), a TRPV4 inhibitor. The TRPV family consists of six members that are subcategorized into four groups: TRPV1/TRPV2, TRPV3, TRPV4 and TRPV5/6. It is reported that TRPV1, 2, 3, 4 are modestly permeable for Ca2+ 38. Various stimulants can cause the activation of TRPV4 such as limited heating. Stimulation of TRPV4 can also be attained by anandamide and arachidonic acid after the conversion to 5, 6‐EET.37, 38 It has been reported that in vascular smooth muscle and endothelial cells, TRPV4 channels are involved in mediating the inflow of calcium ions, and therefore, TRPV4 are considered as essential moderators of the tone in the vascular beds38. Furthermore, it was shown that 11, 12‐EET activated TRPV4 leading to hyperpolarization of the smooth muscle cells’ membrane, causing vasodilation of mesenteric arteries obtained from WT mice.39 On the contrary, 11, 12‐EET did not have any action in the mesenteric arteries from TRPV4 (KO) knockout mice.40 Additional reinforcement for the involvement of TRPV4 channels in the responses to 11, 12‐EET, Earley et al.39 found that RuR completely overturned smooth muscle hyperpolarization induced by 11, 12‐EET in arteries from WT mice. Ding et al.41 reported that in the endothelial cell membrane, a G(s)‐coupled receptor responds to 11(R),12(S)‐EET and mediates the PKA‐dependent translocation and stimulation of TRPC6 channels. Based on the above‐mentioned data together with our results, we suggest that activation of TRPV4 channels is essential for 11, 12‐EET‐dependent vasodilation in the perfused mesenteric vasculature obtained from SD male rats. Further, an inconsistent contribution of TRPV4 has been recently revealed in the cerebral circulation. Early et al.38 suggested a novel linkage among EDHF, 11, 12‐EET, TRPV4 considering hyperpolarization occurring in the vascular smooth muscle and subsequent relaxant responses.38 Their findings indicated that 11, 12‐EET and TRPV4 agonist 4α‐PDD when applied exogenously, TRPV4 currents were stimulated leading to hyperpolarization and relaxation of pressurized cerebral artery. However, administration of ruthenium red, the TRPV4 inhibitor, as well as an antisense knockout of endogenous TRPV4, resulted in development of responses that were profoundly reduced. The stimulation of TRPV4 results in an enhancement in [Ca2+]i, and therefore produces vasoconstriction alternative to relaxation as predicted. A new system integrating this ambiguity depends on a complicated chain of many distinct mechanisms. This includes stimulation of inflow of Ca2+ by TRPV4 channels by 11, 12‐ET, which causes an augmentation in Ca2+ release from intracellular stores. Thus, it causes the stimulation of large‐conductance Ca2+‐dependent K+ channel (BKCa) leading to hyperpolarization of the membrane. Therefore, this effect leads to reduced Ca2+ influx through voltage‐dependent calcium channel (VDCCs), decrease in [Ca2+]i and subsequent vasorelaxation. Another possible mechanism by which EETs activate TRPV4 receptor was provided by Campbell and Fleming.42 They concluded that EETs act on cells of the endothelium to enhance the influx of Ca2+ by TRPV4. Calcium then stimulates the two types of K channels, namely small‐conductance (SK) and intermediate‐conductance (IK) KCa channels to hyperpolarize the endothelial cells and cause the efflux of K+ ions towards the endothelial space below. Following that, sodium‐potassium ATPase or inward rectifying (Kir) K channel is activated in response to potassium ions. This results in a hyperpolarizing and relaxing effect on VSMCs due to transmission of the hyperpolarizing current from endothelial cells into VSMCs.
The results of this study suggest that 11, 12‐EET or blockage of its breakdown and/or metabolism may provide a valuable tool in preventing vascular dysfunction and in metabolic diseases such as diabetes. Inhibition of sEH is a possible method to augment the protection of vascular beds through intimate EETs, which may prove to be effective in the management of many cardiovascular and metabolic problems. Therefore, 11, 12‐EET and sEH inhibitors have the characteristics of potential therapeutic targets to avert the abnormal vascular function related to diabetes.
Development of EET mimetics and antagonists, as well as potent selective sEH inhibitors, is now available to further explore functional effects of EETs. There is a promising future for sEH inhibitors and EET analogues as novel therapies for treatment of hypertension and to stop the progression of chronic kidney disease.42
Conflict of interest
The authors have declared no competing interests.
Bihzad SM, Yousif MHM. 11,12‐Epoxyeicosatrienoic acid induces vasodilator response in the rat perfused mesenteric vasculature. Auton Autacoid Pharmacol. 2017;37:3‐12. https://doi.org/10.1111/aap.12052
Funding Information
This study was funded by Kuwait University, Research Administration, Project No. MR04/09.
References
- 1. Spector AA. Arachidonic acid cytochrome P450 epoxygenases pathway. J Lipid Res. 2009;50:S52‐S56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pfister SL, Gauthier KM, Campbell WB. Vascular pharmacology of epoxyeicosatrienoic acids. Adv Pharmacol. 2010;60:27‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roman JR. P‐450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131‐185. [DOI] [PubMed] [Google Scholar]
- 4. Yang J, Dong H, Hammock D. EETs and OXO‐ETE in airway diseases In: Ray A, ed. Obstructive Airway Diseases. Role of Lipid Mediators. New York, NY: CRS Press; 2012:130‐131. [Google Scholar]
- 5. Hesselink JMK. The terms ‘autacoid’, ‘hormone’ and ‘chalone’ and how they have shifted with time. Auton Autacoid Pharmacol. 2015;35:51‐58. [DOI] [PubMed] [Google Scholar]
- 6. Yousif MH. Phosphoinositide 3‐kinase contributes to diabetes‐induced abnormal vascular reactivity in rat perfused mesenteric bed. Cell Biochem Funct. 2008;26:451‐458. [DOI] [PubMed] [Google Scholar]
- 7. Yousif MH, Benter IF. Role of cytochrome P450 metabolites of arachidonic acid in regulation of corporal smooth muscle tone in diabetic and older rats. Vascul Pharmacol. 2007;47:281‐287. [DOI] [PubMed] [Google Scholar]
- 8. Yousif MH, Benter IF, Dunn KM, Dahly‐Vernon AJ, Akhtar S, Roman RJ. Role of 20‐hydroxyeicosatetraenoic acid in altering vascular reactivity in diabetes. Auton Autacoid Pharmacol. 2009;29:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yousif MH, Benter IF, Abraham S, Akhtar S. Inhibition of Ras‐GTPase improves diabetes‐induced abnormal vascular reactivity in the rat perfused mesenteric vascular bed. Med Princ Pract. 2004;13:57‐62. [DOI] [PubMed] [Google Scholar]
- 10. Yousif MH, Kehinde EO, Benter IF. Different response to angiotensin‐(1‐7) in young, aged and diabetic rabbit corpus cavernosum. Pharmacol Res. 2007;56:209‐216. [DOI] [PubMed] [Google Scholar]
- 11. Benter IF, Yousif MH, Cojocel C, Al‐Maghrebi M, Diz DI. Angiotensin‐(1‐7) prevents diabetes‐induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H666‐72. [DOI] [PubMed] [Google Scholar]
- 12. Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discovery. 2009;8:794‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foley RN, Collins AJ. End‐stage renal disease in the United States: an update from the United States Renal Data System. J Am Soc Nephrol. 2007;18:2644‐2648. [DOI] [PubMed] [Google Scholar]
- 14. Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor a blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt‐sensitive hypertension. Hypertension. 2006;47:557‐562. [DOI] [PubMed] [Google Scholar]
- 15. Node K, Huo Y, Ruan X, et al. Anti‐inflammatory properties of cytochrome P450 epoxygenase‐derived eicosanoids. Science. 1999;285:1276‐1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Falck JR, Reddy L M, Reddy Y K, et al. 11,12‐epoxyeicosatrienoic acid (11,12‐EET): structural determinants for inhibition of TNF‐ ‐induced VCAM‐1 expression. Bioorg Med Chem Lett. 2003;13:4011‐4014. [DOI] [PubMed] [Google Scholar]
- 17. Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS‐induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79:2311‐2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao X, Yamamoto T, Newman JW, et al. Soluble epoxide hydrolase inhibition protects the kidney from hypertension‐induced damage. J Am Soc Nephrol. 2004;15:1244‐1253. [PubMed] [Google Scholar]
- 19. Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Attenuation of tobacco smoke‐induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc Natl Acad Sci USA. 2005;102:2186‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55‐90. [DOI] [PubMed] [Google Scholar]
- 21. Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496‐F503. [DOI] [PubMed] [Google Scholar]
- 22. Wong SM, Vanhoutte MP. COX‐mediated endothelium‐dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin. 2010;31:1095‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Furchgott RF, Vanhoutte PM. Endothelium‐derived relaxing and contracting factors. FASEB J. 1989;3:2007‐2018. [PubMed] [Google Scholar]
- 24. Michel FS, Man GS, Man RY, Vanhoutte PM. Hypertension and the absence of EDHF‐mediated responses favour endothelium dependent contractions in renal arteries of the rat. Br J Pharmacol. 2008;155:217‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vanhoutte PM, Tang EH. Endothelium‐dependent contractions: when a good guy turns bad!. J Physiol. 2008;586:5295‐5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luscher T, Boulanger C, Dohi Y, Yang Z. Endothelium‐derived contracting factors. Hypertension. 1992;19:117‐130. [DOI] [PubMed] [Google Scholar]
- 27. Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium‐derived hyperpolarizing factors. Circ Res. 1996;78:415‐423. [DOI] [PubMed] [Google Scholar]
- 28. Cowan CL, Cohen RA. Two mechanisms mediate relaxation by bradykinin of pig coronary artery: NO‐dependent and‐independent responses. Am J Physiol Heart Circ Physiol. 1991;261:H830‐H835. [DOI] [PubMed] [Google Scholar]
- 29. Eckman DM, Weinert JS, Buxton LO, Keef KD. Cyclic GMP‐independent relaxation and hyperpolarization with acetylcholine in guinea‐pig coronary artery. Br J Pharmacol. 1994;111:1053‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pratt PF, Li P, Hillard CJ, Kurian J, Campbell WB. Endothelium‐independent, ouabain‐sensitive relaxation of bovine coronary arteries by EETs. Am J Physiol Heart Circ Physiol. 2001;280:H1113‐H1121. [DOI] [PubMed] [Google Scholar]
- 31. Larsen BT, Miura H, Hatoum OA, et al. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BKCa channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol Heart Circ Physiol. 2006;290:H491‐H499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dimitropoulou C, West L, Field MB, et al. Protein phosphatase 2A and Ca2+‐ activated K+ channels contribute to 11, 12‐epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat. 2007;83:50‐61. [DOI] [PubMed] [Google Scholar]
- 33. Wang XL, Lu T, Cao S, Shah VH, Lee HC. Inhibition of ATP binding to the carboxyl terminus of Kir 6.2 by epoxyeicosatrienoic acids. Biochim Biophys Acta. 2006;1761:1041‐1049. [DOI] [PubMed] [Google Scholar]
- 34. Hercule HC, Schunck WH, Gross V, et al. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009;29:54‐60. [DOI] [PubMed] [Google Scholar]
- 35. Campbell WB, Deeter C, Gauthier KM, Ingraham RH, Falck JR, Li PL. 14,15 Dihydroxyeicosatrienoic acid relaxes bovine coronary arteries by activation of KCa channels. Am J Physiol Heart Circ Physiol. 2002;282:H1656‐H1664. [DOI] [PubMed] [Google Scholar]
- 36. Sacerdoti D, Bolognesi M, Di PM, et al. Rat mesenteric arterial dilator response to 11, 12‐epoxyeicosatrienoic acid is mediated by activating hemeoxygenase. Am J Physiol Heart Circ Physiol. 2006;291:H1999‐H2002. [DOI] [PubMed] [Google Scholar]
- 37. Dragoni S, Guerra G, Pla AF, et al. A functional transient receptor potential vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J Cell Physiol. 2014;230:95‐104. [DOI] [PubMed] [Google Scholar]
- 38. Zhang P, Mao A, Sun C, et al. Translocation of PKG1α acts on TRPV4‐C1 heteromeric channels to inhibit endothelial Ca2+ entry. Acta Pharmacol Sin. 2016;37:1199‐1207. doi:10.1038/aps.2016.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. TRPV4‐dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol. 2009;297:H1096‐H1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270‐1279. [DOI] [PubMed] [Google Scholar]
- 41. Ding Y, Frömel T, Popp R, Falck JR, Schunck WH, Fleming I. The biological actions of 11,12‐epoxyeicosatrienoic acid in endothelial cells are specific to the R/S‐enantiomer and require the G(s) protein. J Pharmacol Exp Ther. 2014;350:14‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium‐dependent responses. Pflugers Arch. 2010;459:881‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. John D. Imig. Epoxyeicosatrienoic acids, hypertension, and kidney injury. recent advances in hypertension. Recent advances in hypertension. Hypertension. 2015;65:476‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]