

Summary
Classic cardio-metabolic risk factors such as hypertension, stroke, diabetes and hypercholesterolemia all increase the risk of Alzheimer’s disease. We found increased transcription of β-secretase/BACE1, the rate-limiting enzyme for Aβ generation, in eNOS deficient mouse brains and after feeding mice a high fat high cholesterol diet. Up- or down-regulation of PGC-1α reciprocally regulated BACE1 in vitro and in vivo. Modest fasting in mice reduced BACE1 transcription in the brains which was accompanied by elevated PGC-1 expression and activity. Moreover, the suppressive effect of PGC-1 was dependent on activated PPARγ likely via SIRT1-mediated deacetylation in a ligand-independent manner. The BACE1 promoter contains multiple PPAR/RXR sites and direct interactions among SIRT1-PPARγ-PGC-1 at these sites were enhanced with fasting. The novel interference on the BACE1 gene identified here represents a unique non-canonical mechanism of PPARγ-PGC-1 in transcriptional repression in neurons in response to metabolic signals which may involve recruitment of a corepressor NCoR.
Introduction
Alzheimer’s disease (AD) is one of the most devastating neurodegenerative disorders which is characterized by the two pathological hallmarks of amyloid plaques and neurofibrillary tangles. Amyloid peptides (Aβ), the major constituent of plaques, are generated by sequential proteolytic cleavage of the amyloid precursor protein (APP) via β-secretase (BACE1) and the γ-secretase complex (Hardy and Selkoe 2002). The expression and activity levels of BACE1 are elevated in AD brains and correlate with the specific regions affected by Aβ deposition. Taken together with the observation that BACE1-deficient mice display diminished amyloid pathology (Vassar et al., 2009), current evidence strongly suggests that BACE1 elevation leads to enhanced Aβ production and deposition in AD. Given the central role of Aβ in AD pathogenesis and the fact that BACE1 is the rate-limiting enzyme in APP processing and Aβ generation, BACE1 remains one of the most important therapeutic targets for treating AD.
BACE1 expression is tightly regulated at multiple levels between transcription and post-translation (Rossner et al., 2006). A number of transcription factors (TFs) have been identified that positively or negatively regulate BACE1 gene expression in both basal (Sp1, YY1 and HNF3β) (Ge et al., 2004; Sun et al., 2005) and cell-stress conditions (e.g., HIF-1α during hypoxia and NF-KB and PPARγ with inflammation) (Gulielmotto et al., 2012). We recently reported differential regulation of BACE1 by oxidative and nitrosative signals (Kwak et al., 2011), both of which are common denominators in age-related diseases.
While the molecular mechanism underlying ischemia/hypoxia or reactive oxygen species (ROS)-induced BACE1 activation and APP processing has been extensively studied, relatively little is known about BACE1 regulation by metabolic stress. The vast majority of AD cases are late onset and sporadic (SAD) in origin with age being the most profound risk factor. Multiple environmental factors, such as diet and life style, along with genetic factors are all significant contributors. Epidemiological, clinical and experimental evidence strongly link metabolic defects with functional alterations associated with aging of the brain and with AD pathogenesis. Thus, classic cardio-metabolic risk factors such as hypertension, cerebral hypo-perfusion/stroke, diabetes mellitus and hypercholesterolemia have been shown to increase the risk of SAD (Bhat 2010; Craft 2009; de la Torre 2009; Martins et al., 2006).
In this study, we sought to investigate the potential role of the sirtuin 1 (SIRT1) – peroxisome proliferator-activated receptor gamma (PPARγ) and its coactivator (PGC-1) pathway in regulating BACE1 expression. Using in vitro and in vivo experimental systems mimicking metabolic stress (e.g., glucose depletion, hyperglycemia/high cholesterol/HFC diet and fasting), our work demonstrates the potent suppressive effects of these key regulators on BACE1 transcription in response to cellular metabolic status.
Results
Elevated BACE1 transcription by feeding a HFC diet or eNOS deficiency
Disrupted cholesterol homeostasis causes excessive Aβ generation (Pugielli et al., 2003). Previously we observed that young C57BL/6 mice consuming a HFC diet which contained 21% milk fat and 1.25% cholesterol for 8 weeks demonstrated learning/memory deficits and neuroinflammation (Thirumangalakudi et al., 2008). Since high cholesterol is the biggest risk factor for SAD in middle-aged individuals, we repeated 8-week HFC feeding regimen on middle-aged mice.
BACE1 gene and protein expression levels were found to be over 2-fold increased in the mouse forebrains after HFC feeding (Figures 1A and 1B). Furthermore, we found that both PGC-1α and β mRNA levels were significantly reduced following the HFC (Figure 1C). BACE1 reduction appeared to be at the transcription level, since we did not detect significant changes in the protein level of GGA3, a key regulator of BACE1 trafficking and lysosomal degradation (Tesco et al., 2007) or p-eIF2α (O’Connor et al., 2008) (Figure S1).
Figure 1. HFC feeding or eNOS deficiency increases BACE1 expression.

(A) Western blot analysis of BACE1 protein expression from the forebrain extracts of mice fed HFC and control chow diets (n=5/group; 9 months of age). (B) Quantification of panel A by densitometry. (C) Relative mRNA levels of BACE1 and PGC-1α determined by qRT-PCR from the same forebrain samples as for panel A. (D) Relative forebrain BACE mRNA levels of mice with partial and complete eNOS deficiency (n=5/group). Western blot of eNOS protein expression is shown on the top panel. * indicates P < 0.05.
Recently, we demonstrated that BACE1 is highly susceptible to nitric oxide (NO)-mediated regulation. NO generated by the endothelial NO synthase (eNOS) suppresses BACE1 transcription via cGMP/PKG signaling (Kwak et al., 2011). Consistent with this, BACE1 protein expression in the brains of eNOS-deficient mice was found to be elevated (Austin et al., 2010). We observed that the frontal brains of 4-month-old eNOS-/- mice displayed significantly increased mRNAs of BACE1 (Figure 1D) while heterozygous mice did not show significant alteration.
Fasting reduces BACE1 expression in mice
Calorie restriction can activate the AMPK-SIRT1-PGC-1 pathway (Qin et al., 2006). We therefore examined the effect of fasting on BACE1 levels in mouse brains and found BACE1 expression was significantly reduced at the 24 h time point (Figures 2A and 2B), accompanied by marked activation of AMPK and increased PGC-1α/β (Figures 2C and 2D). This inversed correlation between BACE1 and PGC-1 was also detected at the mRNA levels (Figure 2E). Fasting for 24 h resulted in > 85 mg/dl blood glucose levels which are still within the normal range (Figure S2).
Figure 2. Fasting reduces BACE1 expression in mouse brains.
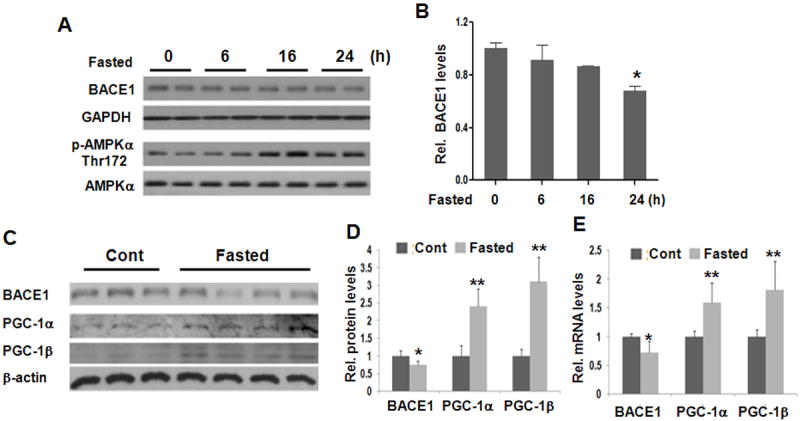
(A and B) Fasting reduces BACE1 expression in mouse forebrains at 24 h time point as determined by Western blot (n=2/time point). (C) Western blot analysis of BACE1 and PGC-1α/β protein in mouse forebrain after 24 hrs of fasting. (D) Quantification of Westerns in panel C. (E) mRNA abundance of BACE1 and PGC-1α/β. Data are represented as means ± S.D., N= 3-5 mice/group; * indicates P< 0.05.
PGC-1 regulates BACE1 in vitro and in vivo
Since AMPK and PGC-1 can both be activated by cGMP/PKG signal transduction (Nisoli et al., 2003), we investigated whether these key metabolic regulators have a direct role in suppressing BACE1. In HEK293 cells, we found that overexpression of PGC-1α suppressed basal transcription of endogenous BACE1 messenger RNA, resulting in a 2-fold reduction of mRNA and protein levels (Figures 3A and 3B). On the other hand, downregulation of either PGC-1α (52 %) or PGC-1β (38 %) gene transcription in rat primary cortical neurons, via specific siRNA delivered from adenoviral transduction, resulted in greater than 2-fold upregulation of BACE1 protein expression (Figures 3C and 3D). A direct role for PGC-1 in suppressing BACE1 expression was also further confirmed by in vivo approaches. Overexpression of PGC-1α (>7-fold) via AAV2-mediated gene transfer severely diminished BACE1 protein expression in the hippocampi of Tg2576 (a FAD model overexpressing an APPSwedish mutant, while downregulation of PGC-1α (1.9-fold) augmented BACE1 expression 2.6-fold WT C57BL/6 mice (Figures 3E and 3F). The effects were confirmed to be on BACE1 transcription as evidenced by the altered mRNA levels (Figure 3G). We found comparable effects of PGC-1α and PGC-1β in suppressing BACE1 upon transfection, indicating that they are interchangeable.
Figure 3. Suppression of BACE1 expression in vitro and in vivo by PGC-1.

(A) Effects of overexpressed myc tagged-PGC-1α on BACE1 protein expression. Protein levels were measured in HEK293 cells 48 h after transient transfection (2 μg) by Western blot analysis. The BACE1 monoclonal antibody (3D5) used was raised in BACE1-null mice. PGC-1α protein was detected by an anti-Myc antibody. Protein levels in the left panel were quantified by densitometry analysis (right panel). (B) Effect of overexpressed PGC-1α on the endogenous levels of BACE1 mRNA as determined by qRT-PCR 48 h after transfection. (C) Downregulation of PGC-1 (α or β) by shRNA upregulates BACE1 expression in primary neurons. Rat primary neurons (DIV10) were infected with PGC-1 shRNA adenovirus for 48 hour; BACE1 protein expression was detected by Western blot and quantified based on data from 4 experiments. Infection efficiency was monitored by the EGFP signals under microscope and by qRT-PCR. (D) PGC-1α and β mRNA levels following knockdown are shown. (E) Effects of PGC-1α overexpression and downregulation on BACE1 expression in vivo. AAV2-PGC-1α or AAV2-EGFP (vehicle, 10 ˆ9 virus particles in 1 μl) were injected into the CA1 region of Tg2576 mouse hippocampus (N=5/group, 5 month-old) and BACE1 protein was determined by Western blot analysis four weeks later. (F) AAV2-PGC-1α-shRNA and AAV2-EGFP (10ˆ10 viruses particles in 1 μl) were injected into the CA1 of C57BL/6 mice (n=5/group; 4 month-old) and BACE1 level was determined four weeks later. (G) The mRNA levels of BACE1 and PGC-1α in hippocampi as determined from the same animals for the studies from panels E and F. No cytotoxicity was detected at the time of the in vitro assays. Data are represented as means ± S.D. from 5-7 independent experiments; * and ** indicate P< 0.05 and P< 0.01, respectively.
PGC-1’s effect on BACE1 requires deacetylation by SIRT1
As with elevating PGC-1 levels, overexpression of SIRT1 suppressed BACE1 transcription while deacetylase inactive (DN) H355A SIRT1 had the opposite effect in HEK293 cells (Figures 4A and 4B). Treatment with the SIRT1 activating agent, resveratrol, repressed BACE1 transcription in a dose-dependent manner (Figure 4C); concentrations greater than 50 μM caused cytotoxicity in primary cultured neurons. On the contrary, the SIRT inhibitor, sirtinol, significantly augmented BACE1 levels at 10 μM (Figure 4D and 4E). BACE1 expression was upregulated 1.8-fold (Figure 4D) by 5 mM nicotinamide. Notably, coexpression of PGC-1 and the DN-SIRT1 plasmid constructs in HEK293 cells completely abolished the suppressive effect mediated by PGC-1 on BACE1 protein levels (Figure 4E). DN-SIRT1 coexpression was found to increase PGC-1 acetylation. Under an extreme “no glucose/NG” condition, which is known to cause neuronal toxicity, BACE1 transcription and expression was found to be elevated, accompanied by decreased expression levels of SIRT1 and PGC-1 as well as PGC-1 activity (increased acetylation); overexpression of PGC-1 completely reversed the detrimental effect of NG on BACE1 (Figure S4).
Figure 4. PGC-1 suppression of BACE1 requires SIRT1 deacetylase activity.
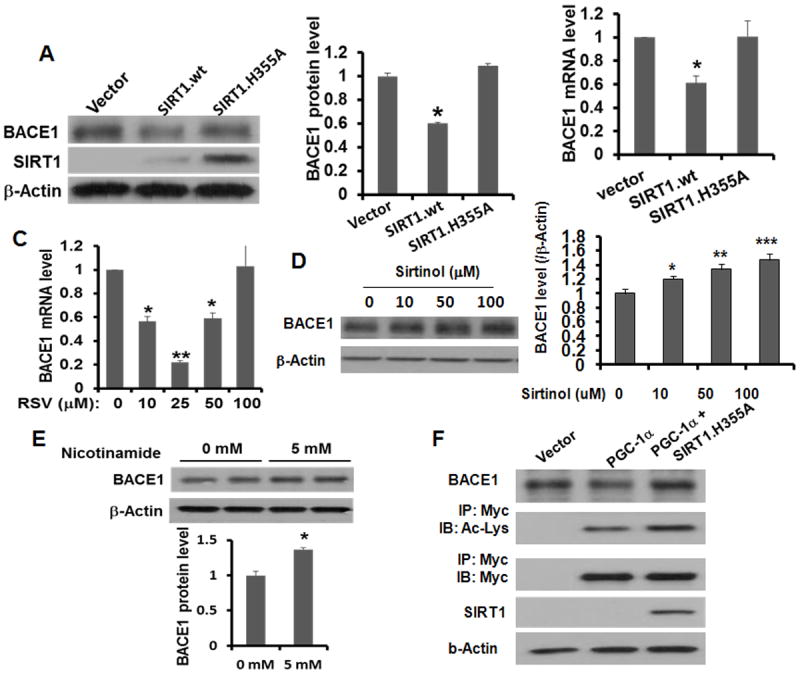
(A) Effects of overexpressed wild type (WT) and a dominant-negative (DN) mutant SIRT1 (SIRT.H355A) on BACE1 protein expression HEK293 cells. Protein levels were detected in HEK293 cells 48 h after transient transfection. (B) The endogenous BACE1 mRNA levels upon SIRT1 transfection were determined by qRT-PCR and quantified based on 3 independent experiments. (C, D, E) Effects of resveratrol, sirtinol and nicotinamide at various concentrations on BACE1 mRNA or protein levels in primary neurons 6 h after treatment. Data are represented as means ± S.D. from 3 independent experiments with * indicating P< 0.05; ** indicating P< 0.01. (F) Effect of SIRT1-mediated deacetylation of PGC-1β on BACE1 suppression. Myc-tagged PGC-1β was cotransfected with DN SIRT1 in HEK293 cells and protein levels were determined 48 h later. PGC-1 acetylation was detected after immunoprecipitation with an anti-Myc antibody and then probed with anti-Acetylated Lys antibody (Cell Signaling).
Recently, SIRT1 was also reported to activate PPARγ via deacetylation on Lys 268 and 293 in “browning” adipocytes (Qiang et al., 2012). Since PGC-1 was classically defined as the co-activator to PPARγ, we also investigated the role of PPARγ and its deacetylation in BACE1 suppression. In cultured neurons, treatment with rosiglitazone (PPARγ ligand) and GW9662 (irreversible antagonist) led to opposing effects on BACE1 expression in dose-dependent manner (Figure 5A). Interestingly, PGC-1’s suppressive effect was largely abolished by GW9662, but unaffected by rosiglitazone, indicating a ligand-independent mechanism (Figure 5B). In 24-h fasted mouse brains, the acetylated forms of both PGC-1 and PPARγ were reduced (Figure 5C). Notably, overexpression of non-acetylated PPARγ (K268T and/or K293T), which represents the active form, repressed BACE1 transcription to a similar degree as PGC-1 or SIRT1 in a lignad-independent mammer (Figure 5D). Surprisingly, overexpression of WT PPARγ did not exert a significant effect on BACE1, indicating that the molecule is not limiting in neurons. In addition, coexpression of SIRT1 with PPARγ resulted in a similar effect as PGC-1 or mutant PPARγ. However, coexpression of PGC-1 with WT SIRT1 did not exert a further additive effect. Similar results were not only observed on the endogenous BACE1 mRNAs, but also on the transfected rBACE1-Luc reporter assay (Figure S5). Taken together, these data strongly indicate that SIRT1 is the upstream regulator of PPARγ and PGC-1.
Figure 5. PGC-1’s suppressive effect requires active PPARγ and involves SIRT1-mediated deacetylation.

(A) Opposing effects of rosiglitazone and GW9662 on BACE1 expression levels. Western blot analysis was conducted at 6 hr after treatment in neurons. (B) PGC-1α effect is dependent on active PPARγ but not on its ligand. Rosiglitazone or GW9662 compounds were added to the HEK293 cells 24 hr after transfection with PGC-1α plasmid and BACE1 gene transcription was determined 6h later by qRT-PCR. Data are represented as means ± S.D. from 3 independent experiments with * indicating P< 0.05. (C) Acetylation status of PPARγ, PGC-1α and PGC-1β was determined by Western. (D) PPARγ represses BACE1 in its active, non-acetylated form. Various plasmids were transfected, alone or in combination, into HEK293 cells and BACE1 qRT-PCR was conducted 24 hr later. For panels C and D, data are represented as means ± S.D. from 3 independent experiments with * indicating P< 0.05 and ** indicating P<0.01. “n.s.” indicates non-statistically significant.
SIRT1, PPARγ and PGC-1 directly interact with the promoter
To investigate the mechanism by which PPARγ-PGC-1α represses BACE1 transcription, we first investigated if these molecules play a suppressive role on BACE1 via the PPARγ-responsive element/PPRE identified previously under inflammatory conditions (Sastre et al., 2006). Analysis of the BACE1 promoter identified four potential PPAR/RXR-responsive elements (Figure S6, Sites 1-4). We generated a series of deletion mutants for the BACE1 promoter and compared their promoter activities. All of the deletions resulted in significantly increased basal promoter activity with an exception on the F9 construct (Figures 6A and 6B). The findings that constructs F7 and F8 resulted in 2-fold increase in BACE1 promoter activity suggested that the -1541/-1209 region contains elements negatively regulate BACE1 (e.g., Sites 1 and 2). The F9 construct, which lacks the HNF-3 binding site, displayed a normal BACE1 promoter activity as compared to WT F1, suggesting that HNF-3 is a strong positive TF. Further deletion (F2) up to -753 region regained increased BACE1 promoter activity, indicating the presence of additional negatively regulatory elements. Notably, the repressive effect of PGC-1 overexpression was lost from any of the deletion promoters, indicating that PGC-1 suppresses in part through the N-terminal region.
Figure 6. PGC-1’s suppressive effect is largely dependent on the first PPRE site.

(A) Schematic diagram of BACE1 promoter (rat, 1.54 kb) in pGL3-basic luciferase construct. Constructs F2-9 represent four BACE1 promoter deletions. (B) Effects of transfecting PGC-1α on BACE1 promoter activity. Relative BACE1 promoter activity from the various deletion constructs is presented as compared to full-length BACE1-Luc (F1). Data are represented as means ± S. D. from 5 independent experiments; * indicates P< 0.05. (C) Effects of PPRE and YY1 mutants on BACE1 promoter activity. PGC-1α was cotransfected with rat BACE1 promoter (wt) or its mutants at PPRE and YY1 sites into HEK293 cells. Luciferasse assay was performed 24 h after transfection. Relative BACE1 promoter activity from the mutants and the effects of PGC-1α is presented as compared to pcDNA3.1 vector cotransfected-BACE1-Luc (wt) activity. (D) Effect of PPRE mutated promoter on PGC-1 in BACE1 suppression. AAV-PGC-1α shRNA was cotransfected with rat BACE1 promoter and its PPRE mutant into HEK293 cells for 24 h. Data are presented as means + S.D. from at least 3 independent experiments.* indicates P<0.05.
Between sites 1 and 2, only the first site displays a typical PPRE motif. Further disruption of the -1357/-1333 PPRE by site-directed mutagenesis led to 36% increased luciferase activity. In addition, mutation of the YY1 site-mutated promoter resulted in 48% reduced reporter activity (Figure 6C and 6D), consistent with the report that YY1 is a key positive TF for BACE1 (Nowak et al., 2006). Interestingly, when either the PPRE or YY1 sites were mutated, PGC-1 overexpression lost its suppressive effect on BACE1 promoter transcription. Downregulation of PGC-1 had no effect on the BACE1-luc with a mutated PPRE. These results strongly indicated that PPRE was the major site at which PGC-1 exerts its regulatory effect on the BACE1 promoter. To seek more direct in vivo evidence of PGC-1’s effect on this PPRE, we conducted a chromatin immunoprecipitation (ChIP) assay on the 24h fasted mouse brains to show that the SIRT1, PGC-1 and PPARγ proteins are associated with the first PPRE site (Figures 7A and S7A). Indeed, similarly enhanced protein-protein interactions between PGC-1 and SIRT1 as well as with PPARγ were detected by coimmunoprecipitation in the fasted brains (Figure 7D).
Figure 7. Enhanced in vivo binding of SIRT1, PPARγ, PGC-1α and corepressor NCoR to multiple sites of the BACE1 promoter region.

(A) In vivo ChIP assay detected increased binding of endogenous PGC-1α, SIRT1 and PPARγ in the protein complex to the first PPRE site in mouse brains upon 24 h-fasting as compared to non-fasted samples. Frontal cortical tissues were pooled from 4 mice per group. ** and *** indicate P< 0.01 and 0.005, respectively. (B) Representative results from the in vivo ChIP assay showing binding on sites 1-4 and (C) on the Sp1, YY1 and HNF-3β sites using the same 24-fasted mouse brains. (D) Enhanced SIRT1-PGC-1 and PPARγ-PGC-1α interactions upon fasting. Fasted and control mouse forebrain lysates were immunoprecipitated by anti-PGC-1α antibody followed by Western blot probing with either SIRT1 or PPARγ antibody.
By in vivo ChIP, we further detected increased binding of SIRT1, PPARγ, and PGC-1 on the other three elements (sites 2-4) in the fasted brains (Figure 7B and S7B), with most prominent enhancement observed on sites 1 and 3. Interestingly, we detected markedly enhanced binding of a nuclear receptor co-repressor (NcoR/NCOR1) also on sites 1 and 3. Based on our preliminary results, we did not see binding of the other major ligand-independent corepressor SMRT/NCoR2 to the BACE1 promoter, suggesting a corepressor specificity of NCoR in terms of negative regulation of BACE1. Further ChIP analysis was conducted on the three positive sites (HNF-3, Sp1 and YY1). As predicted, none of the binding of these positive transcription factors was significantly altered upon fasting (Figure 7C and S7C), while enhanced PGC-1 was found at the HNF-3 and Sp1 sites. SIRT1 binding was either undetected or unaltered.
Discussion
This work was designed to explore mechanisms through which the altered expression of the components of the SIRT1-PPARγ-PGC-1 pathway may regulate BACE1 expression. Herein, we present in vitro and in vivo evidence for the transcriptional regulation of BACE1 by this dominant metabolic signaling pathway under basic and metabolic stress conditions. Our findings provide the first molecular basis for metabolic signals/factors regulating a crucial and rate-limiting enzyme in AD pathogenesis. Given the increasing recognition of several major metabolic-cardio risk factors (e.g., CNS insulin resistance, high cholesterol, ApoE genotype, insufficient eNOS etc) in accelerating AD development and progression, the novel transcriptional regulation of BACE1 identified here may represent a unifying and central mechanism for aberrant amyloidogenesis induced by increased metabolic stress.
PGC-1α was classically defined as a transcription coactivator that interacts with a broad range of TFs that participate in many biological processes including adaptive thermogenesis, mitochondrial biogenesis, glucose/fatty acid metabolism, muscle fiber-type switching, and heart development (Lin et al., 2005). Dysregulated PGC-1α has been implicated in pathogenic conditions such as obesity, type 2 diabetes and cardiomyopathy. The evidence for PGC-1α transcriptional interference in neurodegeneration was initially found by the Huntington-like striatal degeneration in PGC-1 deficient mice (Cui et al., 2006; Lin et al., 2004; Weydt et al., 2006), recently extended to Parkinson’s disease (Clark et al., 2011; Zheng et al., 2010) and to non-neuronal lineages (Tsunemi and La Spada 2012a; Xiang et al. 2011). However, little is known of its role in AD except for reduced expression in the brains of AD patients and Tg2576 mice with insulin resistance induced by a HFD (Ho et al., 2004; Katsouri et al., 2011; Qin et al., 2009). Both PGC-1 isoforms (α and β) are abundantly expressed and widely distributed in the brain and may be interchangeable for certain functions, including neuronal mitochondrial biogenesis (Wareski et al., 2009) and in BACE1 transcriptional regulation as reported here. Recent studies in CaMKII-specific PGC-1α conditional KO mice (Ma et al., 2010) suggested a crucial role for PGC-1 in neuronal function with forebrain neurons being an important integral part of the neural circuitry in governing energy balance.
Caloric restriction has been shown to induce multiple changes in glucose metabolism and to extend lifespan in a broad spectrum of species, possibly attributable to the activated AMPK-SIRT1-PGC-1 signaling pathway. In the liver, SIRT1, the NAD-dependent protein deacetylase, controls the gluconeogenic/glycolytic pathways in response to fasting through interaction with and deacetylation of PGC-1α at specific lysine residues in an NAD(+)-dependent manner (Rodgers et al., 2008). These findings regarding the basic pathways of energy homeostasis in the liver appear to be replicated in the brain in the regulation of BACE1. Under non-fasting conditions, PGC-1α is expressed at very low levels in the brain but PGC-1α is induced by fasting. The expression and activity of PGC-1α in the brain are modulated by transcriptional activation as well as by SIRT-1 mediated deacetylation, which increases PGC-1 activity. Like PGC-1, SIRT1 has emerged as a major regulator of mammalian transcription in response to cellular metabolic status and stress. It has been shown to positively regulate α-secretase promoter transcription/expression, leading to reduced amyloid deposition (Donmez et al., 2010). As with exercise (Lazarov et al., 2005), calorie restriction mitigated excessive amyloidogenesis in Tg2576 brains via activation of α-secretase through SIRT1-mediated transcriptional activation of FoxO3a (Qin et al., 2008).
Transcriptional dysregulation of BACE1 - a crucial mechanism in SAD?
Despite overwhelming evidence of robust transcriptional activation of BACE1 in response to various stress conditions in numerous experimental models including both cellular and rodent AD, the majority of earlier studies failed to detect significant elevation of the mRNA level of BACE in AD patient brains (Gatta et al., 2002; Preece et al., 2003; Yosojima et al., 2001) with one exception (Li et al., 2004). In addition, a recent large scale candidate-gene SNP-expression screen showed comparable BACE1 expression in the cortex of late onset AD/LOAD cases and age-matched controls (10.815 ± 0.038 in 187 cases of control versus 10.763 ± 0.039 in 176 cases of LOAD) (Webster et al., 2009). Since neuron density is much lower in AD cases, a stable expression value actually indicates higher expression per neuron. The discrepancy between experimental models (Tg mice and cultured neurons) versus primary AD brain specimens may also arise in part from the sporadic nature of the majority of AD cases with BACE1 being tightly regulated by multiple mechanisms which are not necessarily mutually exclusive. It should be noted that one recent report found significantly increased BACE1 mRNAs in freshly isolated PBMCs from a large cohort of AD patients as compared to normal controls (Marques et al., 2012).
Although our data clearly indicate transcriptional regulation of BACE1 as the dominant event in response to metabolic stress, we cannot rule out the possibility of translational regulation. In a previous report, the treatment of mice with an acute energy inhibitor only increased BACE1 protein levels but not the mRNA levels, primarily due to elevated eIF2α phosphorylation via a translational control mechanism. Elevated p-eIF2α and BACE1 levels were also observed in 5XFAD transgenic mice and in human AD brains (O’Connor et al., 2008). It should be pointed out that the translational upregulation of BACE1 upon energy depletion using toxins to mitochondrial respiratory chains may be mechanistically different from fasting/calorie restriction used in our studies in terms of elicited changes in signaling pathways though PGC-1. We did not detect significant increase in p-eIF2α in the mouse brains after HFC feeding. Nevertheless, the two major transcriptional and translational mechanisms need not be mutually exclusive and it is unclear which mechanism may play a more important role in sporadic AD pathogenesis. Moreover, metabolic stress may elicit additional regulatory mechanisms post-transcriptionally (miRNA) (Wang et al., 2008) and post-translationally (ubiquitination, S-nitrosylation and oxidation) (Kwak et al. 2011). Together with recently disclosed epigenetic DNA methylation (Marques et al., 2012), BACE1 has proven to be tightly regulated by multiple mechanisms. Lastly, BACE1 protein degradation is emerging as a potentially important regulatory mechanism and PGC-1α was reported to facilitate BACE1 protein degradation via the UPS (Gong et al., 2010), which may also reflect one of the multiple effects of this key regulatory molecule. Interestingly, we also did not detect changes in the GGA3 protein upon metabolic stress, a mechanism discovered in response to cerebral ischemia and brain injury (Walker et al., 2012).
Perhaps due to technical limitations, several earlier studies did not see evidence of BACE1 transcriptional regulation under HFD-induced insulin resistance or by resveratrol (Ho et al., 2004; Vingtdeux et al., 2010). Of particular interest, the anti-amyloidogenic effect of resveratrol was attributed to the AMPK-mediated signal modulation of mTOR-autophagy thereby facilitating BACE1 protein degradation/clearance (Dasgupta and Milbrandt 2007). Our study, based on the different experimental designs (NG, fasting, and HFC), has unequivocally demonstrated BACE1 transcriptional alteration as a key response to metabolic stress. Among the major components of the identified transcription circuitry/network are the PGC-1α-assisted PPARγ transcription complexes which act via direct interaction with the PPRE located in the BACE1 promoter. We have also identified a molecular mechanism whereby SIRT1 functions in glucose homeostasis as a modulator of both PPARγ and PGC-1α. Enhanced functional interaction between SIRT1-PGC-1α along with PPARγ-PGC-1α, has been found in fasting brains as detected by immunoprecipitation of molecular complexes (Figure 7D). These functional interacting partners have been identified as being required to mediate nutrient control of glucose homeostasis in peripheral tissues (Rodgers et al., 2008). Hence, our findings demonstrate that the basic pathways/mechanisms of energy homeostasis can regulate BACE1.
Unique molecular mechanism repressing BACE1 transcription
To our knowledge, this is the first report of transcriptional inhibition by PPARγ-PGC-1α. The standard view of PPARγ-PGC-1-mediated transcription involves transcriptional upregulation of genes in a ligand-dependent manner. However, there are several reports suggesting that increases in PGC-1α levels can negatively impact the expression of certain genes via indirect and poorly understood mechanism(s) (Estall et al., 2009; Jeong et al. 2009; Zhang et al., 2004). One unresolved issue is how PPARγ-PGC-1 is able to repress transcription. There are several possible mechanisms: 1) The PPREs in the BACE1 promoter serve as negative PPREs and convert PPARγ into a transcriptional repressor. 2) PPARγ-PGC-1 act as scaffolding proteins rather than TF/co-activator thereby allowing reverse recruitment of corepressors. Based on literature, PPARγ can either functionally antagonize certain positive TFs to repress transcription or recruit corepressors (Cohen 2006, Perissi et al., 2010). 3) SIRT1 represses PPARγ by docking with corepressors (NCoR and SMRT) as in adipocytes (Picard et al., 2004). 4) There are other critical proteins bound on BACE1 promoter such as HNF-3, YY1 or SP1 that convert PGC-1α into a transcriptional repressor. Our preliminary work has explored these possibilities and has ruled out the PPRE. When two copies of the PPAR/RXR elements (sites 1-4) in the BACE1 promoter were ligated 5’ to the luciferase gene, none of these elements was able to repress expression of the luciferase reporter (unpublished data). With respect to the involvement of corepressors, we found that NCoR was recruited into the promoter complexes with PPARγ, PGC-1 and SIRT1 and this recruitment was independent of rosiglitazone addition. Upon fasting, the association of both PGC-1 and NCoR was enhanced on the BACE1 promoter (Figure 7B). This finding supports the second and third possibilities which are not mutually exclusive. The strongest finding of this work is on the coexistence of both corepressor NCoR and the coactivator PGC-1 in a single complex. Moreover, PPARγ, when activated by deacetylation as demonstrated by the Lys mutants, negatively regulated BACE1 transcription. Since we observed specific effects from the mutant PPARγ at K268T and K293T in suppressing BACE1 transcription and SIRT1 is known to deacetylate from these two residues, SIRT1 is likely responsible for the PPARγ activation in our case. However, it is not clear whether such deacetylation is ligand-dependent as reported in adipocytes (Qiang et al., 2012); our data argue against a ligand-independent mechanism, likely reflecting a cell-type specific mechanism in neurons. It is also unclear whether PPARγ activation by SIRT1 is required to recruit the corepressor NCoR. Although we detected in vivo binding of SIRT1, PPARγ, and PGC-1 on the multiple nuclear receptor-responsive elements, with most prominent binding found on sites 1 and 3, PGC-1 was also found at the HNF-3 and Sp1 sites. There has been one previous report that resveratrol inhibited expression of the angiotensin II type 1 receptor gene through Sp1 raising the possibility that SIRT1 may inhibit BACE1 in part through Sp1 (Miyazaki et al., 2008). Based on the data we collected, we speculate a loop topology as illustrated in the Graphical Abstract in a simplified format: binding of the common factors to the sites 1 and 3 and to HNF-3 may present the recruited corepressor to the positive TF. It is not yet known whether the corepressor NCoR is also recruited to these positive TFs and its recruitment requires or is assisted by PGC-1.
Although the complex regulatory mechanism in terms of the interplay between these molecules warrants further investigation, our work strongly suggests that targeting BACE1 at the transcriptional level may be a viable approach for SAD as demonstrated in a FAD mouse model using lenti-siRNA, leading to a complete reversal of excessive amyloidogenesis and neurodegeneration (Singer et al., 2005). Although our results based on administration of the AAV2-PGC-1α viruses are preliminary due to a small number of Tg2576 mice (Figure 3), it shows promise of the therapeutic value of overexpressing or activating PGC-1 in blunting amyloidogenesis in a familial AD model. Furthermore, virus-mediated gene delivery of SIRT1 and PGC-1 have demonstrated proof-of-concepts in halting neurodegeneration in HD and AD mouse models (Jeong et al., 2011; Kim et al., 2007; Tsunemi et al., 2012a and b). Our major findings on the molecular pathway under various metabolic stress conditions further suggest a promise of generalizing a PGC-1-based approach in SAD. Therefore, further characterization of the transcription network involving PGC-1α, SIRT1 and the BACE1 promoter will further validate the metabolic factors regulating this important gene in Alzheimer’s etiology. More importantly, the study outcomes may be instrumental in future therapeutic design on targeting SAD.
Experimental Procedures
Cell culture, drug treatments, plasmid transfection and virus infection
Rat primary cortical neurons (PRCN) were prepared as described (Chen et al., 2009). Glucose deprivation was performed with DMEM/no glucose (Gibco), or Neurobasal medium/glucose-freeovernight. Treatments with 10-100 μM resveratrol or 5 mM nicotinamide (Sigma) were performed overnight at 37°C. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) with plasmids: pcDNA-PGC-1α and pcDNA-PGC-1β (Addgene); pcDNA4 TO-Myc.His-PGC-1α or β; pcDNA-SIRT1.wt and its mutant SIRT1.H355A. pGL3-Basic luciferase reporter constructs and the site-directed mutagenesis was performed as described in our previous work (Chen et al., 2009; Kwak et al., 2011).
In vivo experiments on mice
All animal care protocols and procedures were performed in according to the Animal Scientific Procedures Act and with the approval of the University of Tennessee Animal Care and Use Committee. High fat high cholesterol (HFC) custom diet containing 21% fat and 1.25% cholesterol (D12079B, Teklad, Harlan Labs) was given to C57BL/6 mice for 2 months from a 5-month of age (n=5/group) and compared to control chow diet. AAV2/5 PGC-1α-shRNA (3.87 × 10 ˆ13 vg/ml, 5-GGTGGATTGAAGTGGTGTAGA-3’) and AAV2/5-EGFP (2.56 × 10 ˆ13 vg/ml) were generated by the Virus Core Facility at the Iowa University while AAV2/1 viruses overexpressing PGC-1α was generated in-house (2 × 10 ˆ 12 vg/ml). All AAV2 viruses were injected into the CA1 region of mouse hippocampus in 1 μl of volume and mice were euthanized 21 days later for analysis.
Immunoprecipitation and immunoblot
The procedures were performed as described (Chen et al., 2009; Kwak et al., 2011) using the following antibodies: mouse anti-BACE1 (3D5); mouse anti-Myc and mouse anti-β-Actin (Sigma); Rabbit anti-SIRT1 (Millipore); mouse anti-PPARγ (81B8) and rabbit polyclonal antibodies against p-AMPKα AMPKα acetylated-lysine, p-eIF2α and eIF2α, GGA3 were all from Cell Signaling; mouse anti-PGC-1α (H-300) (Santa Cruz);
Quantitative RT-PCR on BACE1 and PGC-1 Messages
The procedures were performed as described using the same primers for rat and human bace1 (Chen et al., 2009; Kwak et al., 2011). Rat pgc-1α primers [forward 5’-AAAGGGCCAAGCAGAGAGA-3’ and reverse 5’-GTAAATCACACGGCGCTCTT-3’], rat pgc-1β primers [forward 5’-TTGACAGTGGAGCTTTGTGG-3’and reverse 5’-GGGCTTATATGGAGGTGTGG-3’], human pgc-1α primers [forward 5’-TTATTGGGAAATGCCTCCTG-3’ and reverse 5’-GGGTCATTTGGTGACTCTGG-3’], mouse pgc-1α primers [forward 5’-GAAAGGGCCAAACAGAGAGA-3’ and reverse 5’-GTAAATCACACGGCGCTCTT-3’], and mouse pgc-1β primers [forward 5’-CTCCAGTTCCGGCTCCTC-3’ and reverse 5’-CCCTCTGCTCTCACGTCTG-3’] were used in the present studies. Primers used for rat GAPDH, forward 5’-ACATTGTTGCCATCAACGAC-3’, reverse 5’-CTTGCCGTGGGTAGCGTCAT-3’; human GAPDH primers, forward 5’-AATCCCATCACCATCTTCC-3’ and reverse 5’-GGACTCCACGACGTACTCA-3’; mouse GAPDH primers, forward 5’- GGGTTCCTATAAATACGGACTGC-3’ and reverse 5-CCATTTTGTCTACGGGACGA-3’.
Promoter deletion constructs and Luciferase assay
Deletion constructs and luciferase assays were conducted as described (Chen et al., 2009). PGC-1α or SIRT1 expression vectors, or PGC-1 shRNA plasmids, were cotransfected into HEK293 cells and 24 hours later, cells were collected in passive lysis buffer and analyzed for lucuferase activity. No glucose treatment was performed 5 hours after transfection.
ChIP assays
Chromatin immunoprecipitation (ChIP) assay was performed using the kit from Upstate according to the manufacturer’s instructions. The transfected HEK293 cells were cross-linked with 1.0% formaldehyde and collected in lysis buffer. The following primers were used in PCR assays: for site 1, forward 5’-GAGTAATGTTGGTATGCCTC-3’ and reverse 5’-GGGATGAGAGTATGTCAGTC-3’; for site 2, forward 5’-GCTCCTCCAGTCTCTACTCC-3’ and reverse 5’-GACTACATAGAGAAACTCTG-3’. For detecting in vivo protein bindings, mouse forebrain lysates (20 mg/immunoprecipitation) were used.
Statistics
All quantitative data were presented as means ± S.D. Comparisons between groups were analyzed with T-TEST.
Supplementary Material
Research Highlights.
High fat high cholesterol diet or eNOS deficiency upregulated BACE1 transcription in mice.
Fasting suppresses BACE1 through SIRT1-mediated deacetylation of PPARγ- PGC-1.
Up- or down-regulation of PGC-1α reciprocally regulated BACE1 in vitro and in vivo.
SIRT1-PPARγ- PGC-1 mediated BACE suppression via multiple PPRE sites.
Acknowledgments
We thank Drs. Jiandie Lin and Dimitri Krainc for constructive discussions; Drs. Robert Vassar, Weihong Song, Zhijun Luo, Jiandie Lin and BingZhong Xue for providing anti-BACE1 antibody (3D5), plasmid constructs and PGC-1 adenoviruses. This work was partially supported by the NIH grants (R01 NS054880, R01 AG031893, R21 AG041934 to F-F. L; DK0059368 to EAP; U01 AA016662 and U01 AA013499 to RW; R01NS051575 to NB; R21 NS062886 to HB), the Alzheimer’s Association grant (IIRG-11-204030 to F-F.L.) and the UT Center for Integrative and Translational Genomics to F.-F.L.
References
- Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res. 2010;107:1498–1502. doi: 10.1161/CIRCRESAHA.110.233080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat NR. Linking cardiometabolic disorders to sporadic Alzheimer’s disease: a perspective on potential mechanisms and mediators. J Neurochem. 2010;115:551–562. doi: 10.1111/j.1471-4159.2010.06978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparin L, et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Reddy S, Zheng K, Betensky RA, Simon DK. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson’s disease. BMC Med Genet. 2011;12:69–77. doi: 10.1186/1471-2350-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RN. Nuclear receptor corepressors and PPARγ. Nuc Recept Sig. 2006;4:e003. doi: 10.1621/nrs.04003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA. 2007;104:7217–7122. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses β-amyloid production by activating the a-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Estall JL, Ruas JL, Choi CS, Laznik D, Badman M, Maratos-Flier E, Shulman GI, Spiegelman BM. PGC-1α negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erba axis. Proc Natl Acad Sci USA. 2009;106:22510–22515. doi: 10.1073/pnas.0912533106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta LB, Albertini A, Ravid R, Finazzi D. Levels of beta-secretase BACE and alpha-secretase ADAM10 mRNAs in Alzheimer hippocampus. Neuroreport. 2002;13:2031–2033. doi: 10.1097/00001756-200211150-00008. [DOI] [PubMed] [Google Scholar]
- Ge YW, Maloney B, Sambamurti K, Lahiri DK. Functional characterization of the 5’ flanking region of the BACE gene: identification of a 91 bp fragment involved in basal level of BACE promoter expression. FASEB J. 2004;18:1037–1039. doi: 10.1096/fj.03-1379fje. [DOI] [PubMed] [Google Scholar]
- Gong B, Chen F, Pan Y, Arrieta-Cruz I, Yoshida Y, Haroutunian V, Pasinetti GM. Fbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell. 2010;9:1018–1031. doi: 10.1111/j.1474-9726.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, Catalano MG, Smith MA, Perry G, Danni O, Boccuzzi G, Tabaton M. AGEs/RAGE complex upregulates BACE1 via NF-kappa B pathway activation. Neurobiol Aging. 2012;33:196.e13–27. doi: 10.1016/j.neurobiolaging.2010.05.026. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. Review. [DOI] [PubMed] [Google Scholar]
- Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- Jeong JH, Cho S, Pak YK. Sterol-independent repression of low density lipoprotein receptor promoter by peroxisome proliferator activated receptor gamma coactivator-1α (PGC-1α) Exp Mol Med. 2009;41:406–416. doi: 10.3858/emm.2009.41.6.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, Yates JR, 3rd, Bordone L, Guarente L, Krainc D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2011;18:159–165. doi: 10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M. PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. J Alzheimers Dis. 2011;25:151–162. doi: 10.3233/JAD-2011-101356. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, Liao FF. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener. 2011;6:17–26. doi: 10.1186/1750-1326-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VMY, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS. Environmental enrichment reduces Aβ Levels and amyloid deposition in transgenic mice. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Li R, Linholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci USA. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Ma D, Li S, Lucas EK, Cowell RM, Lin JD. Neuronal inactivation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) protects mice from diet-induced obesity and leads to degenerative lesions. J Biol Chem. 2010;285:39087–39095. doi: 10.1074/jbc.M110.151688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques SC, Lemos R, Ferreiro E, Martins M, de Mendonça A, Santana I, Outeiro TF, Pereira CM. Epigenetic regulation of BACE1 in Alzheimer’s disease patients and in transgenic mice. Neuroscience. 2012;220:256–266. doi: 10.1016/j.neuroscience.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Martins IJ, Hone E, Foster JK, Sunram-Lea SI, Gnjec A, Fuller SJ, Nolan D, Gandy SE, Martins RN. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–736. doi: 10.1038/sj.mp.4001854. [DOI] [PubMed] [Google Scholar]
- Miyazaki R, Ichiki T, Hashimoto T, Inanaga K, Imayama I, Sadoshima J, Sunagawa K. SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2008;28:1263–1269. doi: 10.1161/ATVBAHA.108.166991. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- Nowak K, Lange-Dohna C, Zeitschel U, Günther A, Lüscher B, Robitzki A, Perez-Polo R, Rossner S. The transcription factor Yin Yang 1 is an activator of BACE1 expression. J Neurochem. 2006;96:1696–1707. doi: 10.1111/j.1471-4159.2006.03692.x. [DOI] [PubMed] [Google Scholar]
- O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- Preece P, Virley DJ, Costandi M, Coombes R, Moss SJ, Mudge AW, Jazin E, Cairns NJ. Beta-secretase (BACE) and GSK-3 mRNA levels in Alzheimer’s disease. Brain Res Mol Brain Res. 2003;116:155–158. doi: 10.1016/s0169-328x(03)00233-x. [DOI] [PubMed] [Google Scholar]
- Pugielli A, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Wei Gu W, Farmer SR, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of PPARγ. Cell. 2012;150:620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1 expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Qin W, Zhao W, Ho L, Wang J, Walsh K, Gandy S, Pasinetti GM. Regulation of forkhead transcription factor FoxO3a contributes to calorie restriction-induced prevention of Alzheimer’s disease-type amyloid neuropathology and spatial memory deterioration. Ann N Y Acad Sci. 2008;1147:335–347. doi: 10.1196/annals.1427.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner S, Sastre M, Bourne K, Lichtenthaler SF. Transcriptional and translational regulation of BACE1 expression--implications for Alzheimer’s disease. Prog Neurobiol. 2006;79:95–111. doi: 10.1016/j.pneurobio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Sastre M, Dewacher I, Rossner S, Bogdanovic N, Rosen E, Borghgrae P, Evert BO, Dumitrescu-Ozimek L, Thail DR, Landreth G, et al. Nonsteroid anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPAR gamma. Proc Natl Acad Sci USA. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Sun X, Wang Y, Qing H, Christensen MA, Liu Y, Zhou W, Tong Y, Xiao C, Huang Y, Zhang S, et al. Distinct transcriptional regulation and function of the human BACE2 and BACE1 genes. FASEB J. 2005;19:739–749. doi: 10.1096/fj.04-3426com. [DOI] [PubMed] [Google Scholar]
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumangalakudi L, Prakasam A, Zhang R, Bimonte-Nelson H, Sambamurti K, Kindy MS, Bhat NR. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem. 2008;106:475–485. doi: 10.1111/j.1471-4159.2008.05415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC. Cerebrovascular and cardiovascular pathology in Alzheimer’s disease. Int Rev Neurobiol. 2009;84:35–48. doi: 10.1016/S0074-7742(09)00403-6. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, La Spada AR. PGC-1α at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog Neurobiol. 2012a;97:142–151. doi: 10.1016/j.pneurobio.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012b;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285:9100–9113. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KR, Kang EL, Whalen MJ, Shen Y, Tesco G. Depletion of GGA1 and GGA3 Mediates Postinjury Elevation of BACE1. J Neurosci. 2012;32:10423–10437. doi: 10.1523/JNEUROSCI.5491-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The Expression of MicroRNA miR-107 Decreases Early in Alzheimer’s Disease and May Accelerate Disease Progression through Regulation of β-Site Amyloid Precursor Protein-Cleaving Enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, Kaasik A. PGC 1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J Biol Chem. 2009;284:21379–21385. doi: 10.1074/jbc.M109.018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P, Rohrer K, Zhao A, Marlowe L, Kaleem M, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda V, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neuerodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Valenza M, Cui L, Leoni V, Jeong HK, Brilli E, Zhang J, Peng Q, Duan W, Reeves SA, et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 (alpha) contributes to dysmyelination in experimental models of Huntington’s disease. J Neurosci. 2011;31:9544–9553. doi: 10.1523/JNEUROSCI.1291-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasojima K, McGeer EG, McGeer PL. Relationship between β amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 2001;919:115–121. doi: 10.1016/s0006-8993(01)03008-6. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Castellani LW, Sinal CJ, Gonzalez FJ, Edwards PA. Peroxisome proliferatoractivated receptor-gamma coactivator 1α (PGC-1α) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev. 2004;18:157–169. doi: 10.1101/gad.1138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.