

Abstract
Polyploidy is increasingly recognized as a driver of biological diversity. How and why polyploidization affects gene expression is critical to understanding the link between ploidy elevation and diversification. In polyploid plants, multiple studies have demonstrated that ploidy elevation can confer major but variable consequences for gene expression, ranging from gene-by-gene alterations to entirely silenced genomes. By contrast, animal polyploids remain largely uncharacterized. Accordingly, how animals respond to and manage polyploidy events is not understood. Here, we address this important knowledge gap by analyzing transcriptomes from a triploid hybrid animal, a unisexual Ambystoma salamander, and three sexual Ambystoma species that represent all three parental genomes in the unisexual. We used a novel bioinformatics pipeline that includes competitively mapping triploid sequences to a reference set of orthologous genes in the sexual species to evaluate subgenome expression. Our comparisons of gene expression levels across the three parental genomes revealed that the unisexual triploid displays a pattern of genome balance, where 72% of the genes analyzed were expressed equally among the subgenomes. This result is strikingly different from the genome imbalance typically observed in hybrid polyploid plants. Our analyses represent the first to address gene expression in a triploid hybrid animal and introduce a novel bioinformatic framework for analyzing transcriptomic data.
Keywords: polyploidy, hybridization, gene expression, genome dominance, reproductive mode, salamander
Introduction
Polyploidy has influenced the evolution of organismal diversity on evolutionary and ecological time scales (Dowling and Secor 1997; Van de Peer et al. 2009; Barker et al. 2016). Phenomena ranging from ancient polyploidization events in plants (Vanneste et al. 2014) and other eukaryotes (Van de Peer et al. 2009) to the importance of human-mediated polyploidization in crops (Ramsey and Ramsey 2014) demonstrate the importance of ploidy elevation for the evolution of biological diversity. Phenotypic consequences of polyploidization include higher levels of gene expression (de Godoy et al. 2008; Neiman et al. 2009), increased adaptive potential (Otto and Whitton 2000; Te Beest et al. 2012), and altered nutritional requirements (Neiman et al. 2013). Even though polyploidy is a major contributor to the evolutionary histories of many animal taxa (Gregory and Mable 2005; Otto 2007; Wertheim et al. 2013), most research directed at characterizing the consequences of polyploidy has focused on plants or fungi. Whereas these systems have provided valuable insights into the consequences of ploidy elevation, a more general understanding of the genomic and phenotypic effects of polyploidy requires information from animal polyploids. Here, we fill this information gap by exploring how polyploidy (and the often associated but distinct phenomenon of hybridization) influences gene expression in an animal system.
Polyploidy and Its Consequences for Gene Expression in Plants and Animals
The relationship between ploidy and gene expression is highly variable in both plants (Soltis and Soltis 2000) and animals (Wertheim et al. 2013). For example, whereas increased gene expression associated with ploidy level might generate benefits via larger body sizes or more rapid growth (Birchler et al. 2010), the alteration of expression patterns following ploidy elevation (and/or hybridization) might also reduce the benefits associated with the high gene dosage that polyploids enjoy (Mable et al. 2011). In allopolyploid plants, the challenge posed by epigenetic instability may be addressed through functional diploidization, where genes from one or more genomes are silenced in order to match an ancestral level of expression (Feldman et al. 2012). This hypothesis has received mixed support in allopolyploid plants, which demonstrate expression patterns ranging from additive expression from each allele and expression dominance by a single allele to biases towards a specific parental genome (Madlung 2012). While these patterns may reflect subfunctionalization mechanisms that may be advantageous, we still lack a comprehensive understanding of how polyploidy influences gene expression in plants. We know even less about how gene expression patterns are affected by genome number and identity in polyploid animals despite the critical importance of this knowledge for understanding the general consequences of these large-scale changes in genome composition.
The earliest evaluation of gene expression patterns in polyploid animals came from two polyploid fish taxa, the allotriploid species complex Squalius alburnoides (Alves et al. 2001; Pala et al. 2010) and the synthetic triploid Oryzias latipes (Garcia et al. 2014). Pala et al. (2010) showed that S. alburnoides, via a combination of asexual, hybridogenetic, and normal meiotic reproduction, maintains a diversity of genome combinations that depend on the sympatric sexual species. Transcriptomic data from S. alburnoides indicate that gene copy silencing and genome-specific variation in expression can reduce overall expression to levels similar to diploids (Pala et al. 2008, 2010; Matos et al. 2015). By contrast, the subset of silenced genes identified in O. latipes are clustered within certain chromosomes, which may in turn indicate a chromosome-specific pattern of gene silencing (Garcia et al. 2014). Other asexual polyploid fish (Poecilia formosa, Schedina et al. 2014; Carassius auratus gibelio, Li et al. 2014) also experience differential patterns of expression relative to diploid sexual counterparts. The relatively few gene expression studies in animal polyploids and the variety of observed expression patterns documented prevent generalizations of the effects of polyploidy and hybridization on gene expression in animals.
Studies of gene expression in polyploids present special bioinformatic challenges because of the prevalence of allopolyploidy. In particular, determining the origin of homeologs within a polyploid is especially difficult when >2 parental genomes are present (i.e., in allopolyploids, Garcia et al. 2014). This challenge has been generally addressed through identification of diagnostic single nucleotide polymorphisms (dSNPs) that differentiate homeolog origin amongst the different parental genomes (Garcia et al. 2014). Limitations to this SNP-based approach are imposed by the dependence on genomic regions harboring dSNPs between parental genomes, which requires both a certain level of genetic divergence as well as extensive genomic resources.
A Widespread and Ecologically Successful Vertebrate Polyploid
To study expression in animal polyploids, we compared gene expression among parental genomes within the oldest known unisexual (all female) polyploid vertebrate lineage, unisexual Ambystoma salamanders (Bi and Bogart 2010). Unisexual Ambystoma range from triploid to pentaploid and contain haploid genomes from up to five different sexual Ambystoma species (Bogart et al. 2007, 2009). In contrast to their nuclear genomic diversity, unisexual Ambystoma represent a single monophyletic mitochondrial lineage that originated ∼5 mya (Bi and Bogart 2010). The combination of this ancient mitochondrial lineage with occasional additions, losses, and substitutions of nuclear genomes from sexual species (termed “kleptogenesis”) may have important consequences for nuclear and mitonuclear discordance and epigenetic instability relative to other polyploid vertebrates (Buggs et al. 2008, 2014). A comprehensive characterization of patterns of expression among these “captured” genomes presents a key step in addressing the role of polyploidy and hybridization in the success of unisexual Ambystoma. In particular, a link between differential expression and ecological success within unisexual Ambystoma could provide a mechanism for how unisexual, polyploid, and/or hybrid animals successfully coexist and compete with otherwise similar sexual diploid species in similar niches.
Because unisexual Ambystoma individuals can possess up to five nuclear genomes derived from up to five phylogenetically diverse Ambystoma species, the identification of homeologous sequences presents a bioinformatic challenge. We addressed this issue by developing a novel competitive mapping method that used transcriptomes from potential parental species to compare gene expression levels among individual genomes within a unisexual trihybrid. An overall balanced gene expression profile across the nuclear genomes of a unisexual Ambystoma could be linked to the maintenance of a diverse array of unisexual genome combinations in the natural range of the unisexual salamanders (Bogart et al. 2007). Accordingly, we predicted that subgenomes within a unisexual individual would display relatively similar patterns of expression. More generally, we provide an accessible analytical framework for future gene expression studies in allopolyploids.
Materials and Methods
Sample Collection, Library Preparation, and Sequencing
We collected a single triploid unisexual Ambystoma salamander from Kelley’s Island, Ohio, USA in spring 2011. We used the SNP assay from Greenwald and Gibbs (2012) to identify this individual’s biotype as composed of a single genome from three different sexual species: Ambystoma laterale (“L”), Ambystomatexanum (“T”), and Ambystomatigrinum (“Ti”) (abbreviated as LTTi). We also collected a female individual from each of these three sexual species at sites ∼100 km from Kelley's Island (A. texanum and A. tigrinum from Crawford County, Ohio, and A. laterale from southern Ontario). All field-collected animals were held in captivity for ten months in a 50 °F cold room with a 12 h day/night cycle and with standardized feeding schedules. Unisexual Ambystoma salamanders have not been successfully bred in the laboratory and require significant effort to collect in the field. The collection of this specific LTTi genotype and the associated parental species were done intentionally, with the goal of maximizing our ability to differentiate between subgenomes and perform a rigorous test of the analysis pipeline described below.
We euthanized animals by submersion in MS-222 following an approved IACUC Protocol (Ohio State University IACUC Protocol 2009 A0087). We extracted total RNA from fresh liver tissue using the standard TRIzol protocol (Invitrogen, Carlsbad, CA). After confirming RNA integrity with a Bioanalyzer, we prepared four libraries (one per individual salamander) using Illumina’s TruSeq Stranded mRNA Sample Prep kit following the manufacturer’s instructions. Briefly, mRNA was selected with oligo beads and chemically fragmented to a size of ∼100–400 nt before annealing of random hexamers and first-strand cDNA synthesis. Each barcoded library was then run on half of a lane of an Illumina HiSeq 2000 (single end, 100 bp read length) at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. All raw RNA-seq reads are available via the NCBI Short Read Archive (accession numbers: SRR5346172, SRR5346171, SRR5346170 and SRR5346169) under Bioproject PRJNA378554.
De Novo Transcriptome Assembly
We assembled de novo transcriptomes for each of the four Ambystoma individuals. First, we used the Fastx Toolkit (Gordon and Hannon 2010) to filter out reads that did not have a quality score of ≥20 for at least 75% of bases. We checked for read quality before and after filtering with FASTQC (Andrews 2010). Next, we used Trinity (Grabherr et al. 2011; Haas et al. 2013; version 20131110) for in silico read normalization to reduce the memory requirement for transcriptome assembly by using a maximum kmer coverage of 30× and otherwise default parameters. We then assembled all four transcriptomes individually with Trinity using a group-pairs distance of 350 bases and otherwise default parameters. We used the Trinity plug-in program TransDecoder to identify the most likely coding sequences (CDS) for each assembly (Haas et al. 2013) and used CD-HIT-EST (Huang et al. 2010) to cluster the sequences in each transcriptome, which reduced redundancy of the sequences. Next, we used BlastX via BLAST+ (Camacho et al. 2009) to blast each of the four transcriptomes against the NCBI protein databases. Finally, we used Blast2GO (Conesa et al. 2005) to annotate each transcriptome. We used Blast2GO annotation statistics to determine the extent and quality of annotation for each transcriptome and to compare annotation levels across the transcriptomes. This Blast2GO analysis included quantifying the number of transcripts that received BlastX annotation, the number of transcripts that received both BlastX and GO annotations, and the number of unannotated transcripts relative to the total number of transcripts in each transcriptome. We also determined, on average, how many GO annotations each transcript received and the distribution of taxa represented among top annotation hits (see supplementary tables S1 and S2, Supplementary Material online). Finally, we used the Blast2GO combined graph function to compare GO terms across the transcriptomes (see supplementary fig. S1, Supplementary Material online). All assembled transcriptomes are availalbe via the NCBI Transcriptome Shotgun Assembly Sequence Database on GenBank under Bioproject PRJNA378554.
Generation of Ortholog Reference Set
Our primary question was whether homeologs from the three subgenomes within the unisexual individual were being expressed equally, with the general expectation that genome dominance would be revealed as a pattern whereby one or more of the subgenomes had higher or lower overall gene expression than the other subgenome(s). We also took into account the possibility that other, more complex patterns of expression might exist. For example, the regular genome contributions and substitutions that occur in the unisexual Ambystoma complex might favor co-expression of particular subgenomes versus only relatively small sets of genes with differential homeolog expression. Accordingly, we address homeolog expression on both a genome-wide and gene-by-gene basis and document the presence and extent of each of three outcomes: 1) genome-wide dominance of transcripts from a single species, 2), genome-wide co-expression of all three homeologs, and 3), specific expression patterns for different genes. Because neither a reference genome nor genomic resources are available for this unisexual Ambystoma biotype or for any of the parental species, we use de novo transcriptomes of the sexual species to serve as proxies for the unisexual genomes. This approach also provides a simpler alternative to assembling three separate transcriptomes from the three genomes represented within the unisexual reads, enabling us to readily generate a reference for measuring expression of the unisexual homeologs.
To compare homeolog expression on a gene-by-gene basis, we assembled a set of orthologs from each of the three parental genomes. These three ortholog sets were comprised of sequences that 1) appeared only once in each transcriptome and 2) had an identical annotation for all three transcriptomes. We used custom Python scripts (available at https://github.com/jsharbrough/pathwayGeneExpression; last accessed March 28, 2017) to identify the 3,178 orthologs that satisfied these criteria. This approach to identifying orthologs ensured that we had a representative of each ortholog for each gene to use for our tests of expression bias. Next, we used ClustalΩ (Version 1.2.1) to quantify the pairwise nucleotide distance between each of the three possible pairwise combinations of all three parental sexual species for each set of orthologs (Sievers et al. 2011). We then used a histogram to visualize the distribution of the outcome of each of these pairwise comparisons (see supplementary fig. S2, Supplementary Material online). This histogram revealed a bimodal distribution of pairwise distances: 2,998 putative orthologs with Jukes–Cantor-corrected pairwise distance < 0.6 (mean 0.0497 ± 0.0996 SD) and 180 putative orthologs with a Jukes–Cantor-corrected pairwise distance ≥ 0.6 (mean 0.7131 ± SD 0.0574). We removed this latter set of putative orthologs from subsequent analyses by the logic that the > 0.6 pairwise distance is defined by a local minimum in the distribution of distances and is thus relatively likely to reflect paralogous rather than orthologous relationships. After removing the 180 sequences with a pairwise distance exceeding 0.6 between any two species, we used the remaining 2,998 ortholog triplets for the subsequent expression analyses. We found no significant GO-term enrichment for the reference ortholog set when compared with any of the four complete transcriptomes (Fisher’s Exact Test (FET) with a False Discovery Rate (FDR) of 0.05), indicating that this reference ortholog gene set did not show biased representation of genes from any functional category. This result suggests that our reference ortholog gene set provides a representative sample of the functional types of genes found in the three sexual species.
Testing for Differential Homeolog Expression
Competitive Mapping
To evaluate the relative frequencies of 1) genome co-expression, 2) genome dominance, and 3), gene-by-gene variation in patterns of genome expression, we carried out an expression test that we term “competitive mapping” (fig. 1). This method represented homeolog expression by mapping the RNA-seq reads generated from the unisexual trihybrid to the ortholog gene set composed of representative sexual genomes as a proxy for comparing homeolog expression. By mapping reads from the unisexual to this reference set, we can assess whether reads map more often to a particular ortholog from a particular species relative to the other orthologs from the other two species for a particular gene, with the implications that the mapping can be viewed as a “competition” between orthologs for reads. The reference we used for this competitive mapping step was a combined fasta file that comprised each ortholog from all three parental species (see above). We then performed “competitive mapping” among the orthologs by mapping the unisexual reads to the reference set via Tophat2 (Kim et al. 2013) and used Cufflinks2 to quantify gene expression (Trapnell et al. 2010; Roberts et al. 2011). We used the results of these analyses to evaluate expression patterns for each homeolog.
Fig. 1.—

Pipeline for testing differential genome use. (A) Read processing, transcriptome assembly, and generation of reference ortholog sequence set. (B) Competitive mapping of reads from unisexual against reference ortholog sequence set. (C) Quantification of relative gene expression for three homeologs per gene.
Correcting Expression in the Trihybrid Unisexual
Because some sequence regions will not vary among the orthologs from each unique genome, we accounted for the degree to which the unisexual reads have the same versus different likelihood of mapping to any of the three parental species. The fact that we focused on sequences that appeared once in each transcriptome means that each orthologous set of reads are derived from one location in the genome. Even so, the regions of high sequence similarity between parental species mean that some of the unisexual reads may not map to the “correct” ortholog (e.g., some reads representing expression of the A. texanum genome may map to the A. tigrinum ortholog). To control for this “incorrect” mapping by the unisexual RNA-seq reads, we developed a correction metric that modified the expression value of each unisexual homeolog based on an empirical assessment of the degree to which RNA-seq reads from the parental species map incorrectly to the other two species for each unique triplet of orthologs. This method also accounts for potential biases arising from a situation where incorrect mapping is more likely to represent closely versus relatively distantly related species. To generate our correction metric, we separately mapped reads from each parental species to the ortholog reference set with Tophat2 and quantified gene expression with Cufflinks2. We then used the amount of “incorrect” expression (in fragments per kilobase of transcript per million mapped reads, or FPKM) arising from reads from one species mapping incorrectly to an ortholog sequence from another species (e.g., the FPKM from mapping the A. tigrinum reads to the A texanum sequences) to estimate the baseline contribution of “incorrect” mapping of unisexual reads among the homeologous genomes in the unisexual. This value represents our “correction metric” and is calculated as follows:
A. laterale
Correction metric (ALCM)
((UnisexAti/UnisexTotal) + (UnisexAt/UnisexTotal))/2
Corrected expression value (UnisexAlC)
UnisexAl - (UnisexAl × ALCM) = UnisexAlC
A. tigrinum
Correction metric (TiCM)
((UnisexAl/UnisexTotal) + (UnisexAte/UnisexTotal))/2
Corrected expression value (UnisexAtiC)
UnisexAti - (UnisexAti × AtiCM) = UnisexAtiC
A. texanum
Correction metric (TeCM)
((UnisexAl/UnisexTotal) + (UnisexAti/UnisexTotal))/2
Corrected expression value (UnisexAteC)
UnisexAte - (UnisexAte × AteCM) = UnisexAteC
Evaluating Homeolog Expression Balance: Gene-by-Gene
We used the corrected expression values for unisexuals to test whether each of the three homeologs were expressed at equal versus different levels in the unisexuals. Our null hypothesis was that for each gene, all three homeologs would exhibit a 1:1:1 ratio of expression. We addressed the genome dominance question by performing Holm FDR-corrected χ2 tests to identify genes that deviated from this balanced 1:1:1 expression ratio. First, because this analysis might be more likely to detect differential expression in genes with the highest expression levels, we normalized the corrected expression values for each set of orthologs by dividing the FPKM of each homeolog by the sum of the FPKM for all three of the homeologs. Next, we multiplied those normalized FPKM value by 100 so that unequal expression would be detectable, and equally so, across all ortholog sets, regardless of absolute levels of expression. For each of the 826 genes (28% - 826/2,998 total) for which we detected significantly differentially expressed homeologs, we used the corrected (prior to normalization) FPKM values to rank the expression values of each of the three homeologs per gene as “highest”, “middle”, or “lowest” for that gene. Because there were so few genes for which ties were observed (26/2,998; ∼0.87%), we removed these 26 genes from subsequent analyses. After removing genes with rank ties, we sorted the order of homeolog expression for the remaining 2,972 genes into six categories representing the six possible different combination of the “highest”, “middle”, and “lowest” expression levels among the three homeologs (see supplementary fig. S3, Supplementary Material online). The six different orders represent six distinct hypotheses for how the homeologs for a given gene might be expressed if not co-expressed equally. We then used Blast2GO to test for functional enrichment in all six different gene sets, with a particular focus on whether specific biological functions exhibited any partitioning among the subgenomes. For example, one potential explanation for why unisexual Ambystoma always have an L genome is that the L genome is required for compatibility with the mitochondrial genome, which is monophyletic (in contrast to the nuclear genomes) in unisexual Ambystoma (Bi et al. 2008; Bi and Bogart 2010; Denton et al. 2014). This hypothesis predicts that mitochondrial functions would be especially likely to be enriched in the genes with the highest expression of the A. laterale homeolog.
Evaluating Homeolog Expression Balance: Pathway Analysis
By categorizing differentially expressed homeologs according to the order of homeolog expression, we were also able to test the hypothesis that genes in the same functional pathway exhibit consistent patterns of homeolog expression. As opposed to whole-genome dominance, support for consistent homeolog expression orders would suggest that unisexual Ambystoma have dynamic control over their subgenomes, with the function of certain biochemical processes partitioned out among their homeologous genomes. To evaluate this hypothesis, we used Blast2GO to compare our reference ortholog gene set to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and organize the sequences in this reference set into pathways. We exported these KEGG pathway lists from Blast2GO, identified all of the pathways with at least two genes in the reference ortholog set, and then retained only those 72 pathways with at least two genes having homeologs with differential expression (as determined by Holm FDR-corrected χ2) for further analysis. We then assigned one of six rank orders (see supplementary fig. S3, Supplementary Material online) to each of these genes and used a custom python script (https://github.com/jsharbrough/pathwayGeneExpression; last accessed March 28, 2017) to calculate the proportion of differentially expressed homeologs within each pathway that shared expression rank order as well as the proportion of homeologs within each pathway that shared the same “highest”, “middle”, and “lowest” expressed homeolog. Our null hypothesis was that by chance, 16.7% (1/6, reflecting the six possible expression profile rank orders) of the genes with differential homeolog expression for a given pathway would share the same expression rank order. We then used Mann–Whitney U tests and one-tailed FETs to test 1) whether genes in a given pathway share expression profiles more often than expected by random chance (16.7%), and 2), genes share expression profiles with a higher percentage of genes in the same pathway than with genes from different pathways.
We next tested whether pathways sharing GO terms would exhibit a higher proportion of similar expression profiles compared with random chance (1/6, reflecting the six possible expression profile rank orders) and compared with pathways not sharing GO terms. We first identified the dominant expression profile, if any, for each of the 72 pathways using a custom Python script (https://github.com/jsharbrough/pathwayGeneExpression; last accessed March 28, 2017). The dominant expression profile was determined by whether a particular profile (e.g., T > Ti > L) was more common than all of the other possible profiles and was exhibited by >1 gene for that pathway. We then used Mann–Whitney U tests to compare the median proportion of shared dominant expression profiles of pathways sharing GO terms to the median proportion of shared dominant expression profiles of all pathways not sharing GO terms and used one-tailed FETs to compare the proportion of observed shared dominant profiles of pathways sharing GO terms to expectations under random chance and to pathways not sharing GO terms.
Comparing Transcriptomes between Sexual and Unisexual Ambystoma
In addition to testing for differential homeolog use on a gene-by-gene basis in the unisexual genome, we asked whether these genes were being expressed in a similar manner across the three sexual species and/or the unisexual. To address this question, we compared the rank order of expression for a set of orthologs among the four taxa. We began by assembling a new set of orthologs that included the unisexual transcriptome, using the same criteria implemented to identify orthologs from the three parental species to expand that search to include the transcriptome from the unisexual. This new ortholog set included 2,036 sets of four orthologous sequences. We then used Tophat2 to map reads from each of the four individuals to their own reference set and quantified gene expression for each sequence via Cufflinks2. We then separately rank-ordered sequences by expression for each of the four taxa. We used Spearman’s rank correlation analyses to compare the order of expression between all six possible pairwise comparisons of the four taxa and calculated the residual values from these six comparisons (see supplementary fig. S4 and table S3, Supplementary Material online). We then used these residuals to conduct outlier analyses (ROUT method, Q = 1%, Prism 6) for each of the six pairwise comparisons of gene expression rank for the four taxa analyzed.
Results
Transcriptome Assembly and Annotation
We obtained 17,355, 18,549, 17,017, and 18,016 transcripts for A. texanum, A. tigrinum, A. laterale, and the unisexual, respectively. Of these transcripts, 72.9%, 72.4%, 64.8%, and 74.4% received both BlastX and GO annotations for A. texanum, A. tigrinum, A. laterale, and the unisexual, respectively. We found that the mean transcript length was similar across transcriptomes (range: 740–781 bp), and the mean number of GO annotations per transcript was nearly identical across all four transcriptome assemblies (7.6–7.7). Our BlastX annotation searches revealed striking similarities across the four transcriptomes in the taxa emerging most often as BlastX hits in general and as top BlastX hits (see supplementary tables S1 and S2, Supplementary Material online). Taken together, these results indicate that our transcriptome assemblies and their annotation are all high quality. Comparisons of the types of genes expressed in each transcriptome suggested that whereas the majority (13/16 biological process level 2; 7/9 molecular function level 4; 6/11 cellular compartment level 4) of GO terms identified across the transcriptomes were the same, there was some variation across the sexual transcriptomes (e.g., A. tigrinum and A. laterale but not A. texanum are expressing genes related to immune system processes) (see supplementary fig. S1, Supplementary Material online). The GO terms present in the unisexual transcriptome appear to be a complete combination (e.g., all 16/16 biological process level 2 GO terms) of the three sexual transcriptomes, expressing all of the GO terms present in any one of the sexual transcriptomes (with the exception of the molecular function of transferase activity, which is only expressed in A. laterale).
Genome Expression Balance
We applied a novel test for evaluating differential homeolog expression in a triploid hybrid salamander and found evidence for overall genome balance across the three homeologous genomes. The majority of genes tested (2,172/2,998; 72.5%) was equally expressed by the three genomes, whereas 826 (27.5%) genes exhibited significant differential expression (fig. 2). There was no evidence for enrichment of any GO terms in this latter set of differentially expressed genes. For each differentially expressed gene, we identified which homeolog was expressed at the highest, middle, and lowest levels for each locus (yielding nine distinct sets of genes, see supplementary fig. S3, Supplementary Material online). This analysis revealed that whereas the Ti genome had the most homeologs expressed in the “highest” category (352), no single subgenome was disproportionately represented in any specific expression rank (fig. 3). We tested all nine sets of genes for GO term enrichment via Blast2GO to determine if any biological functions were linked with particular genomes. We only found one example of this type of connection: GO enrichment of the T “high” gene set (i.e., significant differential gene expression in which the A. texanum genome had the highest expression) had an overrepresentation of genes involved with transcription (see supplementary fig. S5, Supplementary Material online). Together, these data suggest that there is not a consistent type of gene that is more likely to be differentially expressed than other gene types or that a particular genome is most often expressed at the highest, lowest, or middle level. On the basis of these observations we cannot reject a scenario of equal expression from all three genomes, and nor can we support the predictions that the L genome would exhibit dominance at the genome-wide level and/or for genes involved with mitochondrial function.
Fig. 2.—

Proportion of differentially expressed genes within reference transcriptome set of 2,998 genes. Differentially expressed genes demonstrate unequal expression (Holm false discovery—corrected χ2q < 0.05) across the three homeologs. The differentially expressed proportion (826/2998) of the reference ortholog set is organized into which homeolog had the highest expression for a given gene (i.e., T, Ti, or L).
Fig. 3.—

The 826 differentially expressed genes organized according to (A) distribution of subgenomes as the highest, lowest, and middle-ranked homeolog per differentially expressed gene and (B) distribution of homeolog expression rank per subgenome.
Consistent Expression Patterns for Genes in the Same Pathway
Next, we tested whether gene expression regulation among subgenomes better fit a simple model of genome-wide dominance versus a more complicated model that involved epistatic interaction. One expectation of epistasis-mediated gene expression regulation is that proteins functioning in the same biochemical pathway should exhibit similar subgenome expression profiles (e.g., a pathway, rather than a single gene, might be dominated by a T > Ti > L expression pattern). We addressed this hypothesis by comparing the expression profiles of genes within a pathway to those of genes from different pathways using a Wilcoxon Signed-Rank test and then used one-tailed FETs to identify which genes and pathways shared expression profiles more often than expected by random chance (16.7%) and/or more often than expression profiles shared between genes from different pathways. On average, differentially expressed genes share expression profiles with 34.3% of other differentially expressed genes in the same pathway but with only 20.0% of differentially expressed genes in other pathways (Mann–Whitney U = 13,219, P < 2.2e-16, fig. 4A). For all 223 differentially expressed genes for which other genes in the same pathway were also differentially expressed, there were 2,094 shared expression profiles out of 6,104 possible within-pathway combinations (34.3%). This frequency of shared expression profiles for genes in the same pathway is significantly higher both than that expected by random chance (1,017/6,104 = 16.7%, FET Odds Ratio (OR) = 2.61, P < 2.2e-16) and the frequency of shared expression profiles between genes from different pathways (9,157/36,698 (20.0%); FET OR = 2.09, P < 2.2e-16). A significantly higher percentage of differentially expressed genes from different pathways (20.0%) shared expression profiles than expected by random chance (16.7%, FET OR = 1.25, P < 2.2e-16), perhaps indicating epistasis between pathways as well. Pathways sharing GO terms exhibited a similar pattern, sharing a dominant expression profile significantly more often than pathways that did not share that GO term (Mann–Whitney U = 51,912, P < 2.2 × 10−16, fig. 4B). For the 27 pathways exhibiting a dominant expression profile, there were a total of 1,605 shared dominant expression profiles within a GO term out of 2,370 possible (67.7%), which was a significantly higher proportion than the 395 expected by random chance (16.7%, FET OR = 10.49, P < 2.2 × 10−16) and the 7,770/66,005 shared dominant expression profiles between pathways not sharing GO terms (11.8%, FET OR = 15.72, P < 2.2 × 10−16). Pathways that did not share GO terms were significantly less likely to share dominant expression profiles than expected by random chance (FET OR = 0.67, P < 2.2 × 10−16). Together, these data indicate that interacting genes and/or pathways are likely expressed by the same genome, but there is no obvious relationship between expression levels of genes/pathways that do not interact and the subgenome by which they are encoded. This result could reflect a mechanism whereby unisexual Ambystoma are able to dynamically regulate gene expression across subgenomes.
Fig. 4.—
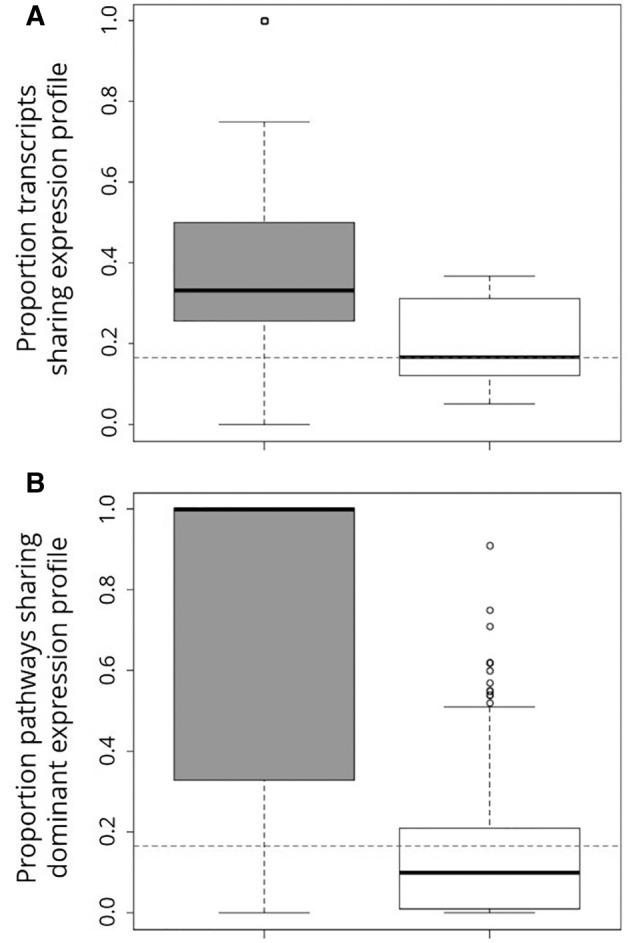
Boxplot depicting relationship between expression profile and shared functional annotation in Ambystoma. (A) Differentially expressed transcripts share expression profiles with a significantly higher proportion of other differentially expressed transcripts within the same biochemical pathway (gray box) than they do with differentially expressed transcripts from other pathways (white box) (Mann–Whitney U = 13,219, P < 2.2 × 10−16). (B) Pathways that share a GO term (gray box) are significantly more likely to share a dominant expression profile than pathways that do not share that GO term (white box) (Mann–Whitney U = 51,912, P < 2.2 × 10−16). For both panels, thick black lines represent median values, boxes represent inner quartile ranges, and whiskers represent outer quartile ranges. The null expectation for genes/pathways sharing an expression rank order by random chance is shown by the dashed horizontal line.
Unisexual Transcriptome Is Expressed Similarly to the Sexual Species
Finally, we addressed whether this unisexual Ambystoma individual exhibited a similar pattern of overall gene expression as the three sexual species. We did this by rank-ordering genes according to expression level for each taxon and using correlation and residual value analyses to compare these ranks. We found a significant and positive correlation between the expression orders of all four taxa (see supplementary fig. S4 and table S3, Supplementary Material online). When we rank ordered these six Spearman’s rho values, the three unisexual–sexual comparisons had higher rho values and lower absolute residual values than the three sexual–sexual comparisons (see supplementary table S3, Supplementary Material online). These results indicate both that unisexual Ambystoma expresses its genes in a similar manner as do its three sexual progenitor species and that gene expression in the unisexual is not more or less similar to any particular sexual species. This result adds support to our conclusion of genome balance among the subgenomes for hybrid unisexual Ambystoma. This same conclusion is suggested by our outlier analysis (ROUT method, Q = 1%, Prism 6) of the residual values for each comparison of gene expression rank order, which did not reveal any outliers that were consistently above or below the regression line for the unisexual versus sexual species comparisons.
Discussion
We applied a novel approach for evaluating differential homeolog expression in a triploid hybrid relative to its three diploid progenitor species and found evidence for a general pattern of genome balance across the three homeologous genomes of a unisexual salamander. Most (∼72%) genes analyzed in the unisexual show equivalent expression, supporting the presence of a gene balance mechanism regulating expression in this allopolyploid animal. Those genes that are differentially expressed are more likely to share their expression pattern with genes in the same pathway than with genes in different pathways. For each subgenome, overall patterns of gene expression in the unisexual individual were similar to those observed in the sexual species. We discuss the evolutionary significance of these results below.
Balanced Gene Expression across Subgenomes
The majority of genes analyzed displayed a pattern of balanced expression, where an equal number of homeologs were expressed from each subgenome of the triploid salamander. This result differs from the few existing reports documenting differential gene expression in other polyploid hybrid animals. For example, whereas the synthetic triploid hybrid fish O.latipes shows a superficially similar percentage of genes expressed equally among subgenomes (82%; Garcia et al. 2014), the remaining differentially expressed genes in O. latipes indicate broad-scale gene silencing. By contrast, our analysis revealed no evidence of subgenome-specific gene silencing in the unisexual Ambystoma and/or that expression is partitioned to particular functional gene sets. Gene silencing is also evident in another hybrid polyploid fish, S.alburnoides, with the silenced genes varying by genomic composition and geographic location of the polyploid (Pala et al. 2008, 2010). Squalius alburnoides is distinguished by a variety of reproductive modes (clonal, gynogenetic, hybridogenetic) and multiple hybrid origins. By contrast, unisexual Ambystoma are a relatively ancient and all-female lineage (∼6 Ma compared with <0.7 Ma; Cunha et al. 2011) with a single hybrid origin and a “leaky” form of gynogenesis. Whereas each of these three vertebrate groups is defined by their polyploidy, none has acquired their genomes in the same manner or at the same rate. One potential explanation for what appears to be an unusual level of genome balance in unisexual salamanders is a relatively high level of gene exchange between the unisexuals and sexual males. The rate of genome introgression from sexual Ambystoma males into the unisexual lineage is estimated to be ∼0.2% genomes/generation (Gibbs and Denton 2016), whereas the frequency of introgression (if any) from sexual to unisexual fish is estimated to be much lower, in the order of thousands of years (Lampert et al. 2005; Sousa-Santos et al. 2006). Because differential expression can result from isolation and neofunctionalization of allopolyploid genome components over evolutionary time scales (Wendel 2000; Wang et al. 2012), the relatively high rate of introgression of foreign genomes into the unisexual Ambystoma lineage may limit the opportunity for extensive genome dominance or silencing to evolve.
Differentially Expressed Genes Are Consistent within Functional Pathways
Genes that were differentially expressed among the subgenomes of the unisexual Ambystoma salamander were more likely to share the same pattern of expression with other genes in the same functional pathway compared with genes in different pathways. This result suggests that epistatic interactions may play a role in gene expression regulation in unisexual Ambystoma. Because allopolyploids like the unisexual Ambystoma cannot purge incompatible gene combinations through Mendelian segregation (reviewed in Rieseberg 2001), the co-expression of genes that make up functional pathways may be favored if this co-expression decreases negative consequences of genomic incompatibilities. A pathway-specific mode of differential expression is consistent with the ability of unisexual Ambystoma to express genomes from a diversity of species (Bogart et al. 2009). Importantly, epistasis-mediated gene expression regulation would require gene expression to be predominantly regulated in trans, possibly due to divergence in transcription factor binding sites. This mechanism could also contribute to the notable longevity of the unisexual Ambystoma lineage by increasing the likelihood of complementary pairing of different nuclear genomes and reducing the likelihood of deleterious mitonuclear mismatch (Bi and Bogart 2010).
Unisexual Subgenomes Show Similar Expression to Sexual Species
Comparative analyses of rank orders showed that the subgenomes within the unisexual individual expressed genes in approximately the same rank order as each sexual species. This result supports a scenario in which sexual genomes, when introgressed into the unisexual lineage, continue to be expressed at similar levels. This result is in contrast to many plant polyploids, in which changes in expression among subgenomes can appear immediately in F1 hybrids (Adams and Wendel 2005) and can be maintained in subsequent generations (Hegarty et al. 2006). Whereas we have no specific information about how many generations the genomes within the unisexual salamander have been isolated in the unisexual lineage, it is likely that the T-genome has been isolated within the specific unisexual salamander in this study for less time than the L- and Ti-genomes because this unisexual was captured from an island population where only unisexuals and A. texanum are present (Kelley’s Island, Ohio; Bogart et al. 1987). Estimates of gene flow from sexual Ambystoma into sympatric unisexual populations can be as high as 0.2% genome/per generation (Gibbs and Denton 2016), suggesting that the T-genome in this unisexual salamander has likely been introgressing from local A. texanum since the formation of Kelley’s Island during the last glacial retreat (∼20,000 ybp) whereas introgression of the other two genomes has been more limited or absent. Even though the amount of time since each of the parental genomes was captured into the unisexual lineage may differ, the subgenomes still maintain a similar rank order of gene expression compared with sexual individuals, suggesting that the expression profiles of captured genomes may be stable over evolutionarily significant periods of time in isolation.
Mitochondrial and nuclear genomes that share cellular environments for greater periods of time are more likely to have evolved relatively efficient mitonuclear interactions when compared with hybrid species with novel combinations of mitochondrial and nuclear genotypes (Sambatti et al. 2008; Arnqvist et al. 2010; Chou and Leu 2015). Whereas relatively long residence time for a genome from a sexual ancestor that is now isolated in the unisexual lineage might predict a greater reliance on that genome for mitonuclear interactions, we found no global or functional differences between expression of the unisexual subgenomes when compared with the sexuals. Because the rate of genome introgression into unisexual Ambystoma is relatively high compared with other unisexual vertebrates, there may be strong selection on unisexuals to maintain the expression profile of introgressed genomes (Charney 2012). In particular, if introgressed genomes initiated sweeping changes in expression (gene/genome dominance or silencing) similar to those observed in many polyploid plant hybrids, the removal of a dominant genome during the kleptogenesis that characterizes unisexual Ambystoma might lead to particularly negative consequences. On the basis of our knowledge of kleptogenesis in Ambystoma and the expression results presented here, unisexual Ambystoma seem to employ a flexible method of expression that could maintain the functionality of “stolen” genomes long after genome exchange occurs.
Analysis of Expression in Polyploid Organisms
Bioinformatic approaches for comparative expression analyses in polyploids have been designed for autopolyploids, allopolyploids harboring no more than two parental genomes (Matos et al. 2015), or trihybrids with diagnostic SNPs (Garcia et al. 2014). None of these existing methods can be applied to the many allopolyploid organisms like unisexual Ambystoma with three or more distinct nuclear genomes. The method described here allows for the comparison of expression profiles (from individual genes to whole transcriptomes) among polyploids with three or more genomes without reliance on diagnostic SNPs. Our approach requires either extensive genomic resources (i.e., high-quality draft genome assembly) or transcriptome data of the sexual species from which the polyploid genomes are derived in order to establish orthologous reference sets. The latter requirement is similar to the application of diagnostic SNP-based methods, which require the data needed to provide the SNPs necessary to differentiate sequences among parental genomes.
To be clear, the new methodology presented here is a measure of relative (rather than absolute) expression differences among subgenomes within a polyploid individual. This limitation means that we cannot yet assess whether equal expression among most genes is scaled to match the expression levels of the same genes in the diploid sexual relatives, a process of decreasing gene redundancy during transcription in polyploids (diploidization) documented in both plants (Birchler and Veitia 2007) and other animals (see Ching et al. 2010 for an example in salmon). Another important limitation of our analysis is its focus on a single unisexual individual, which permits only tentative extrapolation to the diverse array of biotypes in unisexual Ambystoma. Additional transcriptome comparisons between unisexuals with varying numbers of genomes from each sexual species could shed light on the contribution of dosage effects to genome balance in unisexual Ambystoma.
Future Directions
In this study, we provide an intuitive analytical solution for conducting comparisons of gene expression within allopolyploid genomes harboring >2 parental genomes, a scenario most common in relatively understudied vertebrate polyploids. Our results set the stage for addressing multiple additional questions in unisexual Ambystoma. First, evaluating whether patterns in expression vary across the same biotypes will provide important additional insights into the consequences of allopolyploidy for gene expression in animals. A good means of addressing this question will come from the establishment of gene expression patterns of a single, widespread biotype (e.g., two A. laterale genomes and one Ambystomajeffersonianum; Bogart and Klemens 2008) in comparison to the local sympatric sexual species, which would provide a fine-scale picture of how similar individual genomes perform in comparison to the sexual population from which they are derived. Second, does ploidy or genome diversity have greater impact on expression patterns? Expression data from unisexual biotypes that vary in ploidy could reveal if expression patterns change when >1 copy of a particular genome is present (e.g., one A. laterale genome and two A. jeffersonianum versus one A. laterale genome and three A. jeffersonianum genomes). Third, whether the patterns we have revealed in this study will also apply to different genome compositions will also provide critical information regarding the genomic and phenotypic consequences of hybridization and polyploidy and could be rigorously addressed by applying similar analyses to those described here to unisexual Ambystoma across a spectrum of ploidies and genome combinations. Finally, our results do not explain why the A. laterale genome is ubiquitous in the unisexual lineage. The fact that the L genome is present in all unisexuals has led to the suggestion that this genome plays a fundamental role in determining the unisexual phenotype. Such a critical effect of the A. laterale genome could be manifested as dominance in the expression of genes or a reliance on this genome for fundamental cellular processes (Bi et al. 2008). Our results show no support for this explanation: most genes are equally expressed with respect to genome identity, and the limited number of genes that show biased expression are from the A. tigrinum and not A. laterale subgenome. As a first step in further exploring this issue, additional studies of unisexuals with other genome combinations are needed to determine if the dominance by the L genome is a general phenomenon versus being biotype specific.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank K. Greenwald and J. Bogart for help with procuring salamanders, and A. Hernandez, X. Bai, and A. Michel for early discussions about project design. Salamanders were held in captivity under Ohio State IACUC protocol 2012A00000039, and we thank M. Parsley and M. Saccucci for assistance with caring for captive animals. This work was supported by the Ohio State University. Transcriptome sequencing was conducted at the Roy J. Carver Biotechnology Center, University of Illinois using funds from the Ohio State University. This work was also supported by the National Science Foundation under Grant No. NSF-MCB 1122176 to L.B., K.E.M, M.N., J.S., the University of Iowa Graduate College to L.B. and K.E.M., the Evelyn Hart Watson Fellowship to K.E.M, J.S., and the National Center for Science Education to K.E.M.
Literature Cited
- Adams KL, Wendel JF. 2005. Novel patterns of gene expression in polyploid plants. Trends Genet. 21:539–542. [DOI] [PubMed] [Google Scholar]
- Alves MJ, Coelho MM, Collares-Pereira MJ. 2001. Evolution in action through hybridization and polyploidy in an Iberain freshwater fish: a genetic review. Genetica 111:375–385. [DOI] [PubMed] [Google Scholar]
- Arnqvist G, et al. 2010. Genetic architecture of metabolic rate: Environment specific epistasis between mitochondrial and nuclear genes in an insect. Evolution 64:3354–3363. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/; last accessed March 28, 2017.
- Barker MS, Husband BC, Pires JC. 2016. Spreading Winge and flying high: the evolutionary importance of polyploidy after a century of study. Am J Bot. 103:1139–1145. [DOI] [PubMed] [Google Scholar]
- Bi K, Bogart JP. 2010. Time and time again: unisexual salamanders (genus Ambystoma) are the oldest unisexual vertebrates. BMC Evol Biol. 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Bogart JP, Fu J. 2008. The prevalence of genome replacement in unisexual salamanders of the genus Ambystoma (Amphibia, Caudata) revealed by nuclear gene genealogy. BMC Evol Biol. 8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA. 2007. The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell. 19:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Yao H, Chudalayandi S, Vaiman D, Veitia RA. 2010. Heterosis. Plant Cell. 22:2105–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogart JP, Klemens MW. 2008 Nov. Additional distributional records of Ambystoma laterale, A. jeffersonianum (Amphibia: Caudata) and their unisexual kleptogens in northeastern North America. American Mus. 3627:1–58. [Google Scholar]
- Bogart JP, Lowcock L, Zeyl CW. 1987. Genome constitution and reproductive biology of hybrid salamanders, genus Ambystoma, on Kelleys Island in Lake Erie. Can J Zool. 65:2188–2201. [Google Scholar]
- Bogart JP, Bartoszek J, Noble DWA, Bi K. 2009. Sex in unisexual salamanders: discovery of a new sperm donor with ancient affinities. Heredity 103:483–493. [DOI] [PubMed] [Google Scholar]
- Bogart JP, Bi K, Fu J, Noble DWA, Niedzwiecki JH. 2007. Unisexual salamanders (genus Ambystoma) present a new reproductive mode for eukaryotes. Genome 50:119–136. [DOI] [PubMed] [Google Scholar]
- Buggs RJ, Soltis PS, Mavrodiev EV, Symonds VV, Soltis DE. 2008. Does phylogenetic distance between parental genomes govern the success of polyploids? Castanea 73:74–93. [Google Scholar]
- Buggs RJ, et al. 2014. The legacy of diploid progenitors in allopolyploid gene expression patterns. Proc Roy Soc Lond B. 369:20130354.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney ND. 2012. Relating hybrid advantage and genome replacement in unisexual salamanders. Evolution 66:1387–1397. [DOI] [PubMed] [Google Scholar]
- Ching B, Jamieson S, Heath JW, Heath DD, Hubberstey A. 2010. Transcriptional differences between triploid and diploid Chinook salmon (Oncorhynchus tshawytscha) during live Vibrio anguillarum challenge. Heredity 104:224–234. [DOI] [PubMed] [Google Scholar]
- Chou JY, Leu JY. 2015. The Red Queen in mitochondria: cyto-nuclear co-evolution, hybrid breakdown and human disease. Front Genet. 6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3673. [DOI] [PubMed] [Google Scholar]
- Cunha C, Doadrio I, Abrantes J, Coelho MM. 2011. The evolutionary history of the allopolyploid Squalius alburnoides (Cyprinidae) complex in the northern Iberian Peninsula. Heredity 106:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RD, Kenyon LJ, Greenwald KR, Gibbs HL. 2014. Evolutionary basis of mitonuclear discordance between sister species of mole salamanders (Ambystoma sp.). Mol Ecol. 23:2811–2824. [DOI] [PubMed] [Google Scholar]
- de Godoy LMF, et al. 2008. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455:1251–1254. [DOI] [PubMed] [Google Scholar]
- Dowling T, Secor C. 1997. The role of hybridization and introgression in the diversification of animals. Ann Rev Ecol Syst. 28:593–619. [Google Scholar]
- Feldman M, Levy AA, Fahima T, Korol A. 2012. Genomic asymmetry in allopolyploid plants: wheat as a model. J Exp Bot. 63:5045–5059. [DOI] [PubMed] [Google Scholar]
- Garcia TI, et al. 2014. Novel method for analysis of allele specific expression in triploid Oryzias latipes reveals consistent pattern of allele exclusion. PLoS ONE. 9:e100250.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs HL, Denton RD. 2016. Cryptic sex? Estimates of genome replacement in asexual mole salamanders (Ambystoma sp.). Mol Ecol. 25:2805–2815. [DOI] [PubMed] [Google Scholar]
- Gordon A, Hannon G. 2010. Fastx-toolkit. FASTQ/A short-read pre-processing tools (unpublished). http://hannonlab.cshl.edu/fastx_toolkit/.
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald KR, Gibbs HL. 2012. A single nucleotide polymorphism assay for the identification of unisexual Ambystoma salamanders. Mol Ecol. 12:354–362. [DOI] [PubMed] [Google Scholar]
- Gregory TR, Mable BK. 2005. Polyploidy in animals In: Gregory TR, editor. The evolution of the genome. London: Elsevier; p. 427–517. [Google Scholar]
- Haas BJ, et al. 2013. De novo transcript sequence reconstruction from RNA-seq, using the Trinity platform for reference generation and analysis. Nat Protocol. 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty MJ, et al. 2006. Transcriptome shock after interspecific hybridization in Senecio is ameliorated by genome duplication. Curr Biol. 16:1652–1659. [DOI] [PubMed] [Google Scholar]
- Huang Y, Niu B, Gao Y, Fu L, Li W. 2010. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert KP, Lamatsch DK, Epplen JT, Schartl M. 2005. Evidence for a monophyletic origin of triploid clones of the Amazon molly, Poecilia formosa. Evolution 59:881–889. [PubMed] [Google Scholar]
- Li C-Y, et al. 2014. The transcriptomes of the crucian carp complex (Carassius auratus) provide insights into the distinction between unisexual triploids and sexual diploids. Int J Mol Sci. 15:9386–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mable BK, Alexandrou M, Taylor MI. 2011. Genome duplication in amphibians and fish: an extended synthesis. J Zool. 284:151–182. [Google Scholar]
- Madlung A. 2012. Polyploidy and its effect on evolutionary success: old questions revisited with new tools. Heredity 110:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos I, Machado MP, Schartl M, Coelho MM. 2015. Gene expression dosage regulation in an allopolyploid fish. PLoS ONE. 10:e0116309.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman M, Kay AD, Krist AC. 2013. Sensitivity to phosphorus limitation increases with ploidy level in a New Zealand snail. Evolution 67:1511–1517. [DOI] [PubMed] [Google Scholar]
- Neiman M, Theisen KM, Mayry ME, Kay AD. 2009. Can phosphorus limitation contribute to the maintenance of sex? A test of a key assumption. J Evol Biol. 22:1359–1363. [DOI] [PubMed] [Google Scholar]
- Otto SP. 2007. The evolutionary consequences of polyploidy. Cell 131:452–462. [DOI] [PubMed] [Google Scholar]
- Otto SP, Whitton J. 2000. Polyploid incidence and evolution. Ann Rev Genet. 34:401–437. [DOI] [PubMed] [Google Scholar]
- Pala I, Coelho MM, Schartl M. 2008. Dosage compensation by gene-copy silencing in a triploid hybrid fish. Curr Biol. 18:1344–1348. [DOI] [PubMed] [Google Scholar]
- Pala I, Schartl M, Brito M, Malta Vacas J, Coelho MM. 2010. Gene expression regulation and lineage evolution: the North and South tale of the hybrid polyploid Squalius alburnoides complex. Proc Roy Soc Lond B. 277:3519–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey J, Ramsey TS. 2014. Ecological studies of polyploidy in the 100 years following its discovery. Phil Trans Roy Soc Lond B. 369:1–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH. 2001. Polyploid evolution: keeping the peace at genomic reunions. Curr Biol. 11: 925–928. [DOI] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. 2011. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12:R22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambatti JBM, Ortiz-Barrientos D, Baack EJ, Rieseberg LH. 2008. Ecological selection maintains cytonuclear incompatibilities in hybridizing sunflowers. Ecol Lett. 11:1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedina IM, Hartmann S, Groth D, Schlupp I, Tiedemann R. 2014. Comparative analysis of the gonadal transcriptomes of the all-female species Poecilia formosa and its maternal ancestor Poecilia mexicana. BMC Res Note. 7:249.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE. 2000. The role of genetic and genomic attributes in the success of polyploids. Proc Natl Acad Sci U S A. 97:7051–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Santos C, Collares-Pereira M, Almada V. 2006. Evidence of extensive mitochondrial introgression with nearly complete substitution of the typical Squalius pyrenaicus-like mtDNA of the Squalius alburnoides complex (Cyprinidae) in an independent Iberian drainage. J Fish Biol. 68:292–301. [Google Scholar]
- Te Beest M, et al. 2012. The more the better? The role of polyploidy in facilitating plant invasions. Ann Bot. 109:9–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Maere S, Meyer A. 2009. The evolutionary significance of ancient genome duplications. Nat Rev Genet. 10:725–732. [DOI] [PubMed] [Google Scholar]
- Vanneste K, Maere S, Van de Peer Y. 2014. Tangled up in two: a burst of genome duplications at the end of the Cretaceous and the consequences for plant evolution. Phil Trans Roy Soc Lond B. 369:20130353.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang X, Paterson AH. 2012. Genome and gene duplications and gene expression divergence: a view from plants. Ann NY Acad Sci. 1256:1–14. [DOI] [PubMed] [Google Scholar]
- Wendel JF. 2000. Genome evolution in polyploids. Plant Mol Biol. 42:225–249. [PubMed] [Google Scholar]
- Wertheim B, Beukeboom LW, Van De Zande L. 2013. Polyploidy in animals: effects of gene expression on sex determination, evolution and ecology. Cytogenet Gen Res. 140:256–269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.