

Summary
The prevalence of aspergillosis in CF patients has until recently been underestimated, but increasing evidence suggests that it may play an important role in the progression of CF lung disease. In healthy airways, Aspergillus fumigatus can be efficiently removed from the lung by mechanisms such as mucociliary clearance and cough. However, these mechanisms are defective in CF, allowing pathogens such as A. fumigatus to germinate and establish chronic infections within the airways. The precise means by which A. fumigatus contributes to CF lung disease remain largely unclear. As the first point of contact within the lung, and an important component of the innate immune system, it is likely that the mucus barrier plays an important role in this process. Study of the functional interplay between this vital protective barrier, and in particular its principal structural components, the polymeric gel‐forming mucins, and CF pathogens such as A. fumigatus, is at an early stage. A. fumigatus protease activity has been shown to upregulate mucus production by inducing mucin mRNA and protein expression, and A. fumigatus proteases and glycosidases are able to degrade mucins. This may allow A. fumigatus to alter mucus barrier properties to promote fungal colonization of the airways and/or utilize mucins as a nutrient source. Moreover, conidial surface lectin binding to mucin glycans is a key aspect of clearance of Aspergillus from the lung in health but may be an important aspect of colonization, where mucociliary clearance is compromised, as in the CF lung. Here we discuss the nature of the mucus barrier and its mucin components in CF, and how they may be implicated in A. fumigatus infection. Pediatr Pulmonol 2017;52:548–555. © 2016 The Authors. Pediatric Pulmonology. Published by Wiley Periodicals, Inc.
Keywords: Aspergillus fumigatus, cystic fibrosis, mucus, mucins
INTRODUCTION
Cystic fibrosis (CF) lung disease is characterized by chronic progressive airway infection, and clinically by the accumulation of airway secretions. At the level of the small airways, the combination of airway wall inflammation and thick luminal secretions results in airway blockage and remodeling. The role of bacterial pathogens such as Pseudomonas aeruginosa in this process has been the subject of extensive research. Only more recently, however has the impact of fungal organisms such as Aspergillus on CF airways been examined.1, 2 Aspergillus, in particular Aspergillus fumigatus, is one of the main fungal species found in CF airways. This filamentous fungus may act both as an allergen and an opportunistic pathogen in patients with CF, and can be isolated from airway secretions of up to ∼60% of CF patients.3, 4, 5 Chronic infection with A. fumigatus is an independent risk factor for hospital admissions, and sensitization to A. fumigatus has been linked to a greater decline in lung function and increased pulmonary exacerbations.6, 7, 8 However, due to a lack of sensitive culture methods it is likely that the prevalence of A. fumigatus in CF sputum has been underestimated.9
Part of the difficulty in defining the role of A. fumigatus in the progression of CF lung disease lies within the wide range of host responses that result in differing clinical states.9 Historically, A. fumigatus was regarded either as a colonizer of the airways or the cause of an exaggerated host allergic response, causing allergic bronchopulmonary aspergillosis (ABPA). A recent classification, based on a more detailed assessment of host (IgG, IgE) and airway infection (Aspergillus PCR and galactomannan antigen) markers, has provided new insights into the various responses of CF patients to A. fumigatus infection.9, 10 However, these data provide no insight into the long term clinical consequences of these different disease categories, nor about whether or how patients may switch between phenotypes, manifest more than one of these synchronously or resolve a disease state spontaneously. Current treatment for aspergillosis involves the use of antifungal agents such as azoles. However, these are all potentially toxic treatments, with a variable record of therapeutic success.11 Better understanding of the pathophysiology of host‐fungal interactions, particularly in the early colonization steps, may help in the development of new therapeutic strategies.
Airway colonisation by A. fumigatus occurs following inhalation of airborne conidia, asexual spores that are ubiquitous within the environment. These spores are frequently inhaled by humans and their small diameter (2 and 3µm) allows them to easily penetrate the lower airways of the respiratory tract.12 In healthy individuals, inhalation of A. fumigatus conidia is generally harmless, as they are efficiently removed from the airways by entrapment within airway mucus, phagocytosis by airway macrophages and removal by mucociliary clearance.13 Prior to swelling and then germination, they evoke no immune response.14 However, under circumstances where these innate defence mechanisms are compromised, as in CF, conidia persist within the airways. Here they may germinate and form hyphae that can invade the epithelial layer causing tissue damage.12, 13, 15 A. fumigatus secretes the complex carbohydrate galactosaminogalactan over its hyphal surface, which conceals β‐glucan from the immune system and mediates adherence to the galectin‐3 protein on the epithelial surface.16 Swollen conidia and hyphae also secrete allergens and gliotoxin, which have been shown to interfere with phagocytosis of inhaled conidia, preventing their elimination from the airways.17 Gliotoxin has been shown to slow ciliary beating in association with epithelial damage, perhaps through induction of apoptosis,18 with the consequence of reducing clearance of germinating conidia.
In order to colonize the airways effectively, conidia must ultimately adhere to the epithelium or basement membrane of the airway wall, interacting with extracellular matrix components such as laminin and fibrinogen.19 As the first point of contact for A. fumigatus following inhalation however, the mucus barrier is an important site of conidia‐host interactions. Details of these interactions between mucus and A. fumigatus, and how they change in the altered environment of the CF lung remain unclear, but may be key to understanding the susceptibility of CF patients to chronic A. fumigatus infection.
The Mucus Barrier in Cystic Fibrosis
Mucus is a multifaceted secretion composed of water, inorganic salts, proteins, glycoproteins, and nucleic acids, and is a key component of the innate defence system.20 In healthy airways, this dynamic barrier traps inhaled pathogens and other foreign particulates, allowing them to be cleared from the airways by mucociliary transport and/or cough. In CF, the mucus barrier becomes static and adherent, providing an ideal environment for airborne pathogens (viral, bacterial, and fungal) and allowing them to flourish within the airways.21
Exactly how mucus abnormalities contribute to CF lung disease remains to be fully elucidated, as there are many different factors that must be considered. The defective gene in CF encodes for a cyclic adenosine monophosphate regulated chloride channel (CFTR) expressed on the apical surface of lung and other epithelial tissues. CFTR dysfunction creates an ionic imbalance across the airway epithelial layer that affects the hydration, pH and electrolyte and mucus concentration of the airway surface liquid (ASL); all of these factors have been shown to alter mucus properties and negatively impact on mucociliary transport.21, 22, 23, 24 Dehydration of the ASL and the resultant increased mucus concentration affects mucociliary clearance by “osmotically compressing” the periciliary layer and collapsing the cilia, leading to mucus stasis and airway obstruction.21, 22 Data generated from a pig model of CF has shown that decreased pH of the ASL (due to defective bicarbonate secretion through CFTR) and increased calcium concentration negatively impacted on mucociliary transport; decreased pH also directly inhibited the antimicrobial properties of the ASL.24 Moreover, this model has also shown that impaired mucociliary transport can be caused by abnormal tethering of mucus strands to the submucosal glands, with their failure to detach in CF preventing clearance from the airway surface.23
The properties of mucus are largely dependent on polymeric, gel‐forming mucins, which are the major macromolecular constituents of the barrier.25 In healthy airway mucus, MUC5AC and MUC5B are the major gel‐forming mucins, and are secreted mainly by goblet cells and the submucosal glands, respectively (MUC5B is also produced by goblet cells, although in smaller amounts).26, 27 Once secreted, these large O‐linked glycoproteins are responsible for the structural architecture and biophysical properties of mucus. In solution, mucins interact both with one another and with other molecules such as calcium, which plays an important role in mucin cross‐linking.28, 29 Sequence variations in the central repetitive exon of the MUC5AC gene have been associated with severity of CF lung disease,30 perhaps resulting in MUC5AC polymers that have increased capacity to form cross‐links that will influence mucus function in terms of barrier porosity, viscosity, and transport by cilia.31
Mucin polymers are adorned with an array of sugar side chains comprising monosaccharides including N‐acetylgalactosamine (GalNAc), N‐acetylglucosamine (GlcNAc), fucose, sialic acid, and galactose, which can be further modified by sulfation. Charge repulsion between the anionic groups on sulfated galactose and sialic acid residues causes mucins to adopt an expanded conformation, which is central to mucus gel formation and properties.25, 32 These glycans also provide a variety of carbohydrate epitopes that are able to bind pathogens and sequester them within the mucus barrier, as well as providing a protective shield against proteases, helping mucins to resist proteolytic degradation.33
Given the importance of airway secretions in the pathophysiology of CF, it is perhaps not surprising that changes in mucin properties have been observed in CF sputum (for a detailed overview see ref34). In brief, mucins isolated from CF sputum are on average smaller than those from healthy individuals, most likely due to their degradation by the many proteases present in the CF lung. Mucin glycosylation changes have also been observed, with some reports suggesting that a less acidic form of MUC5B predominates in CF sputum,26, 35 while others have reported increased acidity for some mucins in CF.36, 37
Although mucins are the main constituent of healthy mucus, the extent to which they contribute to the biophysical properties of CF sputum has been subject to some debate. Some studies have identified DNA as the main macromolecular constituent of CF sputum, and suggested that it may play a more significant role in defining its biophysical properties.38, 39 Moreover, it has been reported that mucin levels in CF sputum are decreased compared with those of healthy individuals.40 However, these early reports were based on sputum from patients chronically infected with P. aeruginosa. Sputum from patients with no history of P. aeruginosa infection appears to contain mucin levels similar to those of healthy individuals.38 Thus, the apparent low level of mucins measured in infected patients (using antibody based assays) is most likely due to the action of proteases secreted by P. aeruginosa (as well as other airborne pathogens), which degrade mucins38 and reduce their detection by mucin‐specific antibodies. More recently, using size exclusion chromatography and differential refractometry, mucin concentrations have been shown to be higher in CF secretions as compared with healthy secretions.21 Mass spectrometry also revealed that CF mucins become degraded at antibody recognition sites. This highlights the inaccuracy of immunological techniques in measuring mucin concentration in CF secretions, and may explain the seemingly low levels of mucins measured in other studies.21 Indeed, quantitative mass spectrometry investigation of the secretome from CF bronchial epithelial cells has shown an increase in both MUC5AC and MUC5B expression and secretion, even in the absence of inflammation and infection, suggesting that the CF defect may be directly related to increased mucin production.41 In light of these findings, the biophysical properties of CF sputum may be more reliant on the state of mucins than previously reported. Although the precise impact of, and interaction between the different macromolecular components of sputum is not fully understood, it is becoming increasingly clear that mucins are an important constituent of CF airway secretions, both in early and advanced disease.
Interactions Between Aspergillus and Airway Mucins
The alterations in mucus properties and ciliary function in CF airways result in reduced mucus clearance, and this static barrier permits colonization of the airways by inhaled pathogens such as A. fumigatus.42 Initial adhesion of A. fumigatus within the airways involves the binding of specific sugars or lectins on the conidial surface19, 43, 44 (Fig. 1A). The precise mechanistic details of this process are largely unknown, though early studies suggested a role for negatively charged carbohydrates (particularly sialic acid) on the conidial surface.43, 44 More recent work has reported that galactosaminogalactan acts as a fungal adhesin in A. fumigatus, and this polysaccharide has also been shown to suppress host inflammatory responses.16 Tronchin et al. identified a sialic acid‐specific lectin in A. fumigatus conidia, which may also be involved in conidial adhesion to the airway mucosa via components such as laminin and fibrinogen.45 Mucins themselves are likely targets for conidial binding (Fig. 1A), as their glycan chains are often terminated in sialic acid residues. Indeed, sialic acid‐dependent interactions with mucins have been observed in other pathogens such as P. aeruginosa,46 and may represent a common mechanism of pathogen binding prior to colonization of the airways. Subsequent chronic infections are largely associated with the formation of surface‐attached biofilms, though a role for non‐attached aggregates in such infections has also been proposed.47 A. fumigatus may form fungal balls, otherwise know as aspergillomas during colonization of epithelial surfaces such as sinus mucosa or inside the lung cavity. Admixed with fungal hyphae are extensive biofilm materials including glycocalyx, galactomannan, melanin, and extracellular DNA, as well as mucus and cellular debris.48 It is likely but not well substantiated yet that A. fumigatus can form a mixed biofilm with bacteria in the lower airways.49 The role of this potential interaction in facilitating Aspergillus infection is not well established, but it is probable that it also takes place in CF airways.
Figure 1.
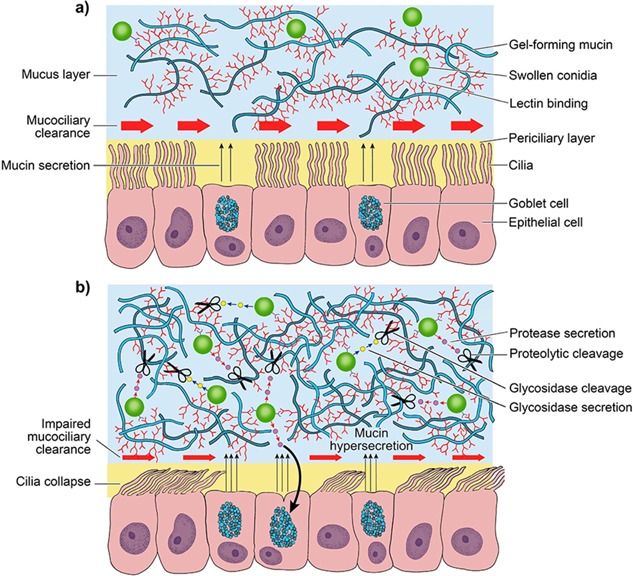
a) Inhaled conidia are efficiently removed from healthy airways by mechanisms such as mucociliary clearance. This is perhaps facilitated by binding of conidial lectins (e.g. FleA) to glycan moieties on gel‐forming mucins (MUC5AC and MUC5B), allowing them to become trapped within the mucus barrier and subsequently cleared from the airways.50 b) In cystic fibrosis mucociliary transport is impaired, preventing the elimination of inhaled pathogens from the airways. Here conidia may secrete proteases and glycosidases that degrade mucin protein and carbohydrate, in order to compromise the protective properties of the mucus barrier. A. fumigatus protease activity has also been shown to induce MUC5AC expression in airway epithelial cells, which may represent a mechanism of host defense against A. fumigatus.
Conidial surface lectins also play key roles in pathogenesis. For example, Aspergillus has been proposed to express a fucose‐specific lectin (FleA, or AFL) on the conidial surface.50, 51 FleA has recently been shown to bind fucosylated glycans on airway mucins, and also appears to be required for binding and phagocytosis of conidia by alveolar macrophages. In a mouse model of A. fumigatus pneumonia, treatment with ΔfleA conidia led to increased lung injury and infection, highlighting a role for FleA as a pathogen‐associated molecular pattern that is important for host defence against A. fumigatus infection.51 Thus, binding of inhaled pathogens to mucins may normally act as a protective mechanism, ensuring they are trapped within the mucus layer and unable to reach the underlying epithelium, before being removed by mucociliary clearance (Fig. 1A). In healthy airways, these interactions may be transient and insufficient to allow A. fumigatus to germinate. However in CF, persistence of A. fumigatus within the airways may lead to germination of conidia and the release of virulence factors (Fig. 1B), making the development of a host immune response more likely. An in vitro study of Aspergillus–epithelial interactions found that CF epithelial cells exhibit defective uptake and killing of A. fumigatus conidia, and excessive conidia‐induced apoptosis.52 Thus in the absence of effective mucociliary clearance, A. fumigatus may have a significant impact upon airway epithelial integrity.
Impact of Aspergillus on Airway Mucins
As well as forming interactions with mucus barrier components, A. fumigatus may also manipulate mucins in order to establish a niche within the airway mucosa. Aspergillus species produce a variety of proteases which are found either intracellularly, tethered to the cell wall or secreted.53 A number of these have been identified as putative virulence factors, and appear to play a role in the development of allergic diseases such as ABPA.17, 54 Proteolytic degradation of mucins has previously been observed in Candida albicans and P. aeruginosa,55, 56 and A. fumigatus has also been shown to degrade both mucin protein and carbohydrate.57 However, this has only been shown using commercially available mucins, which exhibit lower molecular weights and contain more impurities than intact mucins isolated under controlled conditions from “fresh” mucus secretions.58 The action of Aspergillus proteases on intact airway mucins has not yet been explored, but there are a number of reasons why A. fumigatus may benefit from degrading mucin proteins. One possibility is the utilization of mucins as a carbon source, which has been shown to occur in a number of bacteria.59 Degradation of mucins also alters the biophysical properties of mucus, perhaps to provide a more favorable niche, allowing A. fumigatus to penetrate and colonize the mucus barrier more easily. As well as utilizing proteases to degrade mucins, A. fumigatus also secretes glycosidases that degrade mucin glycans57 (Fig. 1B). Degrading mucins in this way could allow A. fumigatus to utilize mucin glycans as a nutrient source, and act as a mechanism for inhibiting entrapment within the airway mucus barrier in order to prevent elimination from the airways. In the rich microbiome of the CF airway the impact of individual proteases and glycosidases cannot be considered in isolation, and infection with A. fumigatus may depend on the action of bacterial pathogens within the lung to compromise airway defenses, or vice versa. Antibiotic treatment of bacterial infections has been shown to cause a significant decrease in the level of Aspergillus in CF sputum, despite having no direct antifungal action.60 However, P. aeruginosa, particularly CF isolates, has been shown to inhibit A. fumigatus biofilm formation.61 Thus, elimination of bacterial infections may in some cases worsen the fungal burden within the CF lung, highlighting the complex nature of the interaction between host, bacteria, and fungi in the lower airways in CF.
Another mechanism by which A. fumigatus proteases can alter mucus barrier properties is by altering mucin gene expression. Using DNA microarray analysis, Oguma and coworkers showed that the serine protease activity of A. fumigatus is able to induce MUC5AC mRNA and protein expression in airway epithelial cells, via the sequential activation of TNF‐α‐converting enzyme (TACE), TGF‐α, and epidermal growth factor receptor (EGFR).54 In line with this, chronic exposure to A. fumigatus has been shown to induce MUC5AC mRNA and protein expression in asthmatic rats.62 Upregulation of MUC5AC in response to A. fumigatus infection may thus be an important host defence mechanism. In support of this contention, upregulation of MUC5AC has been seen in other host–pathogen interactions. For instance, de novo expression of intestinal Muc5ac in mice occurs in response to acute infection with the murine parasitic nematode Trichuris muris, and this response is critical for the prevention of chronic infection.63 This may be in part due to alterations in the structural organization of mucus, as Muc5ac‐deficient mice possess a more porous mucus barrier.63 Similarly, upregulation of MUC5AC in the human lung during A. fumigatus infection may represent a mechanism of host defence.
To date, Aspergillus‐related research has focused mainly on MUC5AC, rather than the other major respiratory mucin MUC5B. Studies have shown that Muc5b is required for airway defence in mice, playing a crucial role in mucociliary clearance and antibacterial responses.64 Thus, it is likely that MUC5B plays a similar role in human airways, and is important for host defence against A. fumigatus. Future work must explore this relationship further, investigating the effects of A. fumigatus on MUC5B and vice versa.
SUMMARY
Airway mucus, and its constituent mucins, comprises the first line of airway defence, and the first component encountered by inhaled fungal spores. Although A. fumigatus and the airway mucus barrier appear to be able to interact and influence each other, many questions remain surrounding the role of this relationship in CF progression. Does A. fumigatus bind directly to mucins? If so, do these interactions promote airway colonisation or do mucins sequester A. fumigatus to hinder its pathological effects? Furthermore, does A. fumigatus degrade components of the mucus barrier as a source of nutrients, or in order to further compromise its protective properties? As A. fumigatus does not colonize CF lungs in isolation, it is also important to consider the interplay that exists between A. fumigatus and other major pathogens such as P. aeruginosa, and how this may also determine its effects on CF airways. Studying these processes in detail will provide us with a valuable understanding of aspergillosis in CF, and may ultimately aid the development of novel therapeutic agents to target such infections and improve patient health.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the help of Helen Carruthers in preparing the artwork for Figure 1. AC is funded by a grant from the North West Lung Centre Charity. DT is funded by a grant from the Cystic Fibrosis Foundation Therapeutics, Inc. (THORNTO7XX0). DD has current grant support from the National Institute of Health Research, Medical Research Council, Global Action Fund for Fungal Infections and the Fungal Infection Trust. AH is supported by a Clinician Scientist award from the UK National Institute for Health Research (NIHRCS012‐13). This report presents independent research funded by the NIHR. The views expressed are those of the authors and not necessarily those of the UK National Health Service, the NIHR or the UK Department of Health.
Conflict of interest: DD holds Founder shares in F2G Ltd, a University of Manchester spin‐out antifungal discovery company, and in Novocyt which markets the Myconostica real‐time molecular assays. He acts or has recently acted as a consultant to Astellas, Sigma Tau, Basilea, Scynexis, Cidara, and Pulmocide. In the last 3 years, he has been paid for talks on behalf of Astellas, Dynamiker, Gilead, Merck, and Pfizer. He is also a member of the Infectious Disease Society of America Aspergillosis Guidelines and European Society for Clinical Microbiology and Infectious Diseases Aspergillosis Guidelines groups. AH has received personal or institutional support for consultancy work with Celtaxys pharmaceuticals, Vertex pharmaceuticals, Chiesi Ltd, and Boehringer Ingelheim.
REFERENCES
- 1. Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev 2002; 15:194–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Middleton PG, Chen SC, Meyer W. Fungal infections and treatment in cystic fibrosis. Curr Opin Pulm Med 2013; 19:670–675. [DOI] [PubMed] [Google Scholar]
- 3. Bakare N, Rickerts V, Bargon J, Just‐Nubling G. Prevalence of Aspergillus fumigatus and other fungal species in the sputum of adult patients with cystic fibrosis. Mycoses 2003; 46:19–23. [DOI] [PubMed] [Google Scholar]
- 4. Mortensen KL, Jensen RH, Johansen HK, Skov M, Pressler T, Howard SJ, Leatherbarrow H, Mellado E, Arendrup MC. Aspergillus species and other molds in respiratory samples from patients with cystic fibrosis: a laboratory‐based study with focus on Aspergillus fumigatus azole resistance. J Clin Microbiol 2011; 49:2243–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson LA, Callerame ML, Schwartz RH. Aspergillosis and atopy in cystic fibrosis. Am Rev Respir Dis 1979; 120:863–873. [DOI] [PubMed] [Google Scholar]
- 6. Amin R, Dupuis A, Aaron SD, Ratjen F. The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest 2010; 137:171–176. [DOI] [PubMed] [Google Scholar]
- 7. Baxter CG, Moore CB, Jones AM, Webb K, Denning DW. IgE‐mediated immune responses and airway detection of Aspergillus and Candida in adult cystic fibrosis. Chest 2013; 143:1351–1357. [DOI] [PubMed] [Google Scholar]
- 8. Kraemer R, Delosea N, Ballinari P, Gallati S, Crameri R. Effect of allergic bronchopulmonary aspergillosis on lung function in children with cystic fibrosis. Am J Respir Crit Care Med 2006; 174:1211–1220. [DOI] [PubMed] [Google Scholar]
- 9. Baxter CG, Dunn G, Jones AM, Webb K, Gore R, Richardson MD, Denning DW. Novel immunologic classification of aspergillosis in adult cystic fibrosis. J Allergy Clin Immunol 2013; 132:560–566.e510. [DOI] [PubMed] [Google Scholar]
- 10. Jones AM, Horsley A, Denning DW. What is the importance of classifying Aspergillus disease in cystic fibrosis patients? Expert Rev Respir Med 2014; 8:389–392. [DOI] [PubMed] [Google Scholar]
- 11. Aaron SD, Vandemheen KL, Freitag A, Pedder L, Cameron W, Lavoie A, Paterson N, Wilcox P, Rabin H, Tullis E, et al. Treatment of Aspergillus fumigatus in patients with cystic fibrosis: a randomized, placebo‐controlled pilot study. PLoS ONE 2012; 7:e36077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Latgé JP. Aspergillus fumigatus and aspergillosis. Clin Microbiol Rev 1999; 12:310–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dagenais TRT, Keller NP. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev 2009; 22:447–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, Pamer EG. Aspergillus fumigatus triggers inflammatory responses by stage‐specific beta‐glucan display. PLoS Pathog 2005; 1:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park SJ, Burdick MD, Mehrad B. Neutrophils mediate maturation and efflux of lung dendritic cells in response to Aspergillus fumigatus germ tubes. Infect Immun 2012; 80:1759–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gravelat FN, Beauvais A, Liu H, Lee MJ, Snarr BD, Chen D, Xu W, Kravtsov I, Hoareau CM, Vanier G, et al. Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta‐glucan from the immune system. PLoS Pathog 2013; 9:1003575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tomee JF, Kauffman HF. Putative virulence factors of Aspergillus fumigatus . Clin Exp Allergy 2000; 30:476–484. [DOI] [PubMed] [Google Scholar]
- 18. Amitani R, Taylor G, Elezis EN, Llewellyn‐Jones C, Mitchell J, Kuze F, Cole PJ, Wilson R. Purification and characterization of factors produced by Aspergillus fumigatus which affect human ciliated respiratory epithelium. Infect Immun 1995; 63:3266–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bouchara JP, Sanchez M, Chevailler A, Marot‐Leblond A, Lissitzky JC, Tronchin G, Chabasse D. Sialic acid‐dependent recognition of laminin and fibrinogen by Aspergillus fumigatus conidia. Infect Immun 1997; 65:2717–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasnain SZ, Gallagher AL, Grencis RK, Thornton DJ. A new role for mucins in immunity: insights from gastrointestinal nematode infection. Int J Biochem Cell Biol 2013; 45:364–374. [DOI] [PubMed] [Google Scholar]
- 21. Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, DeMaria GC, Matsui H, Donaldson SH, Davis CW, et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest 2014; 124:3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, Boucher RC, Rubinstein M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 2012; 337:937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Zabner J, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014; 345:818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, Choudhury B, Varki A, Stoltz DA, Welsh MJ. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest 2016; 126:879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol 2008; 70:459–486. [DOI] [PubMed] [Google Scholar]
- 26. Kirkham S, Sheehan JK, Knight D, Richardson PS, Thornton DJ. Heterogeneity of airways mucus: variations in the amounts and glycoforms of the major oligomeric mucins muc5ac and muc5b. Biochem J 2002; 361:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Groneberg DA, Eynott PR, Oates T, Lim S, Wu R, Carlstedt I, Nicholson AG, Chung KF. Expression of MUC5AC and MUC5B mucins in normal and cystic fibrosis lung. Respir Med 2002; 96:81–86. [DOI] [PubMed] [Google Scholar]
- 28. Raynal BD, Hardingham TE, Sheehan JK, Thornton DJ. Calcium‐dependent protein interactions in muc5b provide reversible cross‐links in salivary mucus. J Biol Chem 2003; 278:28703–28710. [DOI] [PubMed] [Google Scholar]
- 29. Ridley C, Kouvatsos N, Raynal BD, Howard M, Collins RF, Desseyn JL, Jowitt TA, Baldock C, Davis CW, Hardingham TE, et al. Assembly of the respiratory mucin muc5b: a new model for a gel‐forming mucin. J Biol Chem 2014; 289:16409–16420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo X, Pace RG, Stonebraker JR, Commander CW, Dang AT, Drumm ML, Harris A, Zou F, Swallow DM, Wright FA, et al. Mucin variable number tandem repeat polymorphisms and severity of cystic fibrosis lung disease: significant association with MUC5AC. PLoS ONE 2011; 6:e25452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo X, Zheng S, Dang H, Pace RG, Stonebraker JR, Jones CD, Boellmann F, Yuan G, Haridass P, Fedrigo O, et al. Genome reference and sequence variation in the large repetitive central exon of human MUC5AC. Am J Respir Cell Mol Biol 2014; 50:223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stanley P, Schachter H, Taniguchi N. N‐glycans In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of glycobiology. 2nd edition New York: Cold Spring Harbor Laboratory Press; 2008. pp 101–114. [Google Scholar]
- 33. Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunol 2008; 1:183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ehre C, Ridley C, Thornton DJ. Cystic fibrosis: an inherited disease affecting mucin‐producing organs. Int J Biochem Cell Biol 2014; 52:136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davies JR, Svitacheva N, Lannefors L, Kornfalt R, Carlstedt I. Identification of muc5b, muc5ac and small amounts of muc2 mucins in cystic fibrosis airway secretions. Biochem J 1999; 2:321–330. [PMC free article] [PubMed] [Google Scholar]
- 36. Lamblin G, Lafitte JJ, Lhermitte M, Degand P, Roussel P. Mucins from cystic fibrosis sputum. Mod Probl Paediatr 1976; 19:153–164. [PubMed] [Google Scholar]
- 37. Roussel P, Degand P, Lamblin G, Laine A, Lafitte JJ. Biochemical definition of human tracheobronchial mucus. Lung 1978; 154:241–260. [DOI] [PubMed] [Google Scholar]
- 38. Henke MO, John G, Rheineck C, Chillappagari S, Naehrlich L, Rubin BK. Serine proteases degrade airway mucins in cystic fibrosis. Infect Immun 2011; 79:3438–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rubin BK. Mucus structure and properties in cystic fibrosis. Paediatr Respir Rev 2007; 8:4–7. [DOI] [PubMed] [Google Scholar]
- 40. Henke MO, Renner A, Huber RM, Seeds MC, Rubin BK. Muc5ac and muc5b mucins are decreased in cystic fibrosis airway secretions. Am J Respir Cell Mol Biol 2004; 31:86–91. [DOI] [PubMed] [Google Scholar]
- 41. Peters‐Hall JR, Brown KJ, Pillai DK, Tomney A, Garvin LM, Wu X, Rose MC. Quantitative proteomics reveals an altered cystic fibrosis in vitro bronchial epithelial secretome. Am J Respir Cell Mol Biol 2015; 53:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998; 95:1005–1015. [DOI] [PubMed] [Google Scholar]
- 43. Wasylnka JA, Moore MM. Adhesion of aspergillus species to extracellular matrix proteins: evidence for involvement of negatively charged carbohydrates on the conidial surface. Infect Immun 2000; 68:3377–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wasylnka JA, Simmer MI, Moore MM. Differences in sialic acid density in pathogenic and non‐pathogenic aspergillus species. Microbiology 2001; 147:869–877. [DOI] [PubMed] [Google Scholar]
- 45. Tronchin G, Esnault K, Sanchez M, Larcher G, Marot‐Leblond A, Bouchara JP. Purification and partial characterization of a 32‐kilodalton sialic acid‐specific lectin from Aspergillus fumigatus . Infect Immun 2002; 70:6891–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramphal R, Pyle M. Evidence for mucins and sialic acid as receptors for pseudomonas aeruginosa in the lower respiratory tract. Infect Immun 1983; 41:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alhede M, Kragh KN, Qvortrup K, Allesen‐Holm M, van Gennip M, Christensen LD, Jensen PØ, Nielsen AK, Parsek M, Wozniak D, et al. Phenotypes of non‐attached pseudomonas aeruginosa aggregates resemble surface attached biofilm. PLoS ONE 2011; 6:e27943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beauvais A, Latgé JP. Aspergillus biofilm in vitro and in vivo. Microbiol Spectr 2015; 3. doi: 10.1128/microbiolspec.MB‐0017‐2015 [DOI] [PubMed] [Google Scholar]
- 49. Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei‐Cas E, et al. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community–implications for therapeutic management. PLoS ONE 2012; 7:e36313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Houser J, Komarek J, Kostlanova N, Cioci G, Varrot A, Kerr SC, Lahmann M, Balloy V, Fahy JV, Chignard M, et al. A soluble fucose‐specific lectin from Aspergillus fumigatus conidia‐structure, specificity and possible role in fungal pathogenicity. PloS ONE 2013; 8:83077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kerr SC, Fischer GJ, Sinha M, McCabe O, Palmer JM, Choera T, F Yun Lim, Wimmerova, Carrington M,, Yuan S, et al. Flea expression in Aspergillus fumigatus is recognized by fucosylated structures on mucins and macrophages to prevent lung infection. PLoS Pathog 2016; 12:e1005555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chaudhary N, Datta K, Askin FB, Staab JF, Marr KA. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to aspergillus and resultant pulmonary inflammation. Am J Respir Crit Care Med 2012; 185:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ward OP, Rao MB, Kulkarni A. 2009. Proteases, production Encyclopedia of microbiology, 3rd edition Academic Press; pp 495–511. [Google Scholar]
- 54. Oguma T, Asano K, Tomomatsu K, Kodama M, Fukunaga K, Shiomi T, Ohmori N, Ueda S, Takihara T, Shiraishi Y, et al. Induction of mucin and muc5ac expression by the protease activity of Aspergillus fumigatus in airway epithelial cells. J Immunol 2011; 187:999–1005. [DOI] [PubMed] [Google Scholar]
- 55. Aristoteli LP, Willcox MD. Mucin degradation mechanisms by distinct pseudomonas aeruginosa isolates in vitro. Infect Immun 2003; 71:5565–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Colina AR, Aumont F, Deslauriers N, Belhumeur P, de Repentigny L. Evidence for degradation of gastrointestinal mucin by candida albicans secretory aspartyl proteinase. Infect Immun 1996; 64:4514–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. St Leger RJ, Screen SE. In vitro utilization of mucin, lung polymers, plant cell walls and insect cuticle by Aspergillus fumigatus, metarhizium anisopliae and haematonectria haematococca. Mycol Res 2000; 104:463–471. [Google Scholar]
- 58. Jumel K, Fiebrig I, Harding SE. Rapid size distribution and purity analysis of gastric mucus glycoproteins by size exclusion chromatography/multi angle laser light scattering. Int J Biol Macromol 1996; 18:133–139. [DOI] [PubMed] [Google Scholar]
- 59. Salyers AA, Vercellotti JR, West SE, Wilkins TD. Fermentation of mucin and plant polysaccharides by strains of bacteroides from the human colon. Appl Environ Microbiol 1977; 33:319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baxter CG, Rautemaa R, Jones AM, Webb AK, Bull M, Mahenthiralingam E, Denning DW. Intravenous antibiotics reduce the presence of aspergillus in adult cystic fibrosis sputum. Thorax 2013; 68:652–657. [DOI] [PubMed] [Google Scholar]
- 61. Ferreira JA, Penner JC, Moss RB, Haagensen JA, Clemons KV, Spormann AM, Nazik H, Cohen K, Banaei N, Carolino E, et al. Inhibition of Aspergillus fumigatus and its biofilm by pseudomonas aeruginosa is dependent on the source, phenotype and growth conditions of the bacterium. PLoS ONE 2015; 10:e0134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao FS, Gao YY, Liu MJ, Liu YQ. Chronic Aspergillus fumigatus exposure upregulates the expression of mucin 5ac in the airways of asthmatic rats. Exp Lung Res 2012; 38:256–265. [DOI] [PubMed] [Google Scholar]
- 63. Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L, Dickey BF, Wilson MS, Wynn TA, Grencis RK, et al. Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med 2011; 208:893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roy MG, Livraghi‐Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, Alexander SN, Bellinghausen LK, Song AS, Petrova YM, et al. Muc5b is required for airway defence. Nature 2014; 505:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]