

Summary
G6PD deficiency, an enzymopathy affecting 7% of the world population, is caused by over 160 identified amino acid variants in glucose-6-phosphate dehydrogenase (G6PD). The clinical presentation of G6PD deficiency is diverse, likely due to the broad distribution of variants across the protein and the potential for multidimensional biochemical effects. In this study, we use bioinformatic and biochemical analyses to interpret the relationship between G6PD variants and their clinical phenotype. Using structural information and statistical analyses of known G6PD variants, we predict the molecular phenotype of five uncharacterized variants from a reference population database. Through multidimensional analysis of biochemical data, we demonstrate that the clinical phenotypes of G6PD variants are largely determined by a trade-off between protein stability and catalytic activity. This work expands the current understanding of the biochemical underpinnings of G6PD variant pathogenicity, and suggests a promising avenue for correcting G6PD deficiency by targeting essential structural features of G6PD.
Keywords: G6PD, G6PD deficiency, enzymopathy, missense variants, ExAC database, PCA, variants of unknown significance (VUS), protein stability, enzyme activity
eTOC Blurb
G6PD deficiency is one of the most common human enzymopathies, but the relationship between amino acid variant and clinical phenotype is poorly understood. Cunningham et al. find that clinical severity of a G6PD variant is determined by coupling between catalytic activity and protein stability.
Introduction
As the rate-limiting enzyme in the pentose phosphate pathway, glucose-6-phosphate dehydrogenase (G6PD) catalyzes the oxidation of glucose-6-phosphate (G6P) and concomitant reduction of NADP+ to NADPH (Cappellini and Fiorelli, 2008). NADPH then regenerates the essential antioxidant glutathione, and is therefore important in maintaining redox homeostasis, especially in red blood cells, which lack mitochondria (Cappellini and Fiorelli, 2008). Certain single amino acid variants in G6PD lead to G6PD deficiency, one of the most common Mendelian diseases (Cappellini and Fiorelli, 2008). Roughly 7% of the world population is affected, with a geographic distribution that is strongly correlated with malaria prevalence, as G6PD deficiency protects against malaria (Nkhoma et al., 2009; Vulliamy et al., 1992). G6PD deficiency is typically characterized by hemolytic episodes after acute oxidative insults; in rare severe cases, G6PD-deficient patients suffer from chronic non-spherocytic hemolytic anemia (CNSHA) (Cappellini and Fiorelli, 2008).
More than 160 unique missense variants in G6PD have been identified to cause G6PD deficiency, although their effects on G6PD biochemistry and disease phenotype vary widely (Luzzatto, 2006). In an attempt to address the biochemical, clinical, and genetic heterogeneity of G6PD deficiency, the World Health Organization (WHO) stratified patients with G6PD deficiency into four classes based on clinical presentation and G6PD activity in patient blood samples: Class I (<10% activity and CNSHA), II (<10% activity and hemolytic episodes), III (10–60% activity and hemolytic episodes), and IV (60–150% activity and no clinical manifestations) (Luzzatto, 2006; Organization, 1967). However, these classifications are often determined via measurement of G6PD activity in the blood of single subjects, and are possibly influenced by additional genetic, temporal, and environmental factors (Minucci et al., 2009; von Seidlein et al., 2013).
The diverse clinical presentation of G6PD deficiency motivates an equally diverse understanding of the molecular effects of G6PD variants. However, the molecular mechanisms of pathological G6PD variants remain largely unknown. Biochemical characterization of G6PD variants has revealed that pathogenic variants exhibit a range of complex multidimensional effects, including changes in kinetic activity, thermostability, and protein folding (Boonyuen et al., 2016; Gómez-Manzo et al., 2015; Gómez-Manzo et al., 2016; Gómez-Manzo et al., 2014; Huang et al., 2008; Wang and Engel, 2009; Wang et al., 2005, 2006). Crystal structures of human G6PD (Au et al., 2000; Kotaka et al., 2005) identified a dimeric or tetrameric enzyme with two bound NADP+ molecules per subunit: one in the catalytic site, and another in an allosteric site, named the structural NADP+ for its importance in the thermostability and long-term stability of G6PD (Wang et al., 2008). Class I variants often fall near the structural NADP+ site and exhibit decreased thermostability, suggesting that CNSHA associated with G6PD deficiency may result from G6PD instability and subsequent depletion of G6PD in red blood cells (Gómez-Manzo et al., 2014; Wang and Engel, 2009). However, class I variants are also found in many other structural regions of G6PD, and the relationship between the structural or biochemical effects of a G6PD variant and its clinical phenotype remains poorly understood.
To elucidate the biochemical mechanisms underpinning the diverse phenotypes of G6PD variants, we combine statistical analyses with biochemical characterization of clinically relevant G6PD variants and variants identified from ExAC, a sequencing database of multiple large cohorts. We find highly significant relationships between the structural location of a G6PD variant, its effects on enzyme activity and stability, and its clinical outcome. This work provides insight into how competing evolutionary pressures and biological requirements have shaped the biochemical landscape of G6PD variants, predicts the phenotype of uncharacterized G6PD variants that appear in reference population databases, and suggests a promising avenue for treatment of severe G6PD deficiency.
Results
Structural distribution of G6PD variants
We defined structural regions (G6P and NADP+ binding sites and oligomer interfaces) by calculating solvent-accessible surface area using the three available crystal structures of human G6PD (PDB IDs: 1QKI, 2BH9, 2BHL; Methods) (Au et al., 2000; Kotaka et al., 2005). Reported variants (Benmansour et al., 2013; Chaves et al., 2016; Garcia-Magallanes et al., 2014; Jang et al., 2015; Minucci et al., 2012; Warny et al., 2015) and structural regions were then mapped onto a linear representation of G6PD (Fig. 1A,B) and onto the crystal structure (Fig. 1C–H). As previously speculated (Wang et al., 2008), the structural NADP+ binding site is significantly enriched in class I variants (p < 0.05 by Fisher’s exact test). A lack of pathogenic variants at the substrate and cofactor binding sites (p < 0.05) is consistent with the finding that complete loss of G6PD activity is embryonic lethal (Longo et al., 2002). The dimer interface is significantly enriched in pathogenic variants (class I, II, and III, p < 0.005), especially class I variants (p < 0.001). However, there is no significant enrichment of any variants at the tetramer interface. This pattern of variation suggests that while loss of dimerization is detrimental to G6PD activity, tetramerization may not be necessary for enzyme function. Taken together, the structural distribution of pathogenic variants highlights the importance of the dimer interface and structural NADP+ binding site for G6PD function.
Figure 1. G6PD variant classes are distributed differently across the G6PD structure.
A) Linear representation of G6PD showing location of variants and structural regions. Structural regions shown are the G6P, catalytic (cat.) NADP+, and structural (str.) NADP+ binding sites, and dimer and tetramer interfaces. Variants shown are class I–IV and uncharacterized (unchar) variants from the ExAC database.
B) Quantification of the number of variants in each structural region (left) and the percent of amino acids in each region for which a variant has been identified (right).
C) Crystal structure of dimeric G6PD, assembled from PDB IDs 2BH9 and 2BHL.
D–H) Variant locations are shown in spheres on the monomeric structure of G6PD: (D) class I, (E) class II, (F) class III, (G) class IV, (H) uncharacterized variants from the ExAC database.
Structural distribution and pathogenicity prediction of uncharacterized variants from the ExAC database are similar to class IV variants
The recent availability of large sequencing databases provides an excellent opportunity for examining enzyme variation across multiple populations. The Exome Aggregation Consortium (ExAC) database, which catalogs exome sequences from over 60,000 unrelated individuals (Lek et al., 2016), contains 101 single missense variants in G6PD, of which 64 have not been previously reported (Table S1). The ExAC database generally excludes individuals with pediatric illnesses, so as expected none of the 37 previously reported variants in ExAC were class I variants (Table S2). We therefore surmised that the 64 uncharacterized variants are also unlikely to be class I. We also examined allele frequencies of the previously known and uncharacterized variants, as variants of high allele frequency are often benign (Salgado et al., 2016). However, we found high allele frequencies of known pathogenic G6PD mutations (Fig. S1, Tables S1, S2), indicative of the selective advantage of G6PD variants against malaria, making allele frequency of the uncharacterized variants difficult to interpret.
Among these uncharacterized variants identified in the ExAC database, we observed significant enrichment on the surface of the protein (p < 0.001) and depletion in the interior (p < 0.01) and on the dimer interface (p < 0.05). This structural distribution is most similar to the four known class IV mutations, of which two are on the surface and none are in the interior, as well as the class III mutations, of which half are on the surface. Indeed, mutations on the protein surface are less likely to be deleterious than mutations buried in the interior (Ng and Henikoff, 2006); therefore, the uncharacterized variants on the surface of G6PD are likely nonpathogenic.
Because structural location (and therefore sequence position) is a major contributor to mutation severity (Adzhubei et al., 2010; Kumar et al., 2014; Ng and Henikoff, 2003), we examined how many of these uncharacterized variants occur at the same amino acid position as known pathogenic variants. Overlap between uncharacterized and pathogenic variants would suggest that the overlapping uncharacterized variants are also likely to cause pathology. We found only one-quarter as much overlap between uncharacterized and pathogenic variants as expected (p < 0.001 by Fisher’s exact test, Fig 2A). Interestingly, uncharacterized variants overlapped significantly with three of the four known class IV variants (p < 0.001).
Figure 2. Structural distribution and pathogenicity prediction of uncharacterized variants from the ExAC database are similar to class IV variants.
See also Tables S1, S2, and Fig. S1.
A) Odds ratio of the sequence positional overlap between uncharacterized variants and known variants (class I, II, III, or IV) or all pathogenic (I, II and III) variants. An odds ratio of 1 indicates expected overlap; a ratio below 1 indicates less overlap than expected and a ratio above 1 indicates more overlap than expected. p-values are represented as: ** < 0.005; *** < 0.0005.
B,C) Prediction of variant severity using two prediction algorithms: (B) SIFT, which assigns a score between 0 (damaging) and 1 (benign); and (C) PolyPhen2, which assigns a score between 1 (damaging) and 0 (benign). Unchar, uncharacterized variants from ExAC.
D) kcat measurements of wild-type (WT) G6PD and the five uncharacterized variants examined in this study. A line is shown at 60% activity, which delineates the separation between class III and class IV. Data are represented as mean ± SD.
E) T1/2 measurements of wild-type (WT) G6PD and the five uncharacterized variants. kcat and T1/2 measurements both support class IV characterization. Data are represented as mean ± SD.
To further evaluate the pathogenicity of these uncharacterized variants, we used two prediction algorithms: SIFT, which uses evolutionary conservation (Ng and Henikoff, 2003), and PolyPhen2, a machine learning method that combines chemical similarity, sequence information, and 3D structural information (Adzhubei et al., 2010). To test the reliability of these prediction algorithms, we included analysis of the 166 variants for which clinical classification has been previously reported (Fig. 2B,C). Both algorithms showed a trend toward predicting class I variants to be more damaging and class IV variants to be more benign. Using both prediction algorithms, the uncharacterized variants were predicted to be more benign than class I, II, and III variants (p < 0.001 by one-way ANOVA followed by Tukey’s multiple comparison test, Fig. 2B,C), and not significantly different from the class IV variants. However, all of the class groups contained outliers, showing that these prediction algorithms are limited, or that the clinical manifestations of G6PD variants are likely structurally and biochemically complex and may be affected by additional genetic and environmental factors.
Biochemical characterization of G6PD variants reveals diverse effects of different amino acid substitutions
Although sequence position of a protein variant is a major determinant of variant severity, the chemical difference between the original and substituted amino acid is also important (Adzhubei et al., 2010). Of G6PD variant pairs in which two different amino acid variants have been found at the same sequence position, 32 pairs yield the same class of G6PD deficiency, while 19 pairs yield different classes of G6PD deficiency. To further understand the biochemical consequence of amino acid substitutions, we expressed, purified, and biochemically characterized several clinically relevant G6PD variants, focusing on four pairs of variants in which the same amino acid position was changed to two different amino acids. In particular, we chose variant pairs that were assigned the same prediction by SIFT and PolyPhen2, yet yielded two different classes of G6PD deficiency: Y70H (II) and Y70C (III); R198P (I) and R198H (II); E398K (I) and E398G (II); and Q307H (I) and Q307P (III) (Table S3). Q307P yielded too little protein for biochemical characterization, and was excluded from subsequent analyses. For each variant we measured activity parameters (Km, kcat) and stability (T1/2, the temperature at which half of G6PD activity is retained) using WHO standard protocols (Table S3 and Supplemental Experimental Procedures).
To further predict pathogenicity of uncharacterized variants from the ExAC database, we also biochemically characterized five of these variants, sampling a wide range of SIFT and PolyPhen2 prediction scores (Table S3). We found that T1/2 of all five variants was as high as or higher than that of WT G6PD, and kcat of four of these variants was within the class IV activity range of 60–150% (Fig. 2D,E); the fifth had kcat slightly below this range. Thus, in subsequent analysis we treat these five uncharacterized variants as class IV.
Principal component analysis of biochemical data reveals trade-off between activity and stability
Initial inspection of the biochemical data from the selected G6PD variants did not reveal obvious trends, suggesting that there are couplings between biochemical parameters that might be revealed through dimensional reduction. We subjected the data to principal component analysis, using our kinetic and stability measurements. To avoid bias from curve fitting, rather than using fitted kinetic and stability parameters we used a vector containing the raw data from activity and stability measurements (27 data points, Fig. 3A; Methods). Principal components (PCs) 1 and 2 accounted for 50% and 24% of the variance in the data, respectively. Projecting the 13 variants onto these two components, we found that each pair of pathogenic variants at the same amino acid position were biochemically distinct from each other, highlighting the importance of amino acid chemical properties in determining the severity of a protein variant.
Figure 3. Principal component analysis (PCA) of biochemical data reveals biochemical separation between G6PD variant classes.
See also Table S3.
A) Representation of data vectors used for PCA. Normalized median curves of kinetic and biochemical measurements for each G6PD variant are shown in black (wild-type), red (class I), orange (class II), yellow (class III), or green (predicted class IV).
B) Biochemical characterization of 13 G6PD variants projected onto PC 1 and 2.
C,D) Values of principal components (PC) 1 (C) and 2 (D), which represent correlation and anticorrelation, respectively, between activity and stability.
Interestingly, the five uncharacterized variants that we predicted to be class IV clustered together near WT G6PD (Fig. 3B), further validating that these variants are biochemically more similar to WT G6PD than are pathogenic variants. The three class II variants also visually clustered together, and separated from WT/class IV (Fig. 3B). To probe the basis of this separation, we examined the biochemical signatures encoded by PCs 1 and 2 (Fig. 3C,D). PC 1 consisted of mainly positive values, reflecting correlation between activity and stability (i.e. a variant having high activity and high stability, or low activity and low stability). Interestingly, PC 2 contained negative values for the activity measurements and positive values for the stability measurements, reflecting anticorrelation between activity and stability (i.e. a variant having high activity and low stability, or low activity and high stability). This analysis suggests that the clinical phenotype of a G6PD variant is determined by both its overall performance and by a trade-off between catalytic activity and protein stability.
Generalized principal component axes separate G6PD variants into clusters by class
We then generalized the PCs to other previously characterized G6PD variants by plotting the variants from this study and 20 variants from previous work (Boonyuen et al., 2016; Gómez-Manzo et al., 2015; Gómez-Manzo et al., 2016; Gómez-Manzo et al., 2014; Huang et al., 2008; Wang et al., 2008; Wang and Engel, 2009; Wang et al., 2005) (Table S4) onto axes roughly corresponding to the principal components. To reflect correlation and anticorrelation between stability and activity, we compared normalized values of T1/2+kcat and T1/2−kcat, respectively. We found that class II, III, and IV variants segregated visually by class (Fig. 4A, S2) and were quantitatively clustered by silhouette scoring (Fig. 4B). This clustering was not recapitulated by taking into account kcat (Fig. S3A) or T1/2 (Fig. S3B) alone, highlighting the importance of trade-offs between them in determining clinical severity.
Figure 4. PCA-like visualization suggests additional functionality of some class I variants.
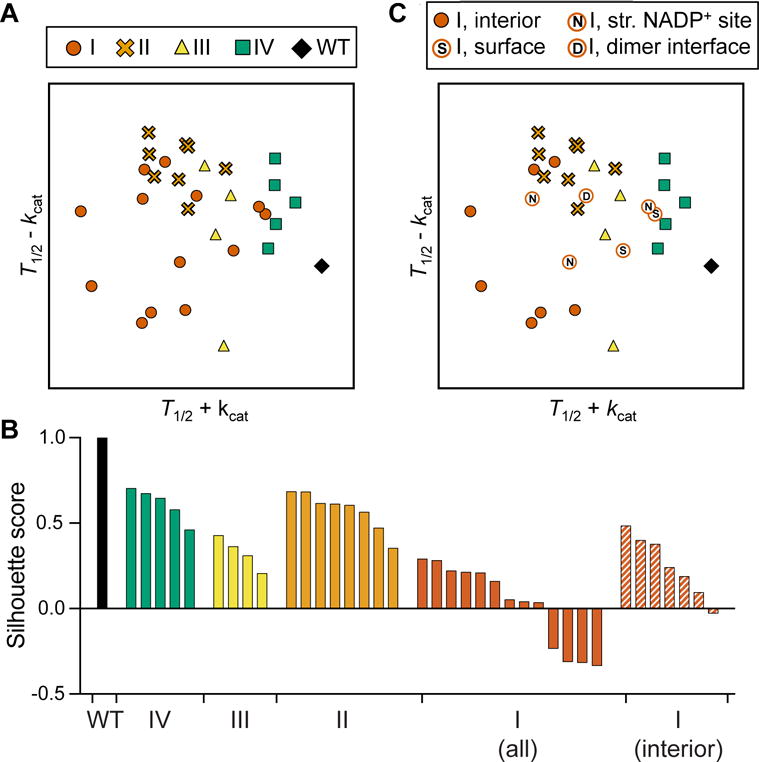
See also Table S4 and Fig. S2, S3.
A) Analysis of 13 G6PD variants from this study and 20 additional variants from previous studies, represented by normalized T1/2+kcat and T1/2−kcat.
B) Silhouette scores of each variant cluster from (A) (WT, IV, II, III, and I) and (C) (class I, excluding variants that are not in the protein interior). A silhouette score ranges from −1 to 1, with a higher score indicating that a point is matched well to its cluster and matched poorly to other clusters.
C) PCA-like plot with class I variants labeled corresponding to their structural location (S: surface, N: structural NADP+, D: dimer interface). Class I variants in solid red circles are located in the protein interior.
In particular, previous work has suggested that class I variants cause CNSHA due to decreased enzyme stability and hence lower enzyme levels in red blood cells. In this study, we observed several class I variants that confirm this finding, but we also show that not all class I variants display reduced stability or enzyme activity in vitro. In the generalized PCA-like plot (Fig. 4A), the class I variants did not cluster significantly; we speculate several possible reasons for this. Because class I mutations are rare, most documented class I mutations are single case study reports. Thus, it is possible that some class I mutations may only mildly disrupt G6PD activity, but could nevertheless contribute to CNSHA when combined with a patient’s unique genetic background. Another possibility is that some class I variants disrupt the function of G6PD in a manner not captured by the assay conditions used in vitro. For example, the high concentrations of Mg2+, NADP+, or G6PD enzyme in vitro may mask the effects of class I variants on dimerization or structural NADP+ binding under physiological conditions. Class I variants on the surface may also disrupt an essential protein-protein interaction in vivo. Indeed, when class I variants located on the protein surface, structural NADP+ site, or dimer interface are excluded from the clustering analysis, the remaining class I variants form a tighter cluster as quantified by silhouette score (Fig. 4B,C).
Discussion
G6PD is ubiquitously expressed and essential for maintaining redox homeostasis in all tissues and cell types. As a result, there are conflicting evolutionary pressures that shape its mutational landscape. Although complete loss of enzyme function is lethal, mild loss of function is advantageous as it protects against malaria. This loss of function can occur through different biochemical mechanisms leading to different clinical outcomes. Based on our PCA of biochemical characterization, unsurprisingly the overall fitness of the enzyme (good stability and activity) is the primary determinant of the clinical outcome of a G6PD variant (PC1, Fig. 3C). Interestingly, as shown by PC2 (Fig. 3D), the clinical outcome is also largely determined by trade-off between stability and activity. The importance of this trade-off is consistent with the necessity for G6PD to retain NADPH-producing activity in all cell types while also remaining stable in red blood cells, which contain no translational machinery, for the lifetime of a red blood cell (up to 110 days).
Taken together, our biochemical characterization and meta-analysis of G6PD variants has clarified the biochemical underpinning that determines the severity of G6PD deficiency. We found that activity or stability alone does not determine or predict the phenotype of a G6PD variant, but rather a combination of both yields significant separation of variants by class. This finding is a crucial advance that informs previous work in which a quantitative model of G6PD kinetics, which did not include protein stability as a parameter, was unable to segregate class I variants into a biochemical space consistent with the CNSHA phenotype (Coelho et al., 2010).
We found that the structural distribution of the uncharacterized and known variants is similar to that of class IV variants (Fig. 2A), and that the predicted pathogenicity of these variants is not significantly different from the class IV variants (Fig. 2B,C). Furthermore, based on the biochemical signatures of five uncharacterized variants from the ExAC database, we predict that these five variants are likely nonpathogenic (class IV), even though one variant (R182W) was predicted to be pathogenic by SIFT (Table S3). However, other uncharacterized variants that were predicted to be severe by SIFT or PolyPhen2, or that lie in the dimer interface, may be outliers in this group and should be further characterized biochemically to predict their phenotype.
Consistent with previous work, we found that G6PD variants with the lowest stability often result in a worse clinical outcome (Fig. 4A). Therefore, we propose that efforts toward identifying a therapy to treat G6PD deficiency should focus on increasing enzyme stability. The structural NADP+ site is known to be important for enzyme stability and we found significant enrichment of class I variants in this site, suggesting that therapies that improve structural NADP+ binding may rescue severe G6PD deficiency. Additionally, we found that the dimer interface is significantly enriched in pathogenic variants, especially class I variants. This suggests enzyme dimerization as a promising target for rescuing G6PD deficiency of any type. Nonetheless, our data indicate that the efficacy of treatment for patients with severe symptoms may be variant-dependent.
Beyond acute and chronic anemia, there are many other pathologies associated with reduced G6PD activity. G6PD deficiency increases the risk of kernicterus and death from neonatal jaundice (Cunningham et al., 2016) and has also been associated with bipolar and schizoaffective disorders (Raj et al., 2014), erectile dysfunction (Morrison et al., 2014), and vitiligo (Namazi, 2015). Because G6PD plays an essential role in maintaining healthspan by protection against oxidative damage (Nóbrega-Pereira et al., 2016), the effects of G6PD deficiency on human health have likely been underestimated and thus it is expected that additional consequences of G6PD deficiency will be identified in the future (Spencer and Stanton, 2016; Stanton, 2012). Our biochemical and informatics-based study suggests a promising avenue for treatment of G6PD deficiency and its sequelae by targeting enzyme stability, structural NADP+ binding, or dimerization.
Experimental Procedures
Definition of structural regions
For each available crystal structure of human G6PD (PDB IDs 1QKI, 2BH9, and 2BHL), solvent accessible surface area (SASA) was calculated using AREAIMOL (Lee and Richards, 1971; Saff and Kuijlaars, 1997) in the CCP4 suite (Winn et al., 2011). Structural regions were defined by changes in SASA; for example, an amino acid was included in the dimer interface if the SASA was reduced in the dimeric structure compared to the monomeric structure. Amino acids that were not at an oligomeric interface or G6P- or NADP+-binding site were designated as “surface” (SASA > 25%) or “interior” (SASA < 25%) (Levy, 2010). For ease of visualization in Fig. 1A, structural regions were approximated by blocks spanning the densest clusters of amino acids in each region. Structure images were generated using PyMol v. 1.7.6.6.
Analysis of Exome Aggregation Consortium (ExAC) database mutations
High-quality missense variants (genotype quality ≥ 20 and depth ≥ 10) in the G6PD transcript ENST00000393564, as of October, 2016, were collected from the ExAC Browser (Lek et al., 2016) and compared with G6PD variants reported in the literature (Benmansour et al., 2013; Chaves et al., 2016; García-Magallanes et al., 2014; Jang et al., 2015; Minucci et al., 2012; Warny et al., 2015). Variants in ExAC that were not previously reported were designated “uncharacterized”. Uncharacterized variants and variants from the literature were submitted by batch query to SIFT (Ng and Henikoff, 2003) (Ensembl protein ID: ENSP00000377194) and PolyPhen2 (Adzhubei et al., 2010) (Uniprot ID: P11413) using the default settings.
Principal component analysis (PCA)
For each G6PD variant, a vector was assembled using the median value for each data point measured: activity measurements across varying [NADP+] (8 data points), activity measurements varying [G6P] (7 data points), and stability measurements (12 data points) (see Enzyme activity and stability measurements in the Supplemental Experimental Procedures for further description of biochemical measurements). The data were then normalized by subtracting the mean and dividing by the standard deviation at each position in the vector. PCA was performed using the Python scikit-learn module (sklearn.decomposition.PCA, v. 0.17.1) (Pedregosa et al., 2011).
Generation of PCA-like plot
T1/2 and kcat data from G6PD variants previously purified and biochemically characterized following WHO standards (Boonyuen et al., 2016; Gómez-Manzo et al., 2015; Gómez-Manzo et al., 2016; Gómez-Manzo et al., 2014; Huang et al., 2008; Wang et al., 2008; Wang and Engel, 2009; Wang et al., 2005) were combined with data from this study, then normalized by subtracting the mean and dividing by the standard deviation. The PCA-like plot (T1/2−kcat versus T1/2+kcat) was then generated using these normalized values.
Silhouette scoring
Silhouette scoring was calculated using a custom Python script. Each cluster was defined as variants in the same clinical class. For each cluster, the nearest cluster was identified by comparing the mean center of each cluster. Then for each point in the cluster, the silhouette score (s) was defined as:
where a is the mean distance from the point to all other points in the cluster and b is the mean distance from the point to all other points in the nearest cluster. A silhouette score ranges from −1 to 1, with a higher score indicating that a point is matched well to its cluster and matched poorly to other clusters.
Supplementary Material
Highlights.
Structural distribution of G6PD variants identifies critical structural regions of G6PD
Five previously uncharacterized G6PD variants from ExAC are likely nonpathogenic
Coupling between catalytic activity and stability determines variant phenotype
Severe G6PD variants affect functions not captured by standard biochemistry protocols
Acknowledgments
This work was supported in part by NIH R01 HD08442 (to D.M.-R.), NSF CAREER Award MCB-1149328 (to K.C.H.), a seed grant from the Stanford Center for Systems Biology under Grant P50-GM107615, Stanford Graduate Fellowships (to A.D.C. and A.C.), a Gerald J. Lieberman Fellowship (to A.C.), and an NSF Graduate Research Fellowship (to A.D.C.). We thank C. Liu, S. Hwang, and A. Raub for their critical readings of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, A.D.C and A.C.; Methodology, A.D.C., A.C., K.C.H, D.M.-R.; Investigation, A.D.C.; Resources, D.M.-R.; Writing — Original Draft, A.D.C; Writing — Review & Editing, A.C., K.C.H., D.M.-R.; Visualization, A.D.C.; Supervision, K.C.H. and D.M.-R.; Funding Acquisition, A.D.C., A.C., K.C.H., D.M.-R.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au SW, Gover S, Lam VM, Adams MJ. Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP(+) molecule and provides insights into enzyme deficiency. Structure (London, England: 1993) 2000;8:293–303. doi: 10.1016/s0969-2126(00)00104-0. [DOI] [PubMed] [Google Scholar]
- Benmansour I, Moradkhani K, Moumni I, Wajcman H, Hafsia R, Ghanem A, Abbès S, Prèhu C. Two new class III G6PD variants [G6PD Tunis (c.920A>C: p.307Gln>Pro) and G6PD Nefza (c.968T>C: p.323 Leu>Pro)] and overview of the spectrum of mutations in Tunisia. Blood Cells, Molecules & Diseases. 2013;50:110–114. doi: 10.1016/j.bcmd.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Boonyuen U, Chamchoy K, Swangsri T, Saralamba N, Day NPJ, Imwong M. Detailed functional analysis of two clinical glucose-6-phosphate dehydrogenase (G6PD) variants, G6PDViangchan and G6PDViangchan+Mahidol: Decreased stability and catalytic efficiency contribute to the clinical phenotype. Molecular Genetics and Metabolism. 2016;118:84–91. doi: 10.1016/j.ymgme.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellini M, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. The Lancet. 2008;371:64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- Chaves A, Eberle SE, Defelipe L, Pepe C, Milanesio B, Aguirre F, Fernandez D, Turjanski A, Feliú-Torres A. Two novel DNA variants associated with glucose-6-phosphate dehydrogenase deficiency found in Argentine pediatric patients. Clinical Biochemistry. 2016;49:10–11. doi: 10.1016/j.clinbiochem.2016.01.018. [DOI] [PubMed] [Google Scholar]
- Coelho PM, Salvador A, Savageau MA. Relating mutant genotype to phenotype via quantitative behavior of the NADPH redox cycle in human erythrocytes. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham AD, Hwang S, Mochly-Rosen D. Glucose-6-Phosphate Dehydrogenase Deficiency and the Need for a Novel Treatment to Prevent Kernicterus. Clinics in Perinatology. 2016;43:341–354. doi: 10.1016/j.clp.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Magallanes N, Luque-Ortega F, Aguilar-Medina EM, Ramos-Payán R, Galaviz-Hernández C, Romero-Quintana JG, Del Pozo-Yauner L, Rangel-Villalobos H, Arámbula-Meraz E. Glucose-6-phosphate dehydrogenase deficiency in northern Mexico and description of a novel mutation. Journal of Genetics. 2014;93:325–330. doi: 10.1007/s12041-014-0366-z. [DOI] [PubMed] [Google Scholar]
- Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, Enríquez-Flores S, De la Mora-De la Mora I, González-Valdez A, García-Torres I, Martínez-Rosas V, Sierra-Palacios E, Lazcano-Pérez F, et al. Mutations of Glucose-6-Phosphate Dehydrogenase Durham, Santa-Maria and A+ Variants Are Associated with Loss Functional and Structural Stability of the Protein. International Journal of Molecular Sciences. 2015;16:28657–28668. doi: 10.3390/ijms161226124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, Serrano-Posada H, González-Valdez A, Martínez-Rosas V, Hernández-Ochoa B, Sierra-Palacios E, Castillo-Rodríguez RA, Cuevas-Cruz M, et al. Functional and Biochemical Characterization of Three Recombinant Human Glucose-6-Phosphate Dehydrogenase Mutants: Zacatecas, Vanua-Lava and Viangchan. International Journal of Molecular Sciences. 2016;17:787. doi: 10.3390/ijms17050787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Manzo S, Terrón-Hernández J, De la Mora-De la Mora I, González-Valdez A, Marcial-Quino J, García-Torres I, Vanoye-Carlo A, López-Velázquez G, Hernández-Alcántara G, Oria-Hernández J, et al. The stability of G6PD is affected by mutations with different clinical phenotypes. International Journal of Molecular Sciences. 2014;15:21179–21201. doi: 10.3390/ijms151121179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Choi MY, Au SWN, Au DMY, Lam VMS, Engel PC. Purification and detailed study of two clinically different human glucose 6-phosphate dehydrogenase variants, G6PD(Plymouth) and G6PD(Mahidol): Evidence for defective protein folding as the basis of disease. Molecular Genetics and Metabolism. 2008;93:44–53. doi: 10.1016/j.ymgme.2007.08.122. [DOI] [PubMed] [Google Scholar]
- Jang MA, Kim JY, Lee KO, Kim SH, Koo HH, Kim HJ. A Novel de novo Mutation in the G6PD Gene in a Korean Boy with Glucose-6-phosphate Dehydrogenase Deficiency: Case Report. Annals of Clinical and Laboratory Science. 2015;45:446–448. [PubMed] [Google Scholar]
- Kotaka M, Gover S, Vandeputte-Rutten L, Au SWN, Lam VMS, Adams MJ. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallographica Section D, Biological Crystallography. 2005;61:495–504. doi: 10.1107/S0907444905002350. [DOI] [PubMed] [Google Scholar]
- Kumar A, Rajendran V, Sethumadhavan R, Shukla P, Tiwari S, Purohit R. Computational SNP analysis: current approaches and future prospects. Cell Biochemistry and Biophysics. 2014;68:233–239. doi: 10.1007/s12013-013-9705-6. [DOI] [PubMed] [Google Scholar]
- Lee B, Richards FM. The interpretation of protein structures: Estimation of static accessibility. Journal of Molecular Biology. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ED. A simple definition of structural regions in proteins and its use in analyzing interface evolution. Journal of Molecular Biology. 2010;403:660–670. doi: 10.1016/j.jmb.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Longo L, Vanegas OC, Patel M, Rosti V, Li H, Waka J, Merghoub T, Pandolfi PP, Notaro R, Manova K, et al. Maternally transmitted severe glucose 6-phosphate dehydrogenase deficiency is an embryonic lethal. The EMBO journal. 2002;21:4229–4239. doi: 10.1093/emboj/cdf426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzatto L. Glucose 6-phosphate dehydrogenase deficiency: from genotype to phenotype. Haematologica. 2006;91:1303–1306. [PubMed] [Google Scholar]
- Minucci A, Giardina B, Zuppi C, Capoluongo E. Glucose-6-phosphate dehydrogenase laboratory assay: How, when, and why? IUBMB life. 2009;61:27–34. doi: 10.1002/iub.137. [DOI] [PubMed] [Google Scholar]
- Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B, Capoluongo E. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the “old” and update of the new mutations. Blood Cells, Molecules & Diseases. 2012;48:154–165. doi: 10.1016/j.bcmd.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Morrison BF, Thompson EB, Shah SD, Wharfe GH. Ischaemic Priapism and Glucose-6-Phosphate Dehydrogenase Deficiency: A Mechanism of Increased Oxidative Stress? The West Indian Medical Journal. 2014;63:658–660. doi: 10.7727/wimj.2013.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namazi MR. What is the important practical implication of detecting decreased G6PD levels in vitiligo? Advanced Biomedical Research. 2015;4:89. doi: 10.4103/2277-9175.156653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annual review of genomics and human genetics. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells, Molecules & Diseases. 2009;42:267–278. doi: 10.1016/j.bcmd.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Nóbrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Viña J, Serrano M. G6PD protects from oxidative damage and improves healthspan in mice. Nature Communications. 2016;7:10894. doi: 10.1038/ncomms10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization, WH. Standardization of procedures for the study of glucose-6-phosphate dehydrogenase: report of a WHO Scientific Group [meeting held in Geneva from 5 to 10 December 1966] 1967 [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- Raj V, Chism K, Minckler MR, Denysenko L. Catatonia and glucose-6-phosphate dehydrogenase deficiency: a report of two cases and a review. Psychosomatics. 2014;55:92–97. doi: 10.1016/j.psym.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Saff EB, Kuijlaars ABJ. Distributing many points on a sphere. The Mathematical Intelligencer. 1997;19:5–11. [Google Scholar]
- Salgado D, Bellgard MI, Desvignes JP, Beroud C. How to Identify Pathogenic Mutations among All Those Variations: Variant Annotation and Filtration in the Genome Sequencing Era. Human Mutation. 2016;37:1272–1282. doi: 10.1002/humu.23110. [DOI] [PubMed] [Google Scholar]
- Spencer NY, Stanton RC. Glucose 6-phosphate dehydrogenase and the kidney. Current opinion in nephrology and hypertension. 2016;26:43–49. doi: 10.1097/MNH.0000000000000294. [DOI] [PubMed] [Google Scholar]
- Stanton RC. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. Iubmb Life. 2012;64:362–369. doi: 10.1002/iub.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Seidlein L, Auburn S, Espino F, Shanks D, Cheng Q, McCarthy J, Baird K, Moyes C, Howes R, Ménard D, et al. Review of key knowledge gaps in glucose-6-phosphate dehydrogenase deficiency detection with regard to the safe clinical deployment of 8-aminoquinoline treatment regimens: a workshop report. Malaria Journal. 2013;12:112. doi: 10.1186/1475-2875-12-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliamy T, Mason P, Luzzatto L. The molecular basis of glucose-6-phosphate dehydrogenase deficiency. Trends in Genetics. 1992;8:138–143. doi: 10.1016/0168-9525(92)90372-B. [DOI] [PubMed] [Google Scholar]
- Wang XT, Chan TF, Lam VMS, Engel PC. What is the role of the second “structural” NADP+-binding site in human glucose 6-phosphate dehydrogenase? Protein Science: A Publication of the Protein Society. 2008;17:1403–1411. doi: 10.1110/ps.035352.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XT, Engel PC. Clinical mutants of human glucose 6-phosphate dehydrogenase: impairment of NADP(+) binding affects both folding and stability. Biochimica Et Biophysica Acta. 2009;1792:804–809. doi: 10.1016/j.bbadis.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Wang XT, Lam VMS, Engel PC. Marked decrease in specific activity contributes to disease phenotype in two human glucose 6-phosphate dehydrogenase mutants, G6PD(Union) and G6PD(Andalus) Human Mutation. 2005;26:284. doi: 10.1002/humu.9367. [DOI] [PubMed] [Google Scholar]
- Wang XT, Lam VMS, Engel PC. Functional properties of two mutants of human glucose 6-phosphate dehydrogenase, R393G and R393H, corresponding to the clinical variants G6PD Wisconsin and Nashville. Biochimica Et Biophysica Acta. 2006;1762:767–774. doi: 10.1016/j.bbadis.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Warny M, Lausen B, Birgens H, Knabe N, Petersen J. Severe G6PD Deficiency Due to a New Missense Mutation in an Infant of Northern European Descent. Journal of Pediatric Hematology/Oncology. 2015;37:e497–499. doi: 10.1097/MPH.0000000000000435. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D: Biological Crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.