

Abstract
Objective
To assess the performance of screening for fetal trisomies 21, 18 and 13 by cell‐free (cf) DNA analysis of maternal blood using a new method based on paired‐end massively parallel shotgun sequencing (MPSS).
Methods
This was a blinded study of plasma samples (1mL) obtained from 1000 women undergoing screening for fetal trisomies 21, 18 and 13 at 11–13 weeks' gestation. The study included 50 cases with confirmed fetal trisomy 21, 30 with trisomy 18, 10 with trisomy 13 and 910 unaffected pregnancies. Paired‐end MPSS with the neoBona® test allowed simultaneous assessment of fetal fraction, cfDNA fragment size distribution and chromosome counting, which were integrated into a new analysis algorithm to calculate trisomy likelihood ratios (t‐score) for each chromosome of interest. Each sample was classified as trisomic or unaffected using chromosome‐specific cut‐offs set at t‐score values of 1.5 for trisomy 21 and 3.0 for trisomies 18 and 13.
Results
Valid results were provided for 988 (98.8%) cases; 12 (1.2%) samples, from nine euploid and three trisomy 21 pregnancies, did not pass quality‐control criteria and were excluded from further analysis. All 47 cases of trisomy 21, all 10 of trisomy 13, 29 of 30 with trisomy 18 and all 901 unaffected cases were classified correctly. Median fetal fraction was 10.5% (range, 0.3–33.8%) and trisomic and unaffected cases with low fetal fractions of < 1% were identified correctly.
Conclusions
This novel method for cfDNA analysis of maternal plasma, which utilizes paired‐end MPSS, can provide accurate prediction of fetal trisomies. Use of a new multicomponent t‐score removes the need to reject samples with fetal fraction < 4%, which potentially extends the benefits of non‐invasive prenatal cfDNA analysis to a larger proportion of pregnancies. © 2016 Authors. Ultrasound in Obstetrics & Gynecology published by John Wiley & Sons Ltd on behalf of International Society of Ultrasound in Obstetrics and Gynecology.
Keywords: cell‐free DNA, paired‐end sequencing, screening, trisomy
INTRODUCTION
Screening for fetal aneuploidy by analysis of cell‐free (cf) DNA in maternal blood was made possible by the advent of massively parallel shotgun sequencing (MPSS) which allows digital counting of cfDNA fragments, either by whole‐genome sequencing1, 2 or targeted approaches3, 4. Detection of fetal trisomy using counting statistics, such as Z‐score or normalized chromosome values (NCV), becomes easier and more robust when the proportion of fetal DNA to total cfDNA in maternal blood is high because of greater separation between normal and aneuploid cases5, 6. The sensitivity of detecting fetal trisomies at low fetal fraction is dependent mostly on the amount of useful counts of the chromosome of interest or sequencing depth6, 7. Inclusion of fetal fraction in analysis algorithms can improve specificity because, in cases of low Z‐scores or NCVs, it helps distinguish between aneuploid cases with low fetal fraction from euploid cases with a higher fetal fraction5.
Paired‐end MPSS allows accurate digital counting while also determining the length of each cfDNA fragment by sequencing both its extremities8. As cfDNA fragments of fetal origin are slightly shorter than maternal ones, size differences can be used to determine fetal fraction8. Additionally, in the case of fetal aneuploidy, counting differences detected from all cfDNA fragments would appear more evident if confirmed on shorter fragments only8, 9.
neoBona® (Labco Diagnostics, Barcelona, Spain) is the first cfDNA‐based screening test to exploit paired‐end MPSS through a novel bioinformatics approach, which has the advantage of combining conventional counting statistics with the distribution of cfDNA fragment size to provide a double check of chromosome counting data. Additionally, by integrating sequencing depth on each chromosome and fetal fraction it allows calculation of a unique trisomy score (t‐score), thereby quantifying the likelihood of fetal trisomy. The objective of this study was to evaluate the performance of this new method on a large blinded set of archived maternal plasma samples, tested without previous knowledge of their outcomes.
METHODS
Study population
Blood samples were collected between April 2006 and February 2015 at King's College Hospital, London, UK, from women with a singleton pregnancy undergoing screening for trisomies 21, 18 and 13 by assessment of a combination of fetal nuchal translucency (NT) thickness and maternal serum free beta human chorionic gonadotropin (β‐hCG) and pregnancy‐associated plasma protein‐A (PAPP‐A) at 11–13 weeks' gestation10. Gestational age was determined from measurement of the fetal crown–rump length11. Women with a high risk from the combined test had chorionic villus sampling (CVS) for fetal karyotyping. Karyotype results obtained from genetic laboratories and details on pregnancy outcome obtained from the maternity computerized records or the general medical practitioners of the women were added into the database as soon as they became available. All patients gave written informed consent to provide samples for research, which was approved by the National Health Service Research Ethics Committee.
Blood samples were collected into EDTA BD vacutainerTM tubes (Becton Dickinson UK limited, Oxford, UK) and centrifuged at 2000 g for 10 min within 15 min of collection (Plasma 1) followed by another 10 min at 16 000 g to further separate cell debris (Plasma 2). Samples of Plasma 1 and 2 were divided into 0.5‐mL aliquots in separate Eppendorf tubes, labelled with a unique patient identifier and stored at −80 °C for up to 9 years until analysis. A total of 50 cases with trisomy 21, 30 with trisomy 18, 10 with trisomy 13 and 910 normal controls with 1 mL of Plasma 1 or 2 were selected for analysis. Cases with aneuploidy were selected at random and each was matched to 11–12 controls that were sampled on the same or next day as the aneuploid case. Normal controls were uncomplicated pregnancies resulting in live birth after 38 weeks' gestation of phenotypically normal neonates assumed to be euploid. None of the samples was previously thawed and refrozen. Plasma samples (two tubes of 0.5 mL per patient) were coded and sent on dry ice from London to the central laboratory of Labco Diagnostics in Barcelona, Spain, where blinded cfDNA analysis was performed using the neoBona test.
Analysis of samples
The only information provided to the laboratory for each sample was the patient‐unique identifier, date of collection and whether it was a Plasma 1 or 2 sample. Each sample was assessed for volume, adequacy of labeling and risk of contamination or sample mixing before evaluation of fetal trisomy. Although the volume was < 1 mL (range, 500–950 μL) in 60 of the 1000 samples, they were included in the analysis. Plasma samples from each patient were collected into 96 deep‐well plates. Plates of Plasma 1 samples underwent a second centrifugation step at 16 000 g before DNA extraction. Samples were processed in batches of 96 using VeriSeq NIPT v1.0 chemistry (Illumina Inc, San Diego, CA, USA) on a fully automated workstation (Hamilton Star, Hamilton, Reno, NV, USA) designed to handle plasma isolation, column‐based DNA extraction, set‐up of sequencing library, quantification, normalization and pooling. Sequencing libraries from each batch of 96 samples were collected in two separate pools of 48 double‐indexed samples which underwent paired‐end MPSS for two sets of 36 cycles using NextSeq 500 and 550 sequencers with TG NextSeq 500/550 High Output Kit v1.2 (Illumina inc). Sequencing outputs were analyzed using the VeriSeq NIPT software v1.0 (Illumina inc).
After de‐multiplexing and filtering, sequence alignment was performed against HG19 for data normalization and interchromosome comparisons7. Regions affected by poor alignment were filtered out and further normalization was applied based on a principal component decomposition as described by Zhao et al.12. Fetal fraction assessment, based on molecular size distributions and differences in coverage between fetal and maternal cfDNA, was complemented with X and Y chromosomes data in cases of male fetuses8, 9, 13. NCVs were calculated for chromosomes 13, 18 and 21, as described previously6, 14. NCV counting statistics are similar in principle to the conventional Z‐score, with a fixed cut‐off of around 3.0 to discriminate between trisomic and unaffected pregnancies, the main difference being that, for NCVs, each chromosome of interest is only normalized against a specific set of chromosomes, optimized for comparable sequencing coverage to minimize variations.
Trisomy likelihood ratios (t‐scores) for each chromosome of interest were calculated for each sample based on the estimated fetal fraction, counting statistics (NCVs) derived from both total and short DNA fragments, and sequencing depth. The likelihood ratio reflects the probability for a sample to be affected, given the observed counting statistics and fetal fraction, versus the probability of a sample to be unaffected, given the same counting data. Thus, using this analysis approach, trisomic samples with low fetal fraction can result in a higher t‐score if they have, for instance, a higher depth of sequencing enabling efficient counting on short DNA fragments which are mostly of fetal origin.
Samples were classified as being compatible with the presence or absence of trisomy 21, 18 or 13 using predefined chromosome specific cut‐offs at t‐score values of 1.5 for trisomy 21 and 3.0 for trisomies 18 and 13.
Quality‐control analyses (QCs) were applied to monitor sequencing depth, the distribution of cfDNA fragment sizes, sequencing coverage for chromosome denominators and for the estimate of fetal fraction. Results were considered valid only for samples passing all QCs.
Results were provided to King's College Hospital in which the classification for each case was compared to pregnancy outcome and detection rates and false‐positive rates were estimated.
RESULTS
The characteristics of the study population are summarized in Table 1. Compared to euploid pregnancies, in pregnancies with trisomy 21, median maternal age, fetal NT and serum free β‐hCG were higher and serum PAPP‐A was lower and in pregnancies with trisomy 18 or 13 median maternal age and fetal NT were higher and serum free β‐hCG and PAPP‐A were lower.
Table 1.
Characteristics of 1000 pregnant women undergoing prenatal screening for fetal trisomies, according to outcome
| Characteristic | Euploid (n = 910) | Trisomy 21 (n = 50) | Trisomy 18 (n = 30) | Trisomy 13 (n = 10) |
|---|---|---|---|---|
| Maternal age (years) | 31.9 (27.3–34.9) | 37.9 (35.3–41.3) | 35.4 (28.8–40.5) | 33.5 (30.0–34.9) |
| Maternal weight (kg) | 65.0 (59.0–75.0) | 68.0 (60.7–75.0) | 66.8 (60.4–75.5) | 64.5 (60.6–64.9) |
| Maternal height (cm) | 165 (160–169) | 166 (161–172) | 166 (160–171) | 167 (164–170) |
| Racial origin | ||||
| Caucasian | 563 (61.9) | 41 (82.0) | 17 (56.7) | 8 (80.0) |
| Afro‐Caribbean | 244 (26.8) | 6 (12.0) | 6 (20.0) | 1 (10.0) |
| South Asian | 32 (3.5) | 1 (2.0) | 3 (10.0) | 0 (0) |
| East Asian | 26 (2.9) | 2 (4.0) | 2 (6.7) | 0 (0) |
| Mixed | 45 (4.9) | 0 (0) | 2 (6.7) | 1 (10.0) |
| Cigarette smoker | 55 (6.0) | 3 (6.0) | 1 (3.3) | 1 (10.0) |
| Method of conception | ||||
| Spontaneous | 880 (96.7) | 46 (92.0) | 26 (86.7) | 10 (100) |
| Ovulation drugs | 9 (1.0) | 3 (6.0) | 2 (6.7) | 0 (0) |
| In‐vitro fertilization | 21 (2.3) | 1 (2.0) | 2 (6.7) | 0 (0) |
| Fetal crown–rump length (mm) | 61.8 (57.0–67.6) | 66.1 (60.0–73.0) | 56.1 (51.9–61.6) | 59.0 (51.1–63.1) |
| GA at screening (weeks) | 12.6 (12.2–13.0) | 12.9 (12.5–13.4) | 12.2 (11.8–12.6) | 12.4 (11.8–12.7) |
| Fetal NT thickness (mm) | 1.7 (1.5–1.9) | 4.4 (3.4–6.2) | 6.5 (3.6–7.9) | 5.2 (2.3–6.3) |
| PAPP‐A MoM | 1.126 (0.766–1.563) | 0.695 (0.441–0.869) | 0.227 (0.135–0.327) | 0.371 (0.282–0.570) |
| Free β‐hCG MoM | 0.995 (0.678–1.582) | 2.259 (1.574–3.109) | 0.293 (0.171–0.362) | 0.314 (0.204–0.747) |
Data are given as median (interquartile range) or n (%).
β‐hCG, beta human chorionic gonadotropin; GA, gestational age; MoM, multiples of the median; NT, nuchal translucency; PAPP‐A, pregnancy‐associated plasma protein‐A.
The cfDNA test provided results for 988 (98.8%) cases. In total, 12 (1.2%) samples, nine from euploid and three from trisomy 21 pregnancies, failed to provide a result and were excluded from further analysis. The reasons for QC failure were size distribution of cfDNA fragments beyond the expected range (n = 6), low sequencing depth for the observed fetal fraction (n = 4), unusually high DNA concentration (n = 1) and insufficient sequencing coverage for determination of fetal fraction (n = 1).
The cfDNA test classified correctly all 47 pregnancies with fetal trisomy 21, all 10 with trisomy 13, 29 (96.7%) of 30 with trisomy 18 and all 901 unaffected pregnancies (Table 2). In one case of trisomy 18, t‐score and NCV values for chromosome 18 were compatible with normal chromosome copy number; in this case the fetal fraction was 11%. One case with trisomy 21 and one unaffected pregnancy had the same NCV of 3.5, but had different t‐scores for trisomy 21 which were 10 and −14, respectively. Therefore, using the predefined cut‐offs of t‐score values of 1.5 for trisomy 21 and 3.0 for both trisomies 18 and 13 resulted in detection rates of 100% for trisomies 21 and 13 and 96.7% for trisomy 18, with false‐positive rate of 0% for all trisomies.
Table 2.
Results of cell‐free DNA analysis by neoBona® test for fetal trisomy screening in 988 women with test result, according to outcome
| Result | Euploid (n = 901) | Trisomy 21 (n = 47) | Trisomy 18 (n = 30) | Trisomy 13 (n = 10) |
|---|---|---|---|---|
| NCV for chromosome 21 | −0.01 (−3.43 to 3.55) | 11.50 (3.59 to 25.67) | 0.32 (−1.88 to 3.48) | −0.048 (−1.36 to 1.25) |
| t‐score for trisomy 21 | −23.2 (−1074.2 to 0.6) | 101.0 (7.2 to 392.1) | −12.0 (−178.4 to −1.1) | −20.5 (−121.8 to −5.2) |
| NCV for chromosome 18 | 0.01 (−3.25 to 6.17) | −0.24 (−2.42 to 2.71) | 12.24 (−1.22* to 36.91) | 0.72 (−1.49 to 2.56) |
| t‐score for trisomy 18 | −31.3 (−1960.6 to 1.0) | −38.6 (−322.2 to −3.6) | 94.5 (−17.9* to 765.2) | −21.9 (−247.2 to −7.2) |
| NCV for chromosome 13 | 0.001 (−4.55 to 4.44) | −0.09 (−1.91 to 2.58) | 0.42 (−2.30 to 2.16) | 14.76 (6.31 to 28.50) |
| t‐score for trisomy 13 | −37.6 (−2591.6 to 0.1) | −48.6 (−449.6 to −1.3) | −15.2 (−442.6 to 2.0) | 209.3 (23.9 to 479.5) |
| Fetal fraction (%) | 10.2 (0.3 to 33.8) | 10.7 (3.8 to 19.8) | 9.6 (0.8 to 23.0) | 7.9 (4.0 to 15.3) |
| Trisomy 21 t‐score > 1.5 | 0 (0) | 47 (100) | 0 (0) | 0 (0) |
| Trisomy 18 t‐score > 3.0 | 0 (0) | 0 (0) | 29 (96.7) | 0 (0) |
| Trisomy 13 t‐score > 3.0 | 0 (0) | 0 (0) | 0 (0) | 10 (100) |
Data are given as median (range) or n (%).
Value from the only discrepant result; case of trisomy 18 with 11% fetal fraction resulted in trisomy likelihood score (t‐score) and normalized chromosome value (NCV) compatible with normal chromosome 18 copy number.
The mean fetal fraction was 10.6% for euploid pregnancies, 11.1% for trisomy 21, 9.4% for trisomy 18 and 8.9% for trisomy 13. One case of trisomy 21, three of trisomy 18 and 58 unaffected pregnancies were identified correctly despite showing fetal fractions below 4%, including one case of trisomy 18 and nine euploid cases with fetal fraction < 1% (Table 2 and Figure 1).
Figure 1.
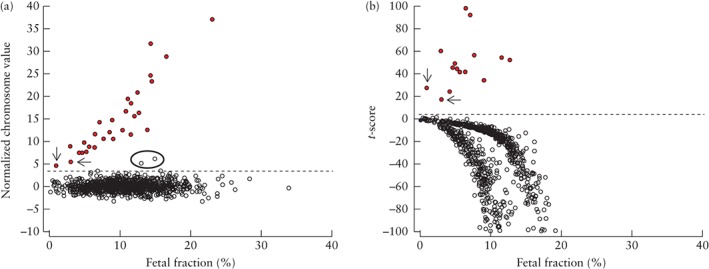
Normalized chromosome
value (NCV) (a) and trisomy likelihood score (t‐score), for values between −100 and 100 (b), for chromosome 18, in 29 pregnancies with trisomy 18 ( ) and 901 unaffected pregnancies (
) and 901 unaffected pregnancies ( ), plotted against fetal fraction. Plot of NCV in (a) shows limitation of this method because, in two cases of trisomy 18 with low fetal fraction (< 4%) (arrows in (a) and (b)), NCV was similar to those of two unaffected cases (circled) with high fetal fraction; these unaffected cases would have been classified wrongly as positive for trisomy 18 as they are above the cut‐off of 3.0 (
), plotted against fetal fraction. Plot of NCV in (a) shows limitation of this method because, in two cases of trisomy 18 with low fetal fraction (< 4%) (arrows in (a) and (b)), NCV was similar to those of two unaffected cases (circled) with high fetal fraction; these unaffected cases would have been classified wrongly as positive for trisomy 18 as they are above the cut‐off of 3.0 ( ). Plot of t‐scores in (b) shows cases of trisomy 18 with score well above cut‐off of 3.0 for trisomy 18 (
). Plot of t‐scores in (b) shows cases of trisomy 18 with score well above cut‐off of 3.0 for trisomy 18 ( ), including one case with fetal fraction < 1%.
), including one case with fetal fraction < 1%.
DISCUSSION
The findings of this study demonstrate the feasibility of a new approach for cfDNA testing of maternal blood in screening for fetal trisomies 21, 18 and 13. Paired‐end MPSS of cfDNA coupled with a novel analysis algorithm provided simultaneous assessment of fetal fraction, distribution of size of DNA fragments and chromosome counting. Trisomy likelihood ratios for each chromosome of interest could then be calculated for each sample based on the estimated fetal fraction, chromosome‐specific counting statistics on total and short fragments and sequencing depth. We used this novel approach to examine stored plasma samples and, at preselected chromosome‐specific cut‐offs of t‐score values of 1.5 for trisomy 21 and 3.0 for trisomies 18 and 13, the test classified correctly all cases of trisomy 21, trisomy 13 and unaffected pregnancies and 29 of 30 cases of trisomy 18. Such high performance of screening is compatible with the best results of previous studies utilizing cfDNA testing to screen for trisomies 21, 18 and 1315.
In the single case of trisomy 18 that was misclassified, the fetal fraction was 11% and is therefore highly unlikely that this error was related to technical issues affecting test sensitivity. Unfortunately, no more sample was available to repeat the test and exclude errors due to laboratory mishandling. In addition, trisomy rescue, generating a normal cell line in the cytotrophoblast, could not be ruled out as the underlying cause of this discrepancy as prenatal diagnosis was only performed on long‐term CVS culture by quantitative fluorescent polymerase chain reaction and karyotype, but not on direct preparation.
The basis for cfDNA testing using counting methods is that, in trisomic pregnancies, the number of molecules derived from the extra fetal chromosome, as a proportion of all sequenced molecules in maternal plasma, is higher than in euploid pregnancies. The ability to detect the small increase in the amount of a given chromosome in maternal plasma in a trisomic compared to a disomic pregnancy is related directly to the fetal fraction and the depth of sequencing3, 16, 17, 18, 19. Trisomy cases with low fetal fraction used to be more difficult to discriminate from normal samples by counting statistics only, as they can produce NCVs with similar values to those occasionally observed in normal samples with higher fetal fraction6, thus reducing test specificity. Also the sensitivity could be affected if, for the sequencing depth used, the proportion of fetal cfDNA is too low to allow discrimination of trisomies by counting statistics only7, 18. For these reasons, when the fetal fraction is below 4%, which occurs in 0.5–6.1% of pregnancies, the cfDNA test is usually presented as a failure and no result is reported15.
Some of the problems due to low fetal fraction have now been overcome by the application of the multicomponent t‐score, as the resolution in discriminating between trisomic and unaffected pregnancies is no longer dependent only on fetal fraction but also on the new possibility of performing additional counting statistics on short DNA fragments, which are mostly of fetal origin. Consequently, trisomic pregnancies with low fetal fraction could result in higher t‐score values than in pregnancies with higher fetal fraction and lower total sequencing depth or less efficient counting statistics on short fragments. This approach proved to be highly efficient at low fetal cfDNA amounts, as all four aneuploid cases with fetal fraction between 0.8% and 3.5% could be detected. The effectiveness of the new multicomponent t‐score to improve overall specificity was evident in one case of trisomy 21 and one unaffected sample that were classified correctly despite generating the same NCV, and thus would be undistinguishable by conventional MPSS analysis algorithms.
Despite testing archived plasma samples, and with suboptimal volumes in 6% of cases, failure to provide a result was only observed in 1.2% of samples. The most common reason for test failure was an abnormal distribution of size of cfDNA fragments, which affected size‐based counting and the measurement of fetal fraction. This artifact was likely to be caused by cfDNA shearing, resulting from sample degradation. It is therefore expected to occur less frequently in routine clinical samples collected in dedicated tubes, designed to prevent cell lysis and stabilize cfDNA. Four more samples failed the QC analysis which combines sequencing metrics and estimated fetal fraction, thus determining if the analysis output has statistical confidence in scoring a sample. Repeating the test on a second aliquot of the same plasma would probably have yielded a valid result. In clinical routine, the test is usually performed within a few days from sampling and with enough volume to be repeated, therefore this technical failure is expected to decrease.
The novel approach presented in this study has the potential of extending the advantages of cfDNA‐based aneuploidy screening to a wider proportion of pregnancies. Complementing conventional counting statistics with size‐based chromosome counting and fetal fraction ensured that accurate prediction of trisomic status was provided in 62 of our cases with fetal fraction < 4%. Consequently, it is no longer necessary to exclude samples from analysis solely because the fetal fraction is < 4% if enough sequencing depth is reached for the corresponding amount of cfDNA and size‐based counting is performed at the same time.
ACKNOWLEDGMENTS
This study was supported by a grant from The Fetal Medicine Foundation (UK Charity No: 1037116). The neoBona test was performed by Labco Diagnostics, Barcelona, Spain with support from Illumina Inc. (San Diego, CA, USA).
REFERENCES
- 1. Chiu RWK, Chan KCA, Gao Y, Lau VYM, Zheng W, Leung TY, Foo CHF, Xie B, Tsui NBY, Lun FMF, Zee BCY, Lau TK, Cantor CR, Lo YMD. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA 2008; 105: 20458–20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA 2008; 105: 16266–162271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sparks AB, Struble CA, Wang ET, Song K, Oliphant A. Noninvasive prenatal detection and selective analysis of cell‐free DNA obtained from maternal blood: evaluation for trisomy 21 and trisomy 18. Am J Obstet Gynecol 2012; 206: 319.e1–9. [DOI] [PubMed] [Google Scholar]
- 4. Nicolaides KH, Syngelaki A, Ashoor G, Birdir C, Touzet G. Noninvasive prenatal testing for fetal trisomies in a routinely screened first‐trimester population. Am J Obstet Gynecol 2012; 207: 374.e1–6. [DOI] [PubMed] [Google Scholar]
- 5. Yeang CH, Ma GC, Hsu HW, Lin YS, Chang SM, Cheng PJ, Chen CA, Ni YH, Chen M. Genome‐wide normalized score: a novel algorithm to detect fetal trisomy 21 during non‐invasive prenatal testing. Ultrasound Obstet Gynecol 2014; 44: 25–30. [DOI] [PubMed] [Google Scholar]
- 6. Rava RP, Srinivasan A, Sehnert AJ, Bianchi DW. Circulating fetal cell‐free DNA fractions differ in autosomal aneuploidies and monosomy X. Clin Chem 2014; 60: 243–250. [DOI] [PubMed] [Google Scholar]
- 7. Fan CH, Quake SR. Sensitivity of Noninvasive Prenatal Detection of Fetal Aneuploidy from Maternal Plasma Using Shotgun Sequencing Is Limited Only by Counting Statistics. PLoS ONE 2010; 5: e10439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu SC, Chan KC, Zheng YW, Jiang P, Liao GJ, Sun H, Akolekar R, Leung TY, Go AT, van Vugt JM, Minekawa R, Oudejans CB, Nicolaides KH, Chiu RW, Lo YM. Size‐based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci USA 2014; 111: 8583–8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim SK, Hannum G, Geis J, Tynan J, Hogg G, Zhao C, Jensen TJ, Mazloom AR, Oeth P, Ehrich M, van den Boom D, Deciu C. Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenat Diagn 2015; 35: 810–815. [DOI] [PubMed] [Google Scholar]
- 10. Nicolaides KH. Screening for fetal aneuploidies at 11 to 13 weeks. Prenat Diagn 2011; 31: 7–15. [DOI] [PubMed] [Google Scholar]
- 11. Robinson HP, Fleming JE. A critical evaluation of sonar crown rump length measurements. Br J Obstet Gynaecol 1975; 82: 702–710. [DOI] [PubMed] [Google Scholar]
- 12. Zhao C, Tynan J, Ehrich M, Hannum G, McCullough R, Saldivar JS, Oeth P, van den Boom D, Deciu C. Detection of fetal subchromosomal abnormalities by sequencing circulating cell‐free DNA from maternal plasma. Clin Chem 2015; 61: 608–616. [DOI] [PubMed] [Google Scholar]
- 13. Fan CH, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Analysis of the size distributions of fetal and maternal cell‐free DNA by paired‐end sequencing. Clin Chem 2010; 56: 1279–1286. [DOI] [PubMed] [Google Scholar]
- 14. Sehnert AJ, Rhees B, Comstock D, de Feo E, Heilek G, Burke J, Rava RP. Optimal detection of fetal chromosomal abnormalities by massively parallel DNA sequencing of cell‐free fetal DNA from maternal blood. Clin Chem 2011; 57: 1042–1049. [DOI] [PubMed] [Google Scholar]
- 15. Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell‐free DNA in maternal blood in screening for fetal aneuploidies: updated meta‐analysis. Ultrasound Obstet Gynecol 2015; 45: 249–266. [DOI] [PubMed] [Google Scholar]
- 16. Palomaki GE, Kloza EM, Lambert‐Messerlian GM, Haddow JE, Neveux LM, Ehrich M, van den Boom D, Bombard AT, Deciu C, Grody WW, Nelson SF, Canick JA. DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet Med 2011; 13: 913–920. [DOI] [PubMed] [Google Scholar]
- 17. Canick JA, Palomaki GE, Kloza EM, Lambert‐Messerlian GM, Haddow JE. The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat Diagn 2013; 33: 667–674. [DOI] [PubMed] [Google Scholar]
- 18. Benn P, Cuckle H. Theoretical performance of non‐invasive prenatal testing for chromosome imbalances using counting of cell‐free DNA fragments in maternal plasma. Prenat Diagn 2014; 34: 778–783. [DOI] [PubMed] [Google Scholar]
- 19. Wright D, Wright A, Nicolaides KH. A unified approach to risk assessment for fetal aneuploidies. Ultrasound Obstet Gynecol 2015; 45: 48–54. [DOI] [PubMed] [Google Scholar]