

Abstract
p53 is a tumor suppressor with a pro-death role in many conditions. However, in some contexts, evidence supports a pro-survival function. p53 has been shown to be activated in acetaminophen (APAP) toxicity but the impact of this on toxicity is uncertain. In the present study, we have found that p53 plays a protective role in APAP-induced liver injury. We inhibited p53 using three different approaches in mice, pifithrin-α (PFTα), knockdown of p53 expression with antisense oligonucleotide, and p53 knockout. Mice were treated with APAP (300 mg/kg) i.p. and after 24h in all three conditions, the liver injury was more severe as reflected in higher ALT levels and great area of necrosis in histology of the liver. Conversely, a p53 activator, nutlin-3a, decreased the liver injury induced by APAP. In the p53 inhibition models, enhanced sustained JNK activation was seen in the early time course, while the JNK was suppressed with the p53 activator. In conclusion, p53 plays a novel protective role in APAP induced liver injury through inhibiting the activation of JNK, a key mediator in APAP-induced oxidative stress.
Keywords: Acetaminophen, p53, JNK, liver injury
Graphical Abstract
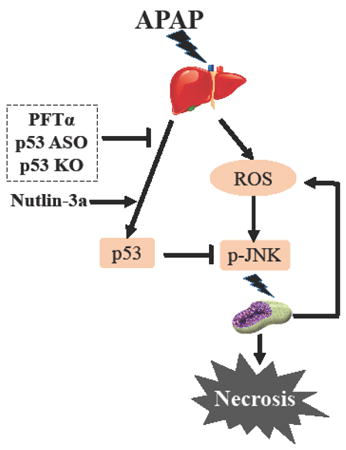
Introduction
Acetaminophen (APAP)-mediated hepatotoxicity has become the leading cause of acute liver failure in United States and most of European countries since first reported in 1960s (1). It is well established that APAP induces hepatotoxicity through mechanisms involved in production of reactive metabolite N-acetyl-p-benzoquinoneimine (NAPQI), depletion of glutathione (GSH), formation of NAPQI-protein adduct, generation of reactive oxygen species (ROS), sustained activation of JNK, disruption of mitochondrial membrane potential (MMP), and ultimately induction of necrosis of hepatocytes (2, 3).
p53 is the first identified and the best known tumor suppressor. The role of p53 in the regulation of cell death induction is well established (4). Activation of p53 generally serves as pro-death signal to trigger cytotoxicity. However, an increasing body of evidence supports a pro-survival function of p53 activation in certain conditions (5-7). As mentioned above, oxidative stress plays a pivotal role in hepatotoxicity of APAP. Induction of oxidative stress can trigger p53 activation. Indeed, previous studies have shown that p53 is activated in response to APAP exposure in both cell culture and animal models (8). However, the functional role of p53 activation in APAP-induced hepatotoxicity has not been systematically addressed. In the present study, an APAP-induced liver injury mouse model was employed to evaluate the role of p53 in the regulation of cell death induction of hepatocytes. The results demonstrated for the first time that p53 activation protected against APAP-induced hepatotoxicity accompanied by suppression of sustained JNK activation.
Materials and Methods
Reagents
Antibodies for p53, p-JNK, Bax, JNK, β-actin were purchased from Cell Signaling Technology (Denvers, MA, USA). Anti-cyp2e1 was purchased from Millipore (Billerica, MA, USA). Antisera for NAPQI adducts was a kind gift of Laura James (University of Arkansas). Acetaminophen, pifithrin α (PFTα), nutilin-3a were purchased from Sigma-Aldrich (St. Louis, MO, USA)
Animal experiments
Male C57BL/6N wildtype mice (19-20g) were purchased from Harlan Bioproducts for Science Inc. (Indianapolis, IN, USA). These mice were administrated by antisense oligonucleotide. These experiments were performed at the Kaplowitz lab, University of Southern California. The remainder of in vivo studies were performed in China Agricultural University, Beijing. Male C57BL/6N or J substrain p53 knockout and paired wildtype mice (19-20g) were purchased from Beijing Vitalstar Biotechnology Co. Ltd (Beijing, China). All mice were housed in a temperature-, light-, and humidity- controlled environment that is accredited by the Associated for Assessment and Accreditation of Laboratory Animal Care. Mice were maintained on standard laboratory chow and had free access to water. All mice were overnight fasted. PFTα (2.2mg/kg, i.p.), dissolved in PBS: DMSO (9:1) and nutilin-3a, (10mg/kg, by gavage), dissolved in 2% Klucel, 0.5% Tween 80, were pretreated 1h before APAP administration. Controls received vehicles. APAP was dissolved in warm PBS and given at 300mg/kg, i.p. Serum and liver tissue were collected 1h, 2h, 4h and 24h after APAP (300mg/kg, i.p).
Antisense oligonucleotide treatment of mice
Antisense oligonucleotide (ASO) was provided by Ionis Phamarceuticals, Carlsbad, CA. Control ASO (CCTTCCCTGAAGGTTCCTCC) and p53 ASO (GTGACTCCTCCATGGCAGTC) Oligonucleotides were synthesized as 20-nt uniform phosphorothioate chimeric oligonucleotides and purified. Oligonucleotides were chimeric containing five nuclease resistant 2′-O- methoxyethylribose-modified phosphorothioate residues on the 5′ and 3′ -ends, flanking a 2′- deoxyribonucleotide/phosphorothioate region that supports RNase H-based cleavage of the targeted mRNA. 50mg/kg control ASO and p53 ASO were i.p. injected every other day for 2 weeks.
Genotyping
The protocol for this study was approved by the Animal Care and Utilization Committee of China Agricultural University. Genotypes of the p53-deficient mice were examined by Polymerase chain reaction (PCR). Three primers for detection are listed as follows. Wild type primer (named p53-4) is CTgATCgTTACTCggCTTgTCC, Common primer (named p53-6) is CTgTCTTCCAgATACTCgggATAC, and mutant primer (named TWB-12) is ATCgCCTTCATACgCCTTCTTgACgAgTTC. PCR were carried out for 1 cycles (98°C, 1 min; 68°C, 1 min20 seconds) and 32 cycles (94°C, 30 seconds; 60°C, 30 seconds; and 72°C, 1min). The expected results were as follows: mutant=286bp (primers of p53-6 & TWB-12); wild type=381bp (primers of p53-4 & p53-6); heterozygote=286bp and 381bp.
Western Blotting
The cell lysate and tissues were prepared in ice-cold radioimmunoprecipitation (RIPA) assay buffer. SDS-PAGE electrophoresis was performed and then protein was transfer to 0.2μm nitrocellulose (NC) membrane (Millipore, Bedford, MA, USA). After blocking in 5% milk, the blots were probed with primary antibody followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies. The signal was visualized by enhanced chemiluminescence (Fisher/Pierce, Rockford. IL, USA) and recorded on an X-ray film (Estman Kodak Company, Rochester, NY, USA).
Primary mouse hepatocyte culture
Primary mouse hepatocytes were isolated from control ASO and p53 ASO treated mice as described (9). The viability of cells exceeded 88%. After plating, the medium was replaced with DMEM/F12 containing 10mM APAP. Whole cell lysate was collected after 1h, 2h and 4h of exposure to APAP.
Histology
After fixed in formalin for 24h, the tissue was paraffin-embedded. And then the tissue paraffin embedded blocks was cut for 5μm thick. The liver sections were stained with hematoxylin and eosin (H&E).
GSH Measurement
Liver GSH was measured using colorimetric kits (#A006-2, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The tissue was homogenized in mataphosphoric acid, and centrifuged at 3000g at 4°C for 10min. The upper aqueous layer was collected for measurement and mixed with 20μL sample and 120μL DTNB: Glutathian Reductase=1:1 solution; after 30s, 60μL B-NADPH was added and the absorbance at 412 nm was immediately read in microplate reader. The rate of 2-nitro-5-thiobenzoic acid formation was then calculated from a standard curve.
ALT measurements
Serum ALT was measured using colorimetric kits (#C009-2, Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Serum (5μL) and ALT substrate (25μL) were added to each well and placed in a 37°C heating bath. After exact 30 minutes, 25 μL of ALT Color Reagent was added to each well, maintaining the timed interval sequence. After an additional 10 minutes incubation at 37 °C, 200 μL of ALT Color Developer was added (maintaining the same timed intervals). After 15 min at room temperature, absorbance of all wells was measured at 510nm.
Statistical analysis
Data are presented as the mean ± SD. T-test was used for comparison of two groups. P<0.05 was considered statistically significant.
Results
p53 inhibition by pharmacological approach triggered more severe liver injury in APAP model
It was previously reported that p53 is activated after APAP administration as reflected in activation of p53 response genes at 6 hours after APAP, but no clear evidence exists as to whether p53 plays a damaging or protective role. (8). PFTα, a small molecule p53 inhibitor, effectively blocks p53-dependent transcriptional activation (10). We pretreated mice with PFTα (2.2mg/kg, i.p) 1h before APAP administration and serum and liver were collected after 24h. Bax, a downstream target of p53, was decreased by PFTα pretreatment providing evidence of successful inhibition of p53 (Fig. 1A). Nevertheless, APAP-induced elevation in serum ALT (Fig. 1B) was higher and hepatic necrosis were more severe (Fig. 1C) in PFTα plus APAP group than APAP alone group.
Fig.1.
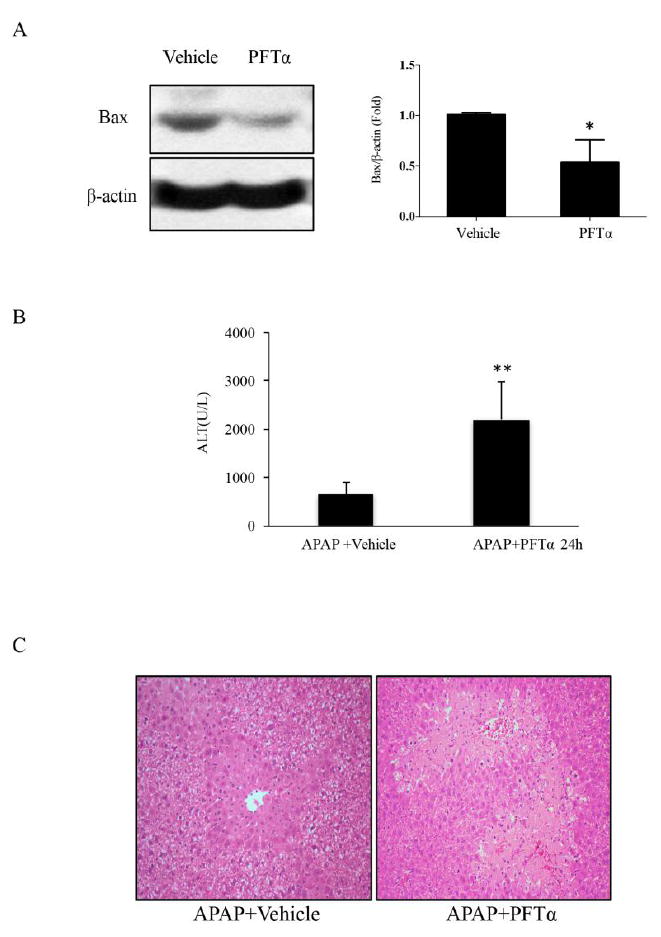
The APAP-induced liver damage after PFTα treatment in vivo. Mice (C57BL/6J) received PFTα (2.2mg/kg, i.p) 1h before APAP (300mg/kg, i.p). A. The expression of Bax 1h after PFTα treatment in vivo ; the right panel shows densitometry of Bax/β-actin of n=3, *p<0.05. B. Serum ALT level at 24h after APAP. C. Liver H&E staining at 24h after APAP. (n=6; **p<0.01)
p53 knockdown or knockout enhanced APAP-induced liver injury
p53 was successfully knocked-down by ASO pretreatment 7 times every other day in vivo (58% decrease) (Fig. 2A). APAP was administrated on the second day after completing treatment. Serum and liver tissue was collected 24h later. As shown in Fig. 2B & C, knockdown of p53 lead to increased injury as reflected in serum ALT and histology.
Fig.2.
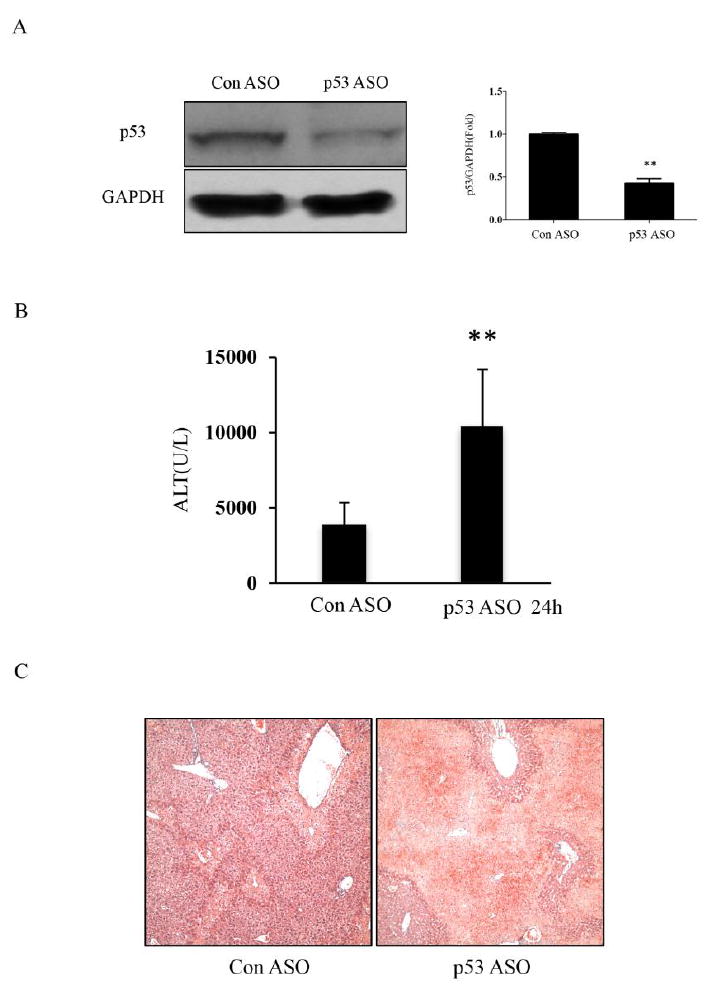
The APAP-induced liver injury after p53 knockdown by ASO in C57BL/6N mice. A. The p53 expression after ASO treatment; the right panel shows densitometry of p53/β-actin of n=3, *p<0.05. B, C The serum ALT level and H&E histology in control and p53 ASO treated mice at 24h. (n=5; **p<0.01)
To further explore the role of p53 in APAP-induced liver injury, p53-/- mice and strain matched WT mice were treated with 300mg/kg APAP i.p. As shown in Fig. 3A, genotyping p53 KO mice confirmed homozygous deletion of p53. Notably, the amplitude of elevation of ALT in p53 KO mice was markedly higher than WT mice (Fig. 3B). Consistent with serological analysis, H&E histological analysis also showed more severe liver injury in p53 deficiency (Fig. 3C).
Fig.3.
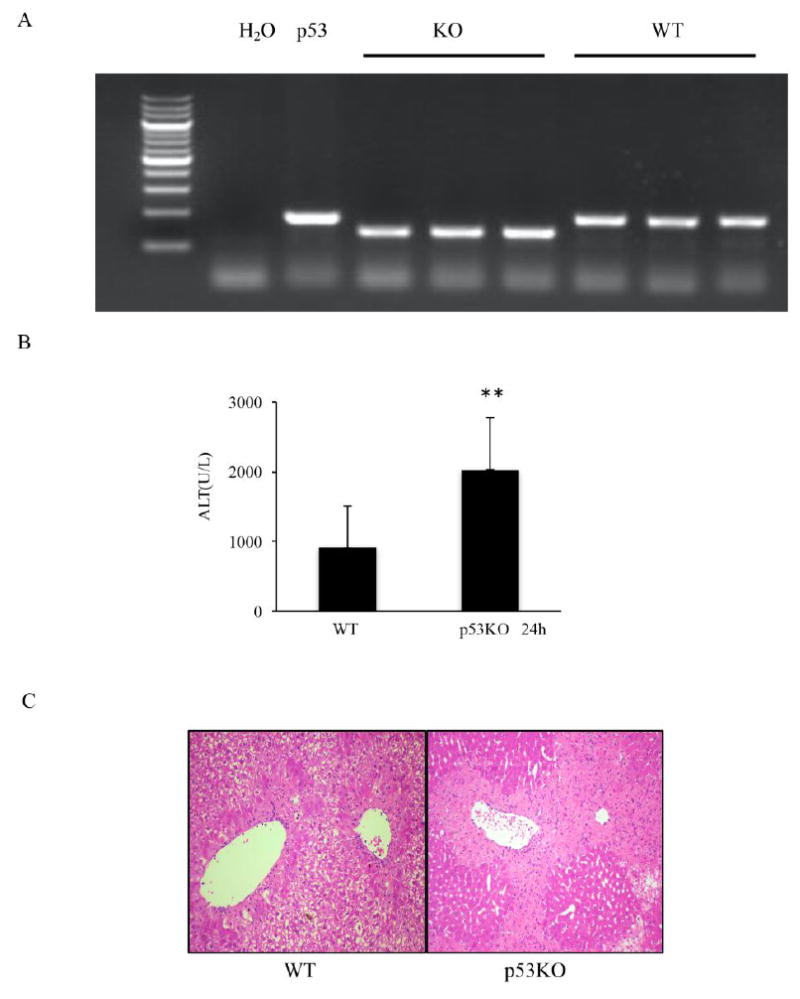
The APAP-induced liver damage in WT and p53 KO mice (C57BL/6J). A. Genotyping in WT and p53 KO mice. B. Serum ALT level in serum and C. H&E histology at 24h. (n=5; **p<0.01).
Taken together, p53 deficiency either by chemical inhibitor, ASO knockdown or gene knockout rendered mice more susceptible to APAP hepatotoxicity, suggesting that p53 plays a protective role in dampening the hepatotoxicity induced by APAP.
The suppression of p53 did not affect the metabolism of APAP
To decipher the molecular basis of the role of p53 in APAP model, we measured the levels of the important APAP metabolism enzyme cyp2e1 in both p53 KO and PFTα-pretreated mice. As shown in Fig. 4A&B, there was no difference with p53 deficiency. In addition, GSH was almost completely depleted at 2h after APAP in both WT and p53 KO mice. p53 deficiency had no influence on the decline of GSH or its recovery (Fig. 4C) and the generation of NAPQI adducts (Fig. 4D). This result suggested that the protective role of p53 was not due to a change of APAP toxic metabolite production.
Fig.4.
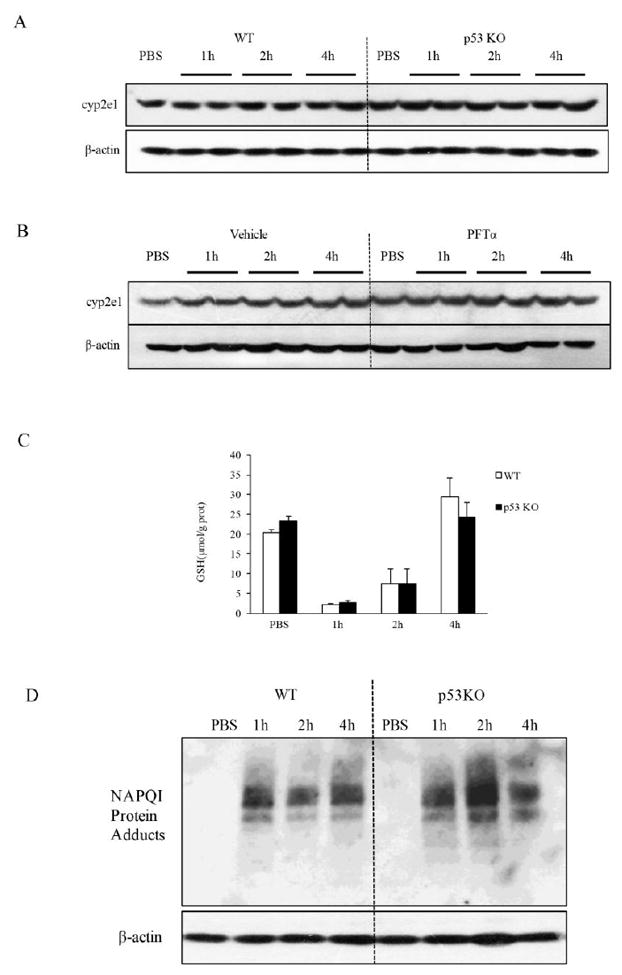
Effect of p53 knockdown or knockout on APAP metabolism. A. Immunoblotting of cyp2e1 after PFTα treatment. B. Immunoblotting of cyp2e1 in both WT and p53 KO mice. C. The levels of GSH in the liver of both WT and p53 KO mice. Comparison of WT and p53 KO group showed no significant differences. D. Immunoblot of NAPQI adducts. Times shown are after APAP dosing.
The suppression of p53 increased the activation of p-JNK
It is well established that APAP induces mitochondrial superoxide which then leads to sustained JNK activation (p-JNK) in the early time course prior to necrosis. APAP administration alone caused upregulation of p-JNK level as expected, while PFTα-pretreated and p53 knockout mice showed markedly enhanced expression of p-JNK (Fig. 5A&B). In order to confirm the specific function of p53 in hepatocytes, we isolated primary mouse hepatocytes after p53 ASO treatment. The p-JNK level in p53 deficient hepatocytes isolated from p53 ASO treated mice was significantly enhanced by 10mM APAP treatment at 4h compared to control ASO hepatocytes (Fig. 5C). These data suggest that endogenous p53 was associated with dampening of sustained p-JNK.
Fig.5.
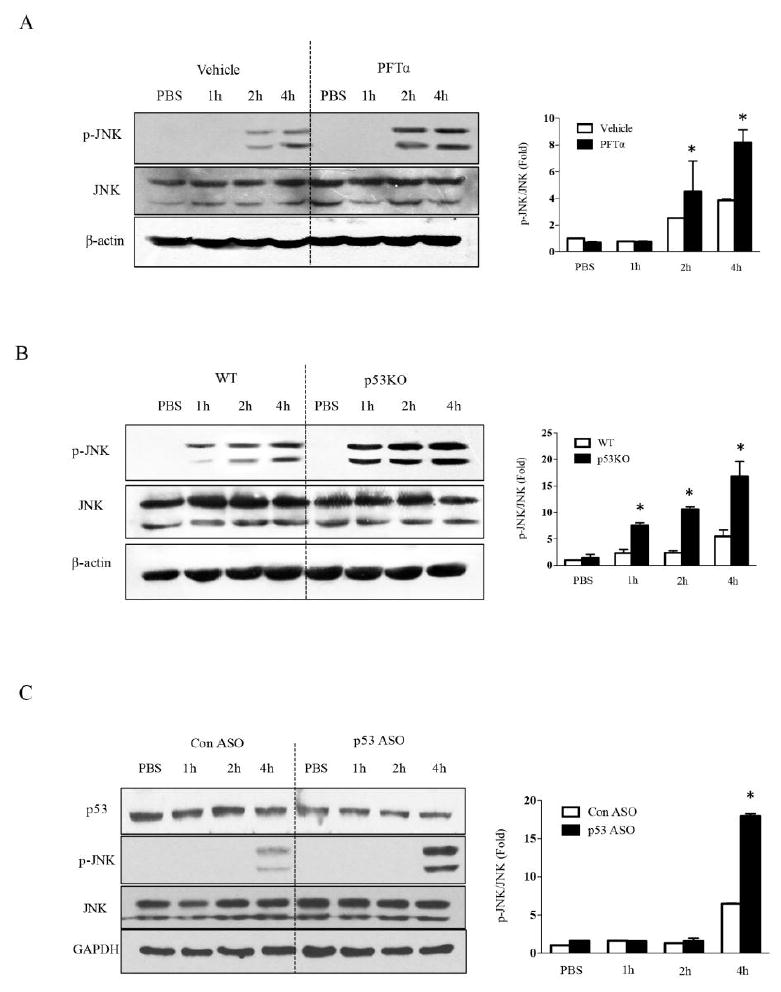
Effect of p53 knockout, knockdown and inhibition on JNK activation. A. The p-JNK level after inhibition of p53 by PFTα. B. The p-JNK level after p53 knockout. C. The p-JNK level induced by APAP following p53 knockdown by ASO in primary mouse hepatocytes. Bar graphs in right panels shows densitometry of p-JNK/JNK of n =3, *p<0.05.
Activation of p53 attenuated the liver injury in APAP model
To ascertain whether pharmacological stimulation of p53 could attenuate the liver injury induced by APAP, we pretreated the mice with 10mg/kg nutlin-3a by gavage (1h before APAP i.p injection) (11). Nutlin 3a is a small-molecule antagonist of MDM2 which stabilizes p53 (12). As shown in Fig. 6A, p53 was increased in nutlin-3a-treated mice compared to vehicle group. Nutlin-3a pretreatment decreased APAP-induced elevation of ALT at 24 hours (Fig. 6B). In line with ALT results, histological analysis also revealed that nutlin-3a alleviated the centrilobular necrosis of the liver after APAP administration (Fig. 6C). These results showed that pharmacological induction of p53 may be beneficial for APAP-induced liver injury. p53 activation had no influence on the decline of GSH (Fig. 6D). To confirm the regulation of p-JNK by p53, we measured the expression of p-JNK after nutlin-3a. p-JNK protein level was downregulated at 6h after APAP (Fig. 6E), suggesting that the protective role of p53 was due to the decrease sustained phosphorylation of JNK induced by APAP.
Fig.6.
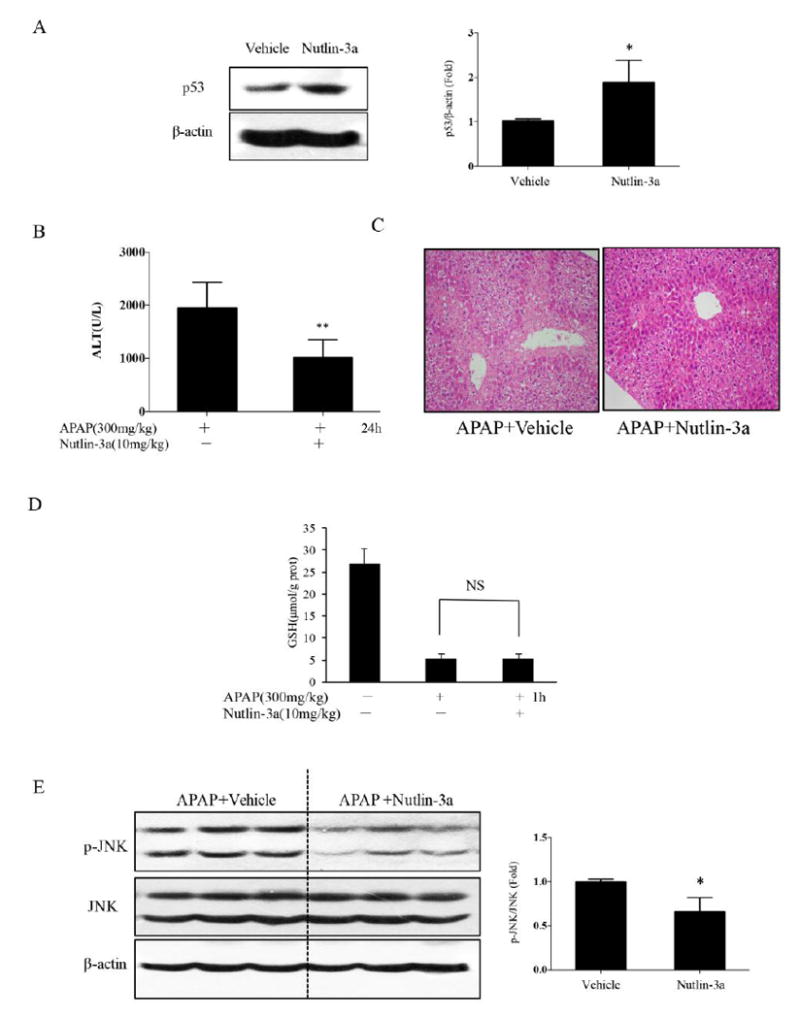
Effect of nutlin-3a induced p53 on APAP toxicity (C57BL/6J). A. Increased p53 6h after nutlin-3a treatment in vivo; the right panel shows densitometry of /β-actin of n=3, *p<0.05. B, C The ALT level and H&E histology at 24h. (n=10 mice, **P<0.01) D. The levels of GSH in the liver after nutlin-3a treatment. (n=3 mice) E. The activation of JNK at 6h after APAP injection in mice after nutlin-3a 1h pretreatment. Bar graphs at right shows densitometry of p-JNK/JNK of n =3, *p<0.05.
Discussion
Activation of p53 can function in either pro-death or pro-survival signaling in response to oxidative stress. Oxidative stress has been shown to play a critical role in APAP-induced hepatotoxicity(13). The objective of the present studies was designed to address the functional role of p53 in APAP-induced acute liver injury. The results of the present study showed that inactivation of p53 by either genetic approach or a chemical inhibitor led to a significant enhanced APAP-induced hepatotoxicity, whereas activation of p53 by its activator resulted in an attenuated liver injury by APAP, indicating a pro-survival function of p53 in response to APAP exposure. The findings of the present study suggest that manipulation of p53 may be a useful approach to protect against APAP-induced acute liver injury.
Regarding the mechanisms underlying the protective role of p53 in APAP-induced liver injury, we first investigated influence of p53 inactivation on APAP-induced cyp2e1 and GSH depletion, and the results indicated that inhibition of p53 affected neither of these parameters, suggesting the toxic metabolism stage of APAP is not the target for p53 to exert its protective activity. We next assessed the effect of p53 inactivation on the changes of APAP-induced sustained JNK activation, a key event downstream of APAP metabolism but upstream of APAP-induced necrosis. The data revealed that JNK phosphorylation was enhanced when p53 was inactivated by either knockout or knockdown or pharmacological approach. Conversely, JNK phosphorylation was decreased when p53 was activated by nutlin-3a, an inhibitor of MDM2 which then inhibits p53 degradation. Together, these results suggest that p53 can counteract APAP-mediated toxic JNK activation. The findings provide a mechanistic support for the protective role of p53 in APAP-induced liver injury.
p53 plays a dual role not only in the regulation of cell death, but also in the modulation of redox (14). The pro-oxidant activity of p53 is found to contribute to its pro-death action, whereas p53’s anti-oxidant activity is correlated with its pro-survival function (15). It has been shown that JNK is initially activated by oxidative stress possibly through activating MAP3K and inhibiting MAPK phosphatases (16, 17). Phosphorylated JNK then translocates to the mitochondria and binds to Sab, a scaffold protein, leading to disruption of mitochondrial electron transport chain and the production of ROS, which causes continuously activated MAPK cascade through a self-sustaining MLK3/ASK1→p-MKK4→p-JNK→Sab→ROS pathway (18, 19). Based on the relationship between ROS generation and JNK activation in response to APAP exposure, we speculate that p53 activation by APAP exerts anti-oxidant activity possibly through up-regulation of a number of anti-oxidant enzymes to inhibit ROS-JNK-ROS loop. An alternative hypothesis is that p53 directly inhibits p-JNK. p53 has been shown to bind to p-JNK and inhibits its activity which would therefore potentially interrupt the p-JNK→Sab→ROS→→p-JNK loop sustaining pathway. These hypotheses need to be tested in future studies.
In summary, p53 was activated in response to APAP-mediated oxidative stress and p53 protected against APAP-induced hepatotoxicity accompanied by suppression of sustained JNK activation, leading to the hypothesis that p53 interference with the JNK activation pathway accounts for its protective role in APAP-induced liver injury.
Highlights.
Inactivation of p53 by either genetic approach or a chemical inhibitor leads to a significant enhanced APAP-induced hepatotoxicity
Activation of p53 by its activator results in an attenuated liver injury by APAP
p53 plays a novel protective role in APAP induced liver injury through inhibiting the activation of JNK, a key mediator in APAP-induced oxidative stress
Acknowledgments
This work was supported by a grant from the Ministry of Science and Technology of China (2012BAD33B09) and National Natural Science Foundation of China (NSFC31671945) and by NIH grant R01DK067215 (NK) and Microscopy, Histology, and Cell Separation and Culture Core of the USC Research Center (P30DK48522).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boyd EM, Bereczky GM. Liver necrosis from paracetamol. Br J Pharmacol Chemother. 1966;26:606–614. doi: 10.1111/j.1476-5381.1966.tb01841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung C, et al. The cyp2e1-humanized transgenic mouse: role of cyp2e1 in acetaminophen hepatotoxicity. Drug metabolism and disposition: the biological fate of chemicals. 2005;33:449–457. doi: 10.1124/dmd.104.002402. [DOI] [PubMed] [Google Scholar]
- 3.McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30:2174–2187. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 5.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- 6.H S-Z, Pang-Kuo Lo, Hsiang-Chin Chen, Fung-Fang Wang. The prosurvival activity of p53 protects cells from UV-induced apoptosis by inhibiting c-Jun NH2-terminal kinase activity and mitochondrial death signaling. Cancer research. 2004;64:8736–8745. doi: 10.1158/0008-5472.CAN-04-2584. [DOI] [PubMed] [Google Scholar]
- 7.Singh B, et al. p53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes Dev. 2002;16:984–993. doi: 10.1101/gad.973602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stamper BD, et al. p53 Contributes to Differentiating Gene Expression Following Exposure to Acetaminophen and Its Less Hepatotoxic Regioisomer Both In Vitro and In Vivo. Gene Regul Syst Bio. 2015;9:1–14. doi: 10.4137/GRSB.S25388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng G, Kaplowitz N. Colchicine protects mice from the lethal effect of an agonistic anti-Fas antibody. J Clin Invest. 2000;105:329–339. doi: 10.1172/JCI7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 11.Li X, et al. The MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic beta-cells. Nat Commun. 2016;7:11740. doi: 10.1038/ncomms11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 13.Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicology and applied pharmacology. 2010;246:8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang MY, et al. The critical role of catalase in prooxidant and antioxidant function of p53. Cell death and differentiation. 2013;20:117–129. doi: 10.1038/cdd.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borras C, Gomez-Cabrera MC, Vina J. The dual role of p53: DNA protection and antioxidant. Free radical research. 2011;45:643–652. doi: 10.3109/10715762.2011.571685. [DOI] [PubMed] [Google Scholar]
- 16.Palit S, Kar S, Sharma G, Das PK. Hesperetin Induces Apoptosis in Breast Carcinoma by Triggering Accumulation of ROS and Activation of ASK1/JNK Pathway. J Cell Physiol. 2015;230:1729–1739. doi: 10.1002/jcp.24818. [DOI] [PubMed] [Google Scholar]
- 17.Kamata H, et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 18.Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. The Journal of biological chemistry. 2011;286:35071–35078. doi: 10.1074/jbc.M111.276089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Win S, Than TA, Min RW, Aghajan M, Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology. 2016;63:1987–2003. doi: 10.1002/hep.28486. [DOI] [PMC free article] [PubMed] [Google Scholar]