

Abstract
Epigenetic silencing of tumor suppressor genes is a phenomenon frequently observed in multiple cancers. Ras-association domain family 1 isoform A (RASSF1A) is a well-characterized tumor suppressor that belongs to the Ras-association domain family. Several studies have demonstrated that hypermethylation of the RASSF1A promoter is frequently observed in lung, prostate, and breast cancers. Phenethyl isothiocyanate (PEITC), a phytochemical abundant in cruciferous vegetables, possesses chemopreventive activities; however, its potential involvement in epigenetic mechanisms remains elusive. The present study aimed to examine the role of PEITC in the epigenetic reactivation of RASSF1A and the induction of apoptosis in LNCaP cells. LNCaP cells were treated for 5 days with 0.01% DMSO, 2.5 or 5 μM PETIC or 2.5 μM azadeoxycytidine (5-Aza) with 0.5 μM trichostatin A (TSA). We evaluated the effects of these treatments on CpG demethylation using methylation-specific polymerase chain reaction (MSP) and bisulfite genomic sequencing (BGS). CpG demethylation was significantly enhanced in cells treated with 5 μM PEITC and 5-Aza+TSA; therefore, the latter treatment was used as a positive control in subsequent experiments. The decrease in RASSF1A promoter methylation correlated with an increase in expression of the RASSF1A gene in a dose-dependent manner. To confirm that promoter demethylation was mediated by DNA methyltransferases (DNMTs), we analyzed the expression levels of DNMTs and histone deacetylases (HDACs) at the gene and protein levels. PEITC reduced DNMT1, 3A and 3B protein levels in a dose-dependent manner, and 5 μM PEITC significantly reduced DNMT3A and 3B protein levels. HDAC1, 2, 4 and 6 protein expression was also inhibited by 5 μM PEITC. The combination of 5-Aza and TSA, a DNMT inhibitor and a HDAC inhibitor, respectively, was used as a positive control as this treatment significantly inhibited both HDACs and DNMTs. The function of RASSF1A reactivation in promoting apoptosis and inducing G2/M cell cycle arrest was analyzed using flow-cytometry analysis with Annexin V and propidium iodide (PI). Growth inhibition effect on LNCaP cells were investigated by colony formation assay. In addition, we analyzed p21, caspase-3 and 7, Bax, and Cyclin B1 protein levels. Flow-cytometry analysis of cells stained with PI alone demonstrated that 5 μM PEITC promotes early apoptosis and G2/M cell cycle arrest. Flow cytometry analysis of cells stained with Annexin V and PI also demonstrated an increased proportion of cells in early apoptosis in cells treated with 5 μM PEITC or 5-Aza with TSA. PEITC and efficiently inhibit colony numbers and total area. In addition, 5 μM PEITC significantly enhanced p21, caspase-3, 7 and Bax levels and reduced Cyclin B1 expression compared with the control group. Collectively, the results of our study suggest that PEITC induces apoptosis in LNCaP cells potentially by reactivating RASSF1A via epigenetic mechanisms.
Keywords: RASSF1A, Epigenetics, Demethylation, Histone deacetylation, Apoptosis, Phenenthyl Isothiocyanate, Tumor suppressor, Prostate cancer
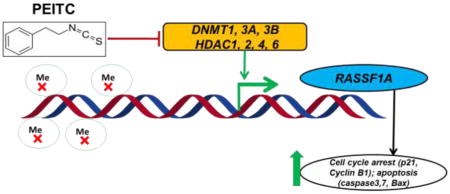
Graphical abstract
Schematic illustrating the role of PEITC in epigenetically regulating the transcription of RASSF1A and its role in cell cycle and apoptosis.
1. Introduction
Prostate cancer (PCa) is one of the most frequent causes of cancer-related deaths in men in the United States. The most recent data from 2015 indicate that greater than 220,800 new cases of PCa are diagnosed and an estimated 27,540 cases of prostate cancer-related deaths occur in men in the US each year[1, 2]. Lifestyle factors, such as diet and nutrition, play a significant role in the pathogenesis of PCa, and multiple studies have revealed that intensive nutrition and lifestyle changes might modulate gene expression in the prostate. These findings highlight the potential role of dietary phytochemicals in the treatment of PCa[3–5]. Studies evaluating epigenetic modifications in cancer, specifically those that lead to the inactivation or silencing of key regulatory genes, such as tumor suppressor genes, has led to the realization that genetic and epigenetic changes play a critical role in tumorigenesis[6]. Recent advances in the field of epigenetics, which are partly due to next generation sequencing techniques, have redirected the focus of cancer researchers. These advances have led to the realization that the development of this deadly disease involves interplay between genetic alterations and epigenetic aberrations. DNA methylation, histone modifications, nucleosome positioning and non-coding RNAs, such as microRNAs, are specific epigenetic aberrations that induce the expression of oncogenes or silence the expression of tumor suppressor genes[7].
In recent years, epigenetic silencing of tumor suppressor genes has gained much attention in cancer research, and the significance of this phenomenon in driving tumorigenesis is now well established. Recent developments in this field have led to the hypothesis that reactivating tumor suppressor genes that have been silenced by promoter methylation might be an effective targeted therapy for treating cancer [8]. Several studies have demonstrated that the epigenetic silencing of several genes, including RARβ2, RARβ4, RASSF1A, CDH13, APC, CDH1, FHIT, CDKN2A and Nrf2, is due to increased promoter methylation exclusively in prostate tumor tissues[9–13]. Various changes in DNA methylation patterns can be detected before PCa becomes invasive, suggesting that epigenetic changes are pivotal events in tumor progression and that epigenomic changes play a critical role in PCa[14].
The RASSF1A gene, a well-characterized tumor suppressor, belongs to the Ras-association domain family of genes and is located on chromosome at 3p21.3. The RASSF1 gene encodes several isoforms, including RASSF1A, RASSF1B, and RASSF1C, all of which are generated from alternative mRNA splicing and promoter usage[15]. RASSF1A transcriptional silencing is frequently observed feature in multiple types of cancer as a result of promoter hypermethylation; however, few reports are available describing the mechanisms underlying this phenomenon[16–19]. RASSF1A transcription is suppressed by hypermethylation in breast, ovarian, gastric, nasopharyngeal, and bladder cancers as well as neuroblastoma and renal cell lines[20–25]. Hypermethylation of the RASSF1A gene at CpG islands has been observed in 70% of prostate cancer cases, and the restoration of RASSF1A expression inhibited tumorigenesis in prostate and renal tumor cell lines [26, 27]. The tumor suppressor function of RASSF1A has been confirmed by studies demonstrating that the exogenous expression RASSF1A decreased colony formation, suppressed anchorage-independent growth and reduced tumor formation in lung, kidney and breast cancers in vivo. In light of the tumor suppression functions of RASSF1A and the observation that restoring RASSF1A expression in tumor cell lines decreases tumorigenicity, restoring RASSF1A expression and elucidating the mechanism by which hypomethylation occurs have significant implications in the context of preventing tumor growth[22, 26, 28]. Recent meta-analyses have revealed that RASSF1A methylation in tumor tissues was directly correlated with the Gleason Score (GS), serum prostate-specific antigen (PSA) level and tumor stage in PCa, indicating that RASSF1A promoter methylation might be a potential biomarker for prostate cancer prognosis and therapy[29, 30]. Several studies have described the effect of phytochemicals, such as curcumin and mahanine, in reversing RASSF1A promoter hypermethylation and restoring RASSF1A expression in breast cancer and prostate cancer cells[31, 32].
Phenethyl isothiocyanate (PEITC), an abundant component of cruciferous vegetables, such as broccoli and watercress, exerts chemoprevention activity by modulating epigenetic modifications[33, 34]. PEITC is a novel epigenetic regulator of both histone deacetylases (HDACs) and DNA methyltransferases (DNMTs), factors that govern the expression and suppression of several genes, including GSTP1 and p21 [35–37]. Recent studies demonstrated that PEITC is an effective HDAC inhibitor and reduces HDAC enzyme activity. These events induce growth arrest and apoptosis in cancer cells both in vitro and in vivo [35, 38]. A recent study revealed that PEITC regulated H3-acetylation (H3-Ac) and site-specific H3 lysine methylations (H3K27me3 and H3K9me2), which are epigenetic modifications associated with attenuating cell proliferation in a concentration- and time-dependent manner in human colon cancer cells[39].
We hypothesized that PEITC induces RASSF1A promoter demethylation in LNCaP cells and that epigenetic reactivation of RASSF1A induces apoptosis in LNCaP cells. To test this hypothesis, we treated LNCaP cells with 0.01% DMSO, 2.5 and 5 μM PEITC or 2.5 μM azadeoxycytidine (5-Aza) with 0.5 μM trichostatin A (TSA). The demethylating effect of the test compounds was assessed using the methylation-specific polymerase chain reaction (MSP) and bisulfite genomic sequencing (BGS). We also evaluated the reactivation of RASSF1A expression and the role of RASSF1A activation in promoting apoptosis. The results of our BGS and protein expression analyses indicate that PEITC reactivates RASSF1A expression by promoting demethylation and inhibiting HDAC activation.
2. Materials and methods
2.1. Cell culture, reagents and cell viability assay
The human prostate cancer cell line LNCaP was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). LNCaP cells were maintained in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY). The cells were grown at 37 °C in a humidified atmosphere with 5% CO2. PEITC, 5-Aza, TSA, bacteriological agar, puromycin, and ethidium bromide were purchased from Sigma–Aldrich (St. Louis, MO, USA). The Annexin V and propidium iodide (PI) apoptosis kit was purchased from Life Technologies. Then, 3.5 × 104 LNCaP cells/ml were seeded in 10-cm tissue culture plates for 24 h and treated with DMSO 0.01% (negative control), PEITC (2.5 μM or 5 μM) or 5-Aza with TSA (2.5 μM and 0.5 μM, respectively) (positive control) for 5 days. The drugs and media were replenished every 2 days. TSA (0.5 μM) was added to the 5-Aza treatment group 20 h prior to harvesting the cells.
LNCaP cells were seeded in 96-well plates at a density of 1,000 cells per well. After 24 h, the cells were then treated with PEITC (1–10μM) for 1 and 5 days. The medium was changed every other day for the 5 day group. MTS assay was performed using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay Kit as described previously (results shown in supporting information).
2.2. DNA methylation analysis
LNCaP cells were plated in 10-cm plates for 24 h and subsequently treated with 0.1% DMSO (control), 2.5 μM 5-Aza and 500 nM TSA, or 2.5 or 5 μM PEITC for 5 days. The medium was changed every other day. For cells treated with 5-Aza and TSA, 500 nM TSA was added 20 h before the cells were harvested. On day 5, the cells were harvested for further analyses. Genomic DNA was isolated from the treated cells using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). Next, 600 ng of genomic DNA was used as a template for bisulfite conversion using the EZ DNA Methylation Gold Kit (Zymo Research Corp., Orange, CA, USA) following the manufacturer's instructions. To obtain products for sequencing, the converted DNA was amplified using semi-nested PCR as previously described by Dammann et al. [18]. Briefly, the DNA sequences were amplified by mixing bisulfite-converted DNA (500 ng) with primer MU379 (50 pmoles) and primer ML730 (50 pmoles) in reaction buffer (20 μl) containing dNTPs (200 μM each dNTP) using Platinum PCR Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA). The reaction conditions consisted of 30 cycles of 95 °C for 1 min, 55 °C for 1 min and 74 °C for 2 min. A semi-nested PCR using the amplified products at a 1:50 dilution, the internal primer ML561 and primer MU379 was performed using similar PCR conditions as described above. The PCR products containing bisulfite-resistant cytosines were ligated into the pCR2.1 vector (Invitrogen), and several clones were sequenced to confirm the presence of the amplified DNA. A minimum of 10 clones per biological replicate from each treatment group were randomly selected and sequenced (Genewiz, Piscataway, NJ, USA). The percentage of methylated CpG sites was calculated by dividing the number of methylated CpG sites by the total number of CpG sites in the DNA sequence. The primer sequences used for BGS are listed in Table 1.
Table 1.
Sequences of the primers used for quantitative PCR, methylation-specific PCR and bisulfite genomic sequencing.
| qPCR primers | ||
|---|---|---|
| Forward | Reverse | |
| DNMT1 | 5’-CCGAGTTGGTGATGGTGTGTAC-3’ | 5’-AGGTTGATGTCTGCGTGGTAGC-3’ |
| DNMT3A | 5’-TATTGATGAGCGCACAAGAGAGC-3’ | 5’-GGGTGTTCCAGGGTAACATTGAG-3’ |
| DNMT3B | 5’-GACTTGGTGATTGGCGGAA-3’ | 5’-GGCCCTGTGAGCAGCAGA-3’ |
| RASSF1A | 5’-CCTCTGTGGCGACTTCATCTG-3’ | 5’-CAACAGTCCAGGCAGACGAG-3’ |
| MSP primers | ||
| RASSF1A (M) | 5’-GTGTTAACGCGTTGCGTATC-3’ | 5’-AACCCCGCGAACTAAAAACGA-3’ |
| RASSF1A (UM) | 5’-TTTGGTTGGAGTGTGTTAATGTG-3’ | 5’-CAAACCCCACAAACTAAAAACAA-3’ |
| BGS primers | ||
| RASSF1A MU730 | 5’-ACCCTCTTCCTCTAACACAATAAAACTAACC-3’ | |
| RASSF1A MU561 | 5’-CCCCACAATCCCTACACCCAAAT-3’ | |
| RASSF1A MU379 | 5’-GTTTTGGTAGTTTAATGAGTTTAGGTTTTTT-3’ | |
Methylation-specific PCR (MSP) was performed using the bisulfite-converted genomic DNA as template. The primer sequences used for the PCR amplification of the methylated (M) and unmethylated regions of the RASSF1A gene are listed in Table 1. The amplification products were separated on a 2% agarose gel by electrophoresis and visualized using ethidium bromide staining and a Gel Documentation 2000 system (Bio-Rad, Hercules, CA, USA). The densities of the bands were quantified using ImageJ software (Version 1.48d; NIH, Bethesda, Maryland, USA).
2.3. RNA isolation and qPCR
Total RNA from the treatment and control groups was extracted using the RNeasy Mini Kit (Carlsbad, CA), and the mRNA was quanti ed using a NanoDrop 2000. Approximately 600 ng of mRNA was reverse-transcribed into cDNA using Taqman RT reagents. The relative expression levels of RASSF1A, DNMT1, DNMT3A and DNMT3B mRNA were determined using qPCR with the cDNA template and the Power SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) in an ABI7900HT system (Applied Biosystems). The sequences of the forward and reverse primers used to analyze these genes are listed in Table 1.
2.4. Protein lysate preparation and Western blot assays
Protein lysates were prepared using radio immunoprecipitation assay (RIPA) buffer (Sigma–Aldrich, St. Louis, MO, USA) supplemented with a protein inhibitor cocktail (Sigma–Aldrich). Total protein was quantified using the bicinchoninic acid (BCA) method (Pierce, Rockford, IL, USA). Briefly, 20 μg of total protein were separated using 4 to 15% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA, USA) and electro-transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). After blocking with 5% BSA (bovine serum albumin; Fisher Scientific, Pittsburgh, PA, USA) in Tris-buffered saline with 0.1% Tween 20 (TBST) (Boston Bioproducts, Ashland, MA, USA), the membranes were sequentially incubated with specific primary antibodies and subsequently incubated with horseradish peroxidase-conjugated secondary antibodies. The blots were visualized using the Supersignal West Femto Chemiluminescent Substrate (Pierce, Rockford, IL, USA) and visualized using a Gel Documentation 2000 system (Bio-Rad, Hercules, CA, USA). Densitometry of the bands was analyzed using ImageJ (Version 1.48d; NIH). The primary antibodies were obtained from the following sources: anti-β-ACTIN from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-DNMT 3A and 3B from IMGENEX (San Diego, CA, USA); and anti-HDAC1, 2, 4, 6, anti-Cyclin B1 and anti-Bax from Cell Signaling Technology (Boston, MA, USA). The secondary antibodies were purchased from Santa Cruz Biotechnology.
2.5. Flow cytometry analysis of cell cycle distribution and apoptosis
PEITC- or 5-Aza-treated LNCaP cells were stained with PI (Sigma–Aldrich Co., St. Louis, MO) or PI and Annexin V-FITC (Life Technologies) and analyzed by flow cytometry using a Beckman Coulter Gallios Flow Cytometer (Brea, CA) in The Flow Cytometry/Cell Sorting & Confocal Microscopy Core Facility at Rutgers University. Briefly, LNCaP cells (3.5 × 104 cells/ml) were seeded in 10-cm plates, allowed to attach overnight and treated (as previously described) with the indicated drugs. Then, the cells were trypsinized, and the pellet was collected by centrifugation at 700 ×g for 5 min. The cells were fixed with 3 ml of ice-cold 70% ethanol for 48 h at 4 °C prior to the cell cycle analysis. The cells were rinsed twice with phosphate-buffered saline (PBS), treated with RNase A (1 mg/ml) for 30 min at 37 °C, stained with 5 μl of 1 mg/ml PI in the dark at room temperature for 30 min and analyzed according to the manufacturer's instructions. Following the incubation, the samples, including floating and adherent cells, were collected and prepared for the analysis of apoptosis using the Annexin V-FITC/PI apoptosis detection kit according to the manufacturer’s instructions.
2.6 LNCaP Colony Formation Assay
LNCaP cells were seeded at 800cells /well density in 6 well dishes. Cells were treated with PEITC (2.5μM, 5μM, 10μM) or 0.1% DMSO (control) every 3 days after seeding. Two weeks later, medium were removed from each of the dishes by aspiration. Cells in each well were washed with 2 mL PBS and fixed by 0.5 mL methanol for 30 minutes. Methanol was removed and the cells were washed by PBS. The cells were stained with 5 mL 0.01% (w/v) crystal violet in dH2O for 30 minutes. Excess crystal violet was washed out with dH2O and kept dry. Digital images of the colonies were obtained using a Nikon Eclipse E600 microscope (Micron-optics, NJ). The number and total area of colonies containing more than 50 individual cells were counted by ImageJ (Version 1.48d; NIH, Bethesda, Maryland, USA).
2.7. Statistical analysis
The data are presented as the mean ± standard error of the mean (SEM). Statistical analyses were performed using Student’s t-test and one way ANOVA using Info Stat statistical software program. P-values less than 0.05 were considered statistically significant and are indicated with *. P-values less than 0.01 are indicated with #.
3. Results
3.1 PEITC induced RASSF1A expression in LNCaP cells by inducing promoter demethylation
Hypermethylation of the 16 CpG sites in the RASSF1A promoter region has previously been observed in multiple prostate cancer cell lines, including LNCaP cells[40].The reversal of RASSF1A promoter hypermethylation in LNCaP cells treated with PEITC was analyzed using MSP and BGS via the semi-nested PCR method. The extent of methylation in MSP was quantified by analyzing the density of the gel bands. The results of the MSP analysis revealed high levels methylated DNA in untreated LNCaP cells, suggesting that the RASSF1A promoter was hypermethylated in untreated cells. However, in the cells treated with PEITC (2.5 or 5 μM) or 5-Aza (2.5 μM) and TSA (0.5 μM), an increase in unmethylated DNA regions was noted compared with the control (Figure 1A). To further analyze the extent of promoter methylation, we used BGS to analyze the 16 CpG sites (Figure 1B) in fragment containing the 3Sp1 consensus binding sites and the putative transcription and translation initiation sites present in Exon 1α (−197 and +438). The Genbank accession number of the cDNA sequence is AF132675 [18, 41]. In the control group, an average 98.1% of the 16 CpG sites were methylated. However, in cells treated with 2.5 μM or 5 μM PEITC, the percentage of methylated CpG sites decreased to 93.3% and 90.7%, respectively, indicating that CpG demethylation was induced in cells treated with PEITC compared with the control group. The percent of methylated CpG sites in the positive control group treated with 5-Aza (2.5 μM) and TSA (0.5 μM) was 83.6% (Figure 1C). The data obtained from MSP and BGS demonstrated that 5 μM PEITC significantly reduced CpG methylation in the RASSF1A gene. Previous studies have reported that reactivation of RASSF1A expression is mediated by promoter demethylation in various types of cancers [31, 42, 43]. We hypothesized that PEITC-induced RASSF1A promoter demethylation increases RASSF1A expression in LNCaP cells. Quantitative polymerase chain reaction (qPCR) analysis revealed that RASSF1A expression was significantly enhanced following treatment with 2.5 μM and 5 μM PEITC in a dose-dependent manner, suggesting that the reactivation of RASSF1A resulted from promoter demethylation (Figure 1D). Treatment with 5-Aza (2.5 μM) and TSA (0.5 μM) significantly enhanced RASSF1A expression, consistent with the observation that the RASSF1A promoter was demethylated in these cells (Figure 1D).
Figure 1.



Effect of PEITC on RASSF1A promoter demethylation. LNCaP cells (3.5 × 104 cells/ml) were seeded in 10-cm tissue culture plates. LNCaP cells were treated with 0.01% DMSO, PEITC (2.5 or 5 μM), or 5-Aza (2.5 μM) and TSA (0.5 μM) for 5 days. The media containing the drugs was changed on day 3 and day 5. Cells were harvested on day 6 for all the experiments. A) Representative images from the methylation-specific PCR experiments. Genomic DNA was extracted from cells harvested on day 6, and the bisulfite conversion was subsequently performed. M: methylated, U: unmethylated. The relative intensity of the methylated and unmethylated bands was measured using ImageJ software, and the values are presented in the lower panel. B) The methylation pattern of the 16 CpG sites in the fragment of RASSF1A promoter was analyzed using bisulfite genomic sequencing. Solid circles indicate methylated CpG sites, and empty circles indicate unmethylated CpG sites. Ten representative clones from each of the 3 independent experiments were selected for analysis. C) The percentage of CpG methylation is presented. The methylation percentage was calculated from three independent experiments as the number of methylated CpG sites over the total number of CpG sites examined. D) The effect of PEITC on RASSF1A mRNA expression. Total mRNA was isolated and analyzed using quantitative real-time PCR. The data are presented as the mean ± SEM of 3 independent experiments. * P < 0.05 versus the control group. # P < 0.01 versus the control group.
3.2 Regulation of DNA methyltransferases by PEITC
To further investigate PEITC-induced demethylation of the RASSF1A promoter, we evaluated the effect of PEITC on DNMTs, which are enzymes that catalyze the addition of methyl group. Given that we observed a decrease in RASSF1A promoter methylation in cells treated with PEITC, we hypothesized that PEITC and 5-Aza might repress the transcription of DNMTs. The results from the qPCR analysis demonstrated that the expression of DNMT1 (Figure 2A) and DNMT3A mRNA was significantly reduced in cells treated with PEITC (5 μM) or 5-Aza (2.5 μM) and TSA (0.5 μM) (Figure 2B). In addition, the expression of DNMT 3B mRNA transcripts was significantly reduced in cells treated with 5 μM PEITC (Figure 2C). However, the levels of all the DNMTs probed were not significantly affected in cells treated with 2.5 μM PEITC. To determine whether the reduction in gene expression translated to a reduction in protein expression, we evaluated DNMT protein levels using Western blot analysis. Consistent with the reduction in gene expression, the protein levels of the all DNMTs evaluated were significantly reduced in cells treated with 2.5 μM or 5 μM PEITC compared with the control cells. In addition, analysis of densitometry demonstrated that DNMT protein levels were reduced in the positive control group of cells treated with 5-Aza as expected (Figure 2D and 2E).
Figure 2.


Effect of PEITC on DNMT gene and protein expression in LNCaP cells. Total mRNA was isolated and analyzed using quantitative real-time PCR. Total proteins were extracted and analyzed using western blot assays. The relative fold change of protein expression levels was calculated by dividing the intensity of the protein band by the intensity of the control protein band, and the values were normalized to β-actin levels using ImageJ. A) DNMT1 gene expression B) DNMT3A gene expression C) DNMT3B gene expression D) Representative Western blots images of DNMT3A and 3B protein expression. The representative blots are from the same image. The lanes have been rearranged from the same blot to be consistent with the increasing order of PEITC treatment as reported in all the figures. (Original blots have been provided in the supplemental information) E) Data are presented as the mean ± SEM protein expression levels of 3 independent experiments. * P < 0.05 versus the control group, # P < 0.01 versus the control group.
3.3 Regulation of HDAC protein expression by PEITC
HDACs are enzymes that catalyze the removal of acetyl groups, an epigenetic modification associated with the induction of gene expression. PEITC is an HDAC inhibitor[37] that promotes apoptosis. We hypothesized that PEITC treatment would alter HDAC expression, and we used western blot analysis to investigate this hypothesis. Treatment with 5 μM PEITC or 2.5 μM 5-Aza and 0.5 μM TSA significantly reduced HDAC1, HDAC2, HDAC4 and HDAC6 protein levels (Figure 3A and Figure 3B). PEITC at a concentration of 2.5 μM also significantly reduced HDAC 4 and HDAC 6 protein levels (Figure 3A and 3B); however no significant differences in the other HDACs evaluated were noted between the control and the treatment groups (data not shown). These data indicated that the repression of DNMTs and HDAC1, 2, 4 and 6 induced by treatment with PEITC in LNCaP cells led to RASSF1A promoter demethylation and subsequent activation of RASSF1A.
Figure 3.


Effect of PEITC on HDAC protein expression in LNCaP cells. Total proteins were extracted and analyzed using western blot assays. The relative fold change of protein expression levels was calculated by dividing the intensity of the protein band by the intensity of the control protein band, and the values were normalized to β-actin levels using ImageJ. A) Representative western blot images of HDAC1, 2, 4 and 6 protein expressions. The representative blots are from the same image. The lanes have been rearranged to be consistent with the increasing order of PEITC treatment as reported in all the figures. (Original blots have been provided in the supplemental information) B) Data are presented as the mean ± SEM protein expression levels of 3 independent experiments. * P < 0.05 versus the control group. # P < 0.01 versus the control group.
3.4 PEITC-induced RASSF1A expression induces p21, caspase-3, 7 and Bax expression, reduces Cyclin B1 expression and promotes apoptosis
Based on the results of the western blot analysis, we further hypothesized that the increase in RASSF1A expression induced by promoter demethylation promotes cell cycle arrest by increasing expression of the p21 gene (Figure 4A) and promotes apoptosis by increasing caspase-3, Bax and caspase-7 expression and reducing Cyclin B1 expression (Figure 4B). To test this hypothesis, we performed flow cytometry analysis to determine the cell cycle distribution of PI-stained cells. PEITC significantly enhanced the proportion of cells in the sub-G0 phase, indicating that these cells were undergoing early apoptosis. In addition, a significant increase in the proportion of cells in the G2/M phase was noted in the PEITC (5 μM) and 5-AZA (2.5 μM) and TSA (0.5 μM) treatment groups, indicating that these cells were arrested in the G2/M phase of the cell cycle (Figure 4D). To confirm whether the growth inhibition induced by PEITC resulted from apoptosis, we performed flow-cytometry analysis of LNCaP cells stained with Annexin V and PI that were treated with PEITC (2.5 μM and 5 μM) and 5-AZA (2.5 μM) for 5 days. The results of this analysis demonstrated that treatment with PEITC (5 μM), 5-AZA (2.5 μM) and TSA (0.5 μM) increased the number of cells in early apoptosis (Annexin V – positive cells) from 4.6% in control cells to 11.1% in cells treated with PEITC (5 μM) and 53.7% in cells treated with 5AZA (2.5 μM) and TSA (0.5 μM) (Figure 4E). No difference was observed in the cells treated with 2.5 μM PEITC compared with the control group (data not shown). With respect to late apoptosis (PI-positive cells), a similar trend was observed in cells treated with 5 μM PEITC, which exhibited a 2-fold increase in the proportion of cells in late apoptosis compared with the control group (Figure 4E). Together, these results demonstrate that the reactivation of RASSF1A gene expression by PEITC-induced promoter demethylation promoted cell cycle arrest and apoptosis.
Figure 4.




Effect of PEITC on A) p21 gene expression. Total mRNA was isolated and analyzed using quantitative real-time PCR. B) Representative western blot images of p21, caspase-3 caspase-7, Bax and Cyclin B1 protein expression. Total proteins were extracted and analyzed using western blot assays. The relative fold change of protein expression levels was calculated by dividing the intensity of each protein band by the intensity of the control protein band, and the values were normalized to β-actin levels using ImageJ. C) Data are presented as the mean ± SEM protein expression of 3 independent experiments. D) Cell cycle progression in LNCaP cells treated with PEITC or 5-Aza and stained with propidium iodide (PI) was analyzed using flow cytometry with a Beckman Coulter Gallios Flow Cytometer. The legend presents the distribution of cells in the sub G0, G0/G1, S and G2/M phases of the cell cycle. E) Percentage of normal, necrotic and apoptotic cells as determined by flow cytometry of cells stained with PI/Annexin V-FITC. In all panels, the bottom left quadrant represents normal cells, the top right quadrant represents necrotic cells and the bottom right quadrant represents apoptotic cells. The control group is presented in the panel on the left, cells treated with 5 μM PEITC are presented in the center panel, and cells treated with 5-Aza and TSA are presented in the panel on the right.*, P<0.05, # P < 0.01 versus the control group.
3.5 PEITC-inhibited colony formation in LNCaP Cells
As colony formation ability of prostate cancer cells was correlated with its aggressive potential[44], the colony formation assay was used to analyze the inhibition effects of PEITC on the growth of LNCaP cells. LNCaP cells without treatment produced 263±53 colonies, upon treating with PEITC 2.5μM, 5μM and 10μM for 2 weeks, the generated colonies were 159±42, 63±4 and 45±24 respectively, which is significantly reduced from the control group (Figures 5A and 5B). The total colony area were also decreased significantly, after treating with PEITC 2.5μM, 5μM and 10μM for 2 weeks, their relative colony areas were 21.6%, 4.3% and 1.5% respectively (Figure 5C). Taken together, PEITC can effectively inhibit colony formation of LNCaP cells.
Figure 5.

Effect of PEITC on A) LNCaP Colony Formation. LNCaP cells were seeded at 800cells /well density in 6 well dishes. Cells were treated with PEITC (2.5μM, 5μM, 10μM) or 0.1% DMSO (control) every 3 days after seeding for two weeks. The cell colonies were stained with 5 mL 0.01% (w/v) crystal violet. The number and area of colonies containing more than 50 individual cells were counted by ImageJ. B) Number of colonies in different treatment groups. C) Relative total colony area in different groups. Data are presented as the mean ± SEM of 3 independent experiments, # P < 0.01 versus the control group.
4. Discussion
RASSF1A is one of the most frequently hypermethylated genes in human cancers, and RASSF1A hypermethylation is associated with the loss of RASSF1A expression in several types of cancer, including lung, breast, bladder and gastric cancer [21, 22, 24, 45, 46]. In contrast to the RASSF1C isoform, RASSF1A is expressed at higher levels in normal tissues than in lung and breast cancer tumor tissues, suggesting the RASSF1A acts as a tumor suppressor[47]. Previous studies reported that the silencing of RASSF1A expression was a frequently observed phenomenon in advanced stages of prostate cancer[27, 30] and that reversing RASSF1A methylation using phytochemicals, such as mahanine and soy phytoestrogens, reactivated RASSF1A transcription[48, 49]. Several studies that evaluated the mechanism underlying RASSF1A inactivation by analyzing CpG methylation and histone acetylation revealed that reduced histone acetylation or H3K4me2 methylation and increased dimethyl-H3-K9 methylation played a crucial role in RASSF1A promoter methylation and the corresponding inactivation of RASSF1A in prostate cancer [50]. Other studies that investigated the tumor suppressor function of RASSF1A by analyzing microtubule dynamics and apoptosis induction revealed that RASSF1A exerted its tumor suppressor effects primarily by restricting cell cycle progression [51]. The tumor-suppressive function of RASSF1A is through the induction of acetylated α-tubulin and modulation of cell migration [52]. Our study showed that PEITC induced promoter demethylation and induced G2/M cell cycle arrest and apoptosis. It is known that proteins are one of the targets for isothiocyanates. Studies have shown that PEITC covalently binds to tubulin and modifies the cysteine residue forming adducts leading to growth inhibition [53]. It is also shown that increase in RASSF1A expression by 5Aza correlated with an increase in acetylated tubulin [54].
The use of PEITC as a chemopreventive agent has been well studied in prostate cancer and colon cancer by our group and several others [55–57]. Recent investigations into the use of PEITC as a demethylating agent and HDAC inhibitor have provided new insights into its role in regulating epigenetic modifications and regulating genes like p21, GSTP1[36, 37]. PEITC used in combination with conventional chemotherapeutics like Paclitaxel and Doxorubicin has shown synergistic effects [38].Studies have shown that efficacy of Doxorubicin is known to be inhibited by HER2 over expression. However, when treated in combination with PEITC the cell survival was reduced by 50% [58]. Studies have linked HER2 overexpression as well as the over expression of other epidermal growth factors to hyper methylation of RASSF1A [59, 60] . Our hypothesis of RASSF1A demethylation by PEITC could be one of the possible mechanisms by which PEITC enhances the efficacy of doxorubicin. We hypothesized that PEITC induces demethylation of the RASSF1A promoter in LNCaP cells and that reactivating RASSF1A by reversing this epigenetic modification induces apoptosis in LNCaP cells in vitro. Using MSP and BGS analysis, we demonstrated that PEITC promotes the demethylation of the RASSF1A promoter (Figure 1A and 1C). RASSF1A promoter demethylation was also observed in cells treated with 5-Aza and TSA. As treatment with 5-Aza and TSA is known to reverse the methylation of several genes, these agents were used as a positive control [61]. The demethylation of the RASSF1A promoter was partly due to the inhibition of DNMT1, 3A and 3B gene expression that resulted from treatment with PEITC at both concentrations evaluated (2.5 and 5 μM) (Figure 2A, B and C). However, the protein levels of DNMT3A and 3B but not DNMT1 were significantly inhibited by 5 μM PEITC (Figure 2D and 2E). The tumor suppressor function of RASSF1A is attributed to its ability to modulate cell cycle progression and mitotic spindle dynamics [52, 62]. In this study, we observed that PEITC (5 μM) significantly up-regulated RASSF1A gene expression and p21 expression (Figures 1D and 4A), thus resulting in the induction of G2/M cell cycle arrest (Figure 4D). The ability of RASSF1A to induce p21 expression and promote cell cycle arrest at the G1 and G2/M cell cycle phases was previously demonstrated in tumors from immunodeficient mice [62, 63]. Studies have also revealed that the over expression of RASSF1A caused G2/M arrest and caused localization of microtubules and caused mitotic arrest [64].
Several studies demonstrated that the RASSF1 gene exhibits pro-apoptotic activity and that the RASSF1A isoform exhibits stronger pro-apoptotic activity than the RASSF1C isoform. RASSF1A increases caspase-3/7 activation to promote apoptosis in breast, lung and ovarian tumors and simultaneously down-regulates the expression of cyclin D1[47, 65]. RASSF1A was also found to activate the key apoptosis gene Bax[66, 67]. Our study revealed that promoter demethylation activated RASSF1A gene expression in LNCaP cells treated with 5 μM PEITC. This affect might account for the increase in apoptosis we observed as caspase-3, caspase 7 and Bax protein expression were significantly enhanced and Cyclin B1 decreased in these cells (Figure 4B). Cyclin B1 is a key regulatory protein for transition the cells from G2 to M phase, however overexpressed Cyclin B1 can lead to uncontrolled cell growth and is highly related to carcinogenesis of many cancers such as breast cancer[68], pancreatic cancer[69], colorectal cancer[70], bladder cancer[71]. It is found that reduction of Cyclin B1 can cause cancer cell apoptosis by arresting the cells in G2 phase[72]. Besides increasing p21 expression, PEITC (5 μM) were also able to reduce Cyclin B1 expression (Figure 4B), which further demonstrate the mechanism of G2/M arrest and apoptosis by PEITC.
We further analyzed the pro-apoptotic role of PEITC in LNCaP cells using flow cytometry analysis of Annexin V- and PI-stained normal, necrotic and apoptotic cells and observed a 2-fold increase in apoptotic cells following PEITC treatment (5 μM). This effect was stronger than that observed in the positive control group of cells treated with 5-Aza and TSA, which exhibited a 1-fold increase in the number of apoptotic cells (Figure 4E). To investigate the direct inhibition effect of PEITC on human prostate cancer LNCaP cells, we analyzed the colony formation influence by PEITC (2.5μM, 5μM and 10μM) (Figure 5A). It is found that after treatment with all the 3 concentrations of PEITC for 2 weeks, the colony numbers and total colony area were decreased dramatically comparing with control group (Figures 5B and 5C). Hence it suggests PEITC treatment can efficiently inhibit LNCaP cells growth and aggression.
This study is one of the first to demonstrate the role of PEITC in activating RASSF1A and promoting apoptosis in vitro. In conclusion, our findings provide new insight into the role of phytochemicals as epigenetic modulators. Here, 5 μM PEITC significantly induced promoter demethylation and consequently increased RASSF1A gene expression. In addition, PEITC-induced activation of RASSF1A promoted apoptosis and cell cycle arrest. These results further emphasize the DNMT and HDAC inhibitory function of PEITC and its role in promoting apoptosis.
5. Conclusion
Our study revealed that PEITC is a potent regulator of DNA methylation that reverses transcriptionally silenced RASSF1A by inducing promoter demethylation via inhibiting DNMTs and HDACs. RASSF1A activation by PEITC in LNCaP cells induced G2/M cell cycle arrest and apoptosis, indicating that RASSF1A is a potential molecular marker that can be targeted by phytochemicals to prevent prostate cancer.
Supplementary Material
Acknowledgments
We thank all the members of Kong lab for their suggestions and helpful discussion in the preparation of this manuscript. This work was supported in part by institutional funds and by R01-CA118947, R01-CA152826, from the National Cancer Institute (NCI), R01AT007065 from the National Center for Complementary and Alternative Medicines (NCCAM) and the Office of Dietary Supplements (ODS).
Abbreviations
- PEITC
Phenethyl isothiocyanate
- RASSF1A
RAS superfamily protein isoform 1A
- DNMT
DNA methyl transferases
- HDAC
Histone deacetylases
- MSP
methylation specific
- PCR, BGS
bisulfite genomic sequencing
- PCa
Prostate cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Uncategorized References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Abbas A, Patterson W, 3rd, Georgel PT. The epigenetic potentials of dietary polyphenols in prostate cancer management. Biochem Cell Biol. 2013;91(6):361–8. doi: 10.1139/bcb-2012-0044. [DOI] [PubMed] [Google Scholar]
- 4.Ornish D, Magbanua MJ, Weidner G, Weinberg V, Kemp C, Green C, Mattie MD, Marlin R, Simko J, Shinohara K, Haqq CM, Carroll PR. Changes in prostate gene expression in men undergoing an intensive nutrition and lifestyle intervention. Proc Natl Acad Sci U S A. 2008;105(24):8369–74. doi: 10.1073/pnas.0803080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo Y, Su Z-Y, Kong A-NT. Current Perspectives on Epigenetic Modifications by Dietary Chemopreventive and Herbal Phytochemicals. Current Pharmacology Reports. 2015 doi: 10.1007/s40495-015-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saini S, Arora S, Majid S, Shahryari V, Chen Y, Deng G, Yamamura S, Ueno K, Dahiya R. Curcumin modulates microRNA-203-mediated regulation of the Src-Akt axis in bladder cancer. Cancer Prev Res (Phila) 2011;4(10):1698–709. doi: 10.1158/1940-6207.CAPR-11-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zochbauer-Muller S, Farinas AJ, Minna JD, McConnell J, Frenkel EP, Gazdar AF. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res. 2002;8(2):514–9. [PubMed] [Google Scholar]
- 10.Woodson K, Gillespie J, Hanson J, Emmert-Buck M, Phillips JM, Linehan WM, Tangrea JA. Heterogeneous gene methylation patterns among pre-invasive and cancerous lesions of the prostate: a histopathologic study of whole mount prostate specimens. Prostate. 2004;60(1):25–31. doi: 10.1002/pros.20013. [DOI] [PubMed] [Google Scholar]
- 11.Khor TO, Fuentes F, Shu L, Paredes-Gonzalez X, Yang AY, Liu Y, Smiraglia DJ, Yegnasubramanian S, Nelson WG, Kong AN. Epigenetic DNA methylation of antioxidative stress regulator NRF2 in human prostate cancer. Cancer Prev Res (Phila) 2014;7(12):1186–97. doi: 10.1158/1940-6207.CAPR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, Foster BA, Kan YW, Kong AN. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PloS one. 2010;5(1):e8579. doi: 10.1371/journal.pone.0008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majumdar S, Buckles E, Estrada J, Koochekpour S. Aberrant DNA methylation and prostate cancer. Curr Genomics. 2011;12(7):486–505. doi: 10.2174/138920211797904061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry AS, Watson RW, Lawler M, Hollywood D. The epigenome as a therapeutic target in prostate cancer. Nat Rev Urol. 2010;7(12):668–80. doi: 10.1038/nrurol.2010.185. [DOI] [PubMed] [Google Scholar]
- 15.Dammann R, Schagdarsurengin U, Seidel C, Strunnikova M, Rastetter M, Baier K, Pfeifer GP. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol. 2005;20(2):645–63. doi: 10.14670/HH-20.645. [DOI] [PubMed] [Google Scholar]
- 16.Pfeifer GP, Yoon JH, Liu L, Tommasi S, Wilczynski SP, Dammann R. Methylation of the RASSF1A gene in human cancers. Biol Chem. 2002;383(6):907–14. doi: 10.1515/BC.2002.097. [DOI] [PubMed] [Google Scholar]
- 17.Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta. 2009;1796(2):114–28. doi: 10.1016/j.bbcan.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25(3):315–9. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, He J, Li J, Tian D, Gu L, Zhou M. Methylation of RASSF1A gene promoter is regulated by p53 and DAXX. FASEB J. 2013;27(1):232–42. doi: 10.1096/fj.12-215491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon JH, Dammann R, Pfeifer GP. Hypermethylation of the CpG island of the RASSF1A gene in ovarian and renal cell carcinomas. Int J Cancer. 2001;94(2):212–7. doi: 10.1002/ijc.1466. [DOI] [PubMed] [Google Scholar]
- 21.Byun DS, Lee MG, Chae KS, Ryu BG, Chi SG. Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer research. 2001;61(19):7034–8. [PubMed] [Google Scholar]
- 22.Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001;93(9):691–9. doi: 10.1093/jnci/93.9.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo KW, Kwong J, Hui AB, Chan SY, To KF, Chan AS, Chow LS, Teo PM, Johnson PJ, Huang DP. High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer research. 2001;61(10):3877–81. [PubMed] [Google Scholar]
- 24.Lee MG, Kim HY, Byun DS, Lee SJ, Lee CH, Kim JI, Chang SG, Chi SG. Frequent epigenetic inactivation of RASSF1A in human bladder carcinoma. Cancer research. 2001;61(18):6688–92. [PubMed] [Google Scholar]
- 25.Yang Q, Zage P, Kagan D, Tian Y, Seshadri R, Salwen HR, Liu S, Chlenski A, Cohn SL. Association of epigenetic inactivation of RASSF1A with poor outcome in human neuroblastoma. Clin Cancer Res. 2004;10(24):8493–500. doi: 10.1158/1078-0432.CCR-04-1331. [DOI] [PubMed] [Google Scholar]
- 26.Dreijerink K, Braga E, Kuzmin I, Geil L, Duh FM, Angeloni D, Zbar B, Lerman MI, Stanbridge EJ, Minna JD, Protopopov A, Li J, Kashuba V, Klein G, Zabarovsky ER. The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci U S A. 2001;98(13):7504–9. doi: 10.1073/pnas.131216298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuzmin I, Gillespie JW, Protopopov A, Geil L, Dreijerink K, Yang Y, Vocke CD, Duh FM, Zabarovsky E, Minna JD, Rhim JS, Emmert-Buck MR, Linehan WM, Lerman MI. The RASSF1A tumor suppressor gene is inactivated in prostate tumors and suppresses growth of prostate carcinoma cells. Cancer research. 2002;62(12):3498–502. [PubMed] [Google Scholar]
- 28.van der Weyden L, Tachibana KK, Gonzalez MA, Adams DJ, Ng BL, Petty R, Venkitaraman AR, Arends MJ, Bradley A. The RASSF1A isoform of RASSF1 promotes microtubule stability and suppresses tumorigenesis. Molecular and cellular biology. 2005;25(18):8356–67. doi: 10.1128/MCB.25.18.8356-8367.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge YZ, Xu LW, Jia RP, Xu Z, Feng YM, Wu R, Yu P, Zhao Y, Gui ZL, Tan SJ, Song Q. The association between RASSF1A promoter methylation and prostate cancer: evidence from 19 published studies. Tumour Biol. 2014;35(4):3881–90. doi: 10.1007/s13277-013-1515-3. [DOI] [PubMed] [Google Scholar]
- 30.Pan J, Chen J, Zhang B, Chen X, Huang B, Zhuang J, Mo C, Qiu S. Association between RASSF1A promoter methylation and prostate cancer: a systematic review and meta-analysis. PLoS One. 2013;8(9):e75283. doi: 10.1371/journal.pone.0075283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du L, Xie Z, Wu LC, Chiu M, Lin J, Chan KK, Liu S, Liu Z. Reactivation of RASSF1A in breast cancer cells by curcumin. Nutr Cancer. 2012;64(8):1228–35. doi: 10.1080/01635581.2012.717682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal S, Amin KS, Jagadeesh S, Baishay G, Rao PG, Barua NC, Bhattacharya S, Banerjee PP. Mahanine restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in prostate cancer cells. Mol Cancer. 2013;12(1):99. doi: 10.1186/1476-4598-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veeranki OL, Bhattacharya A, Tang L, Marshall JR, Zhang Y. Cruciferous Vegetables, Isothiocyanates, and Prevention of Bladder Cancer. Current Pharmacology Reports. 2015 doi: 10.1007/s40495-015-0024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Royston KJ, Tollefsbol TO. The Epigenetic Impact of Cruciferous Vegetables on Cancer Prevention. Curr Pharmacol Rep. 2015;1(1):46–51. doi: 10.1007/s40495-014-0003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beklemisheva AA, Fang Y, Feng J, Ma X, Dai W, Chiao JW. Epigenetic mechanism of growth inhibition induced by phenylhexyl isothiocyanate in prostate cancer cells. Anticancer Res. 2006;26(2A):1225–30. [PubMed] [Google Scholar]
- 36.Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Molecular carcinogenesis. 2007;46(1):24–31. doi: 10.1002/mc.20258. [DOI] [PubMed] [Google Scholar]
- 37.Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, Feng J, Liu D, Chiao JW. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. International journal of oncology. 2008;33(2):375–80. [PubMed] [Google Scholar]
- 38.Liu K, Cang S, Ma Y, Chiao JW. Synergistic effect of paclitaxel and epigenetic agent phenethyl isothiocyanate on growth inhibition, cell cycle arrest and apoptosis in breast cancer cells. Cancer cell international. 2013;13(1):10. doi: 10.1186/1475-2867-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Chakravarty S, Dey M. Phenethylisothiocyanate alters site- and promoter-specific histone tail modifications in cancer cells. PLoS One. 2013;8(5):e64535. doi: 10.1371/journal.pone.0064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L, Yoon JH, Dammann R, Pfeifer GP. Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene. 2002;21(44):6835–40. doi: 10.1038/sj.onc.1205814. [DOI] [PubMed] [Google Scholar]
- 41.Strunnikova M, Schagdarsurengin U, Kehlen A, Garbe JC, Stampfer MR, Dammann R. Chromatin inactivation precedes de novo DNA methylation during the progressive epigenetic silencing of the RASSF1A promoter. Molecular and cellular biology. 2005;25(10):3923–33. doi: 10.1128/MCB.25.10.3923-3933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montenegro MF, Saez-Ayala M, Pinero-Madrona A, Cabezas-Herrera J, Rodriguez-Lopez JN. Reactivation of the tumour suppressor RASSF1A in breast cancer by simultaneous targeting of DNA and E2F1 methylation. PLoS One. 2012;7(12):e52231. doi: 10.1371/journal.pone.0052231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gray SG, Baird AM, O'Kelly F, Nikolaidis G, Almgren M, Meunier A, Dockry E, Hollywood D, Ekstrom TJ, Perry AS, O'Byrne KJ. Gemcitabine reactivates epigenetically silenced genes and functions as a DNA methyltransferase inhibitor. International journal of molecular medicine. 2012;30(6):1505–11. doi: 10.3892/ijmm.2012.1138. [DOI] [PubMed] [Google Scholar]
- 44.Nair HK, Rao KV, Aalinkeel R, Mahajan S, Chawda R, Schwartz SA. Inhibition of prostate cancer cell colony formation by the flavonoid quercetin correlates with modulation of specific regulatory genes. Clin Diagn Lab Immunol. 2004;11(1):63–9. doi: 10.1128/CDLI.11.1.63-69.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hesson LB, Cooper WN, Latif F. The role of RASSF1A methylation in cancer. Dis Markers. 2007;23(1–2):73–87. doi: 10.1155/2007/291538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mengxi D, Qian W, Nan W, Xiaoguang X, Shijun L. Effect of DNA methylation inhibitor on RASSF1A genes expression in non-small cell lung cancer cell line A549 and A549DDP. Cancer cell international. 2013;13(1):91. doi: 10.1186/1475-2867-13-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reeves ME, Firek M, Chen ST, Amaar Y. The RASSF1 Gene and the Opposing Effects of the RASSF1A and RASSF1C Isoforms on Cell Proliferation and Apoptosis. Molecular biology international. 2013;2013:145096. doi: 10.1155/2013/145096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jagadeesh S, Sinha S, Pal BC, Bhattacharya S, Banerjee PP. Mahanine reverses an epigenetically silenced tumor suppressor gene RASSF1A in human prostate cancer cells. Biochem Biophys Res Commun. 2007;362(1):212–7. doi: 10.1016/j.bbrc.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 49.Vardi A, Bosviel R, Rabiau N, Adjakly M, Satih S, Dechelotte P, Boiteux JP, Fontana L, Bignon YJ, Guy L, Bernard-Gallon DJ. Soy phytoestrogens modify DNA methylation of GSTP1, RASSF1A, EPH2 and BRCA1 promoter in prostate cancer cells. In Vivo. 2010;24(4):393–400. [PubMed] [Google Scholar]
- 50.Kawamoto K, Okino ST, Place RF, Urakami S, Hirata H, Kikuno N, Kawakami T, Tanaka Y, Pookot D, Chen Z, Majid S, Enokida H, Nakagawa M, Dahiya R. Epigenetic modifications of RASSF1A gene through chromatin remodeling in prostate cancer. Clin Cancer Res. 2007;13(9):2541–8. doi: 10.1158/1078-0432.CCR-06-2225. [DOI] [PubMed] [Google Scholar]
- 51.Donninger H, Clark JA, Monaghan MK, Schmidt ML, Vos M, Clark GJ. Cell cycle restriction is more important than apoptosis induction for RASSF1A protein tumor suppression. The Journal of biological chemistry. 2014;289(45):31287–95. doi: 10.1074/jbc.M114.609537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vos MD, Martinez A, Elam C, Dallol A, Taylor BJ, Latif F, Clark GJ. A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer research. 2004;64(12):4244–50. doi: 10.1158/0008-5472.CAN-04-0339. [DOI] [PubMed] [Google Scholar]
- 53.Mi L, Xiao Z, Hood BL, Dakshanamurthy S, Wang X, Govind S, Conrads TP, Veenstra TD, Chung FL. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. The Journal of biological chemistry. 2008;283(32):22136–46. doi: 10.1074/jbc.M802330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jung HY, Jung JS, Whang YM, Kim YH. RASSF1A Suppresses Cell Migration through Inactivation of HDAC6 and Increase of Acetylated alpha-Tubulin. Cancer research and treatment : official journal of Korean Cancer Association. 2013;45(2):134–44. doi: 10.4143/crt.2013.45.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12(1):87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barve A, Khor TO, Hao X, Keum YS, Yang CS, Reddy B, Kong AN. Murine prostate cancer inhibition by dietary phytochemicals--curcumin and phenyethylisothiocyanate. Pharm Res. 2008;25(9):2181–9. doi: 10.1007/s11095-008-9574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khor TO, Cheung WK, Prawan A, Reddy BS, Kong AN. Chemoprevention of familial adenomatous polyposis in Apc(Min/+) mice by phenethyl isothiocyanate (PEITC) Molecular carcinogenesis. 2008;47(5):321–5. doi: 10.1002/mc.20390. [DOI] [PubMed] [Google Scholar]
- 58.Gupta P, Srivastava SK. Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC medicine. 2012;10:80. doi: 10.1186/1741-7015-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho YH, Shen J, Gammon MD, Zhang YJ, Wang Q, Gonzalez K, Xu X, Bradshaw PT, Teitelbaum SL, Garbowski G, Hibshoosh H, Neugut AI, Chen J, Santella RM. Prognostic significance of gene-specific promoter hypermethylation in breast cancer patients. Breast cancer research and treatment. 2012;131(1):197–205. doi: 10.1007/s10549-011-1712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kajabova V, Smolkova B, Zmetakova I, Sebova K, Krivulcik T, Bella V, Kajo K, Machalekova K, Fridrichova I. RASSF1A Promoter Methylation Levels Positively Correlate with Estrogen Receptor Expression in Breast Cancer Patients. Translational oncology. 2013;6(3):297–304. doi: 10.1593/tlo.13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen WJ, Dai DQ, Teng Y, Liu HB. Regulation of demethylation and re-expression of RASSF1A gene in gastric cancer cell lines by combined treatment of 5-Aza–CdR and NaB. World journal of gastroenterology. 2008;14(4):595–600. doi: 10.3748/wjg.14.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rong R, Jin W, Zhang J, Sheikh MS, Huang Y. Tumor suppressor RASSF1A is a microtubule-binding protein that stabilizes microtubules and induces G2/M arrest. Oncogene. 2004;23(50):8216–30. doi: 10.1038/sj.onc.1207901. [DOI] [PubMed] [Google Scholar]
- 63.Thaler S, Hahnel PS, Schad A, Dammann R, Schuler M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer research. 2009;69(5):1748–57. doi: 10.1158/0008-5472.CAN-08-1377. [DOI] [PubMed] [Google Scholar]
- 64.Song MS, Song SJ, Ayad NG, Chang JS, Lee JH, Hong HK, Lee H, Choi N, Kim J, Kim H, Kim JW, Choi EJ, Kirschner MW, Lim DS. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nature cell biology. 2004;6(2):129–37. doi: 10.1038/ncb1091. [DOI] [PubMed] [Google Scholar]
- 65.Fu L, Zhang S. RASSF1A promotes apoptosis and suppresses the proliferation of ovarian cancer cells. International journal of molecular medicine. 2014;33(5):1153–60. doi: 10.3892/ijmm.2014.1671. [DOI] [PubMed] [Google Scholar]
- 66.Baksh S, Tommasi S, Fenton S, Yu VC, Martins LM, Pfeifer GP, Latif F, Downward J, Neel BG. The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Molecular cell. 2005;18(6):637–50. doi: 10.1016/j.molcel.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 67.Vos MD, Dallol A, Eckfeld K, Allen NP, Donninger H, Hesson LB, Calvisi D, Latif F, Clark GJ. The RASSF1A tumor suppressor activates Bax via MOAP-1. The Journal of biological chemistry. 2006;281(8):4557–63. doi: 10.1074/jbc.M512128200. [DOI] [PubMed] [Google Scholar]
- 68.Rajput S, Khera N, Guo Z, Hoog J, Li S, Ma CX. Inhibition of cyclin dependent kinase 9 by dinaciclib suppresses cyclin B1 expression and tumor growth in triple negative breast cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou L, Li J, Zhao YP, Cui QC, Zhou WX, Guo JC, You L, Wu WM, Zhang TP. The prognostic value of Cyclin B1 in pancreatic cancer. Medical oncology (Northwood, London, England) 2014;31(9):107. doi: 10.1007/s12032-014-0107-4. [DOI] [PubMed] [Google Scholar]
- 70.Egeland EV, Boye K, Pettersen SJ, Haugen MH, Oyjord T, Malerod L, Flatmark K, Maelandsmo GM. Enrichment of nuclear S100A4 during G2/M in colorectal cancer cells: possible association with cyclin B1 and centrosomes. Clinical & experimental metastasis. 2015;32(8):755–67. doi: 10.1007/s10585-015-9742-1. [DOI] [PubMed] [Google Scholar]
- 71.Huang L, Peng Y, Zhong G, Xie W, Dong W, Wang B, Chen X, Gu P, He W, Wu S, Lin T, Huang J. PBRM1 suppresses bladder cancer by cyclin B1 induced cell cycle arrest. Oncotarget. 2015;6(18):16366–78. doi: 10.18632/oncotarget.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yuan J, Kramer A, Matthess Y, Yan R, Spankuch B, Gatje R, Knecht R, Kaufmann M, Strebhardt K. Stable gene silencing of cyclin B1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo. Oncogene. 2006;25(12):1753–62. doi: 10.1038/sj.onc.1209202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.