

Abstract
The cell type that normally limits the inflammatory and atherosclerotic process is the vascular endothelial cell (EC) that can be regulated by proinflammatory and various stresses. Toll-like receptor-4 (TLR4) plays an important role in the pathogenesis of atherosclerosis, in part, by activating apoptosis signal-regulating kinase 1 (ASK1) to initiate the activation of MAP kinases pathways and the expression of inflammatory genes. In the present study, we test the hypothesis that AGI-1067 acts as an anti-inflammatory agent by inhibiting the activation of ASK1 in human EC. Pretreatment of human aortic endothelial cells with AGI-1067 inhibits TLR4 ligand (LPS)-induced activation of ASK1 and the downstream p38 and c-Jun N-terminal kinase (JNK) MAP kinases. LPS dissociates two endogenous inhibitors thioredoxin-1 (Trx1) and 14-3-3 from ASK1, leading to ASK1 autoactivation. Interestingly, AGI-1067 inhibits the dissociation of Trx1, but not 14-3-3, from ASK1. However, inhibition of Trx1 dissociation from ASK1 by AGI-1067 is sufficient to suppress LPS-mediated phosphorylation of the transcription factors c-Jun and activating transcription factor 2, and inhibit LPS-induced inflammatory genes including vascular cell adhesion molecule 1, E-selectin, IL-6 and monocyte chemoattractant protein 1. Our findings suggest that AGI-1067 as a unique ASK1 inhibitor to inhibit TLR4-mediated ASK1 activation, contributing to its anti-inflammatory properties.
Keywords: AGI-1067, apoptosis signal-regulating kinase 1, thioredoxin, toll-like receptor 4, mitogen-activated protein kinase
INTRODUCTION
Myocardial infarction caused by atherosclerosis of coronary arteries remains the leading cause of death in the developed countries. Atherosclerosis involves plaque formation in the arterial wall and is characterized by inflammation, lipid accumulation, smooth muscle cell proliferation/neointima expansion, cell death and fibrosis1, 2. Subendothelial retention and accumulation of lipoproteins could be converted to atherogenic remnant lipoproteins and low-density lipoprotein (LDL) 3, 4. A key early inflammatory response to retained lipoproteins is activation of vascular endothelial cells (EC), which may be enhanced by oxidative modifications of these lipoproteins. Activated EC increase expressions of adhesion molecules and chemokines which in turn promote infiltration of immune cells, including macrophages, neutrophils, master cells, dendritic cells and T cells 2, 5–7. Macrophages take up native and oxidized LDL to become lipid-laden foam cells and together with other cells release atherogenic cytokines and other activators such as reactive oxygen species (ROS) that exacerbate EC dysfunction (characterized by a reduction in amount of bioavailable nitric oxide) and EC apoptosis, and promote atherosclerotic plaque development 8. Therefore, the phenotypic changes in EC may be an early event during initiation and progression of atherosclerosis 2, 6, 7, 9, 10.
Toll-like receptor 4 (TLR4), the primary receptor for lipopolysaccharide (LPS), plays an important role in the pathogenesis of atherosclerosis3, 11. Epidemiological data suggest that high plasma endotoxin levels constitute a significant risk factor for the development of atherosclerosis 12–17. TLR4 mRNA and protein levels are dramatically increased in atherosclerotic plaques18. Administration of endotoxin increases atherosclerotic lesion formation in apolipoprotein E-deficient (ApoE−/−) mice and hypercholesterolemic rabbits19, 20. TLR4/ApoE double-null (TLR4−/−ApoE−/−) mice fed with a high cholesterol diet show decreased lesion formation compared to ApoE−/− control mice 21. Furthermore, in addition to LPS, TLR4 recognizes oxidized variation of low density lipoproteins (LDL) also termed minimally modified LDL, heat shock proteins, and fibronectin extra domain A; all of which are found in atherosclerotic lesions22–25.
ASK1 (apoptosis signal-regulating kinase 1), a member of the MAP3K family activating MAP2K-JNK/p38 cascades, can be activated in response to various stress stimuli, including pro-inflammatory cytokines and LPS 26, 27. Studies in overexpression systems and from ASK1 knockout mice have shown that ASK1 is a critical mediator in tumor necrosis factor (TNF), ROS, and LPS-induced inflammatory signaling 28–30. ASK1 is a 170 kD protein that is composed of an inhibitory N-terminal domain, an internal kinase domain, and a C-terminal regulatory domain. Several intracellular proteins bind to different domains of ASK1, keeping it in an inactive state in resting cells. The redox sensor protein thioredoxin-1 (Trx1) in a reduced form binds to the N-terminal domain of ASK1, while glutaredoxin binds to the C-terminal domain, in each case, inhibiting ASK1 kinase activity 31, 32. Phosphoserine-binding protein 14-3-3 associates with ASK1 via the pSer967 site of ASK1 and inhibits ASK1-induced apoptosis 33–37. Stress stimuli activate ASK1, in part by dissociating ASK1 from its inhibitors Trx1 and 14-3-310.
Given the critical role of ASK1 in inflammation and cardiovascular diseases, finding of ASK1 inhibitors has been actively investigated in both Pharmaceutical companies and academic labs. We have recently shown that a selective ASK1 inhibitor GS-444217, which specifically binds to an active form of ASK1 homodimer, can effectively block cardiopyopathy in a mouse model 38. In the present study, we investigate another type of ASK1 AGI-1067. AG-1067: [mono[4-[[1-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl] thio]-1-methylethyl]thio]-2,6-bis (1,1-dimethylethyl) phenyl] ester] (butanedioc acid), is an anti-atherosclerotic agent. Pre-clinical studies establish that AGI-1067 inhibits the development of atherosclerosis in three animal models: hypercholesterolemic cynomolgus monkeys, LDL receptor-deficient mice and ApoE−/−mice 39. AGI-1067 is a potent lipid peroxide antioxidant, capable of reducing basal levels of reactive oxygen species (ROS) in endothelial cells, and inhibiting tumor necrosis factor-alpha (TNF-α) induced vascular cell adhesion molecule-1 (VCAM-1) and monocyte chemoattractant protein-1 (MCP-1) expression without affecting NF-κB activity40. In addition, AGI-1067 inhibits LPS-induced tissue factor production and suppresses the activation of MAP kinases and redox-sensitive ASK1 in both monocytic cells and endothelial cells 41. However, the mechansim by which AGI-1067 inhibts LPS-induced ASK1 has not been determiend.
In the present study, we report that AGI-1067 inhibits LPS/TLR4-mediated expression of VCAM-1, E-selectin, IL-6, and MCP-1 in EC. This correlates with AGI-1067 inhibition of LPS/TLR4-induced activation of ASK1 and the downstream p38/JNK1/2 MAP kinase pathway and transcription factors c-Jun and activating transcription factor 2 (ATF2). Mechanistically, we show that AGI-1067 specifically suppresses the dissociation of ASK1 from Trx1, but not 14-3-3, suggesting that AGI is a unique ASK1 inhibitor.
METHODS
Materials
LPS (Escherichia coli 026:B6) and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). AGI-1067 was synthesized as previously described42. AGI-1067 was dissolved in DMSO and added to cultured cells so that the final DMSO concentration was 0.1%. Antibodies were purchased from a variety of commercial sources: anti-phospho-p38 MAPK (9211), anti-p38 (9217), anti-TLR4 (2246), anti-Phospho-c-Jun (2361), and anti-phospho ASK1 (special order) from Cell Signaling Technology (Beverly, MA ); anti-ASK1 (sc-5294) and anti-β-tubulin (sc-5274) and anti-14-3-3 from Santa Cruz Biotechnologies (Santa Cruz, CA); anti-phospho JNK (44-682G) from BioSource International (Camarillo, CA); anti-pan-JNK/SAPK1 (610628) from BD BioScience (San Diego, CA); anti-phospho-ATF2 Thr69/71 (05-891) from Upstate (Lake Placid, NY); and anti-rabbit IRdye700 (611-130-122) and anti-mouse IRdye800 (610-132-121) from Rockland, Inc (Gilbertsville, PA). Anti-Trx1 antibody was described previously34, 43.
Cell Culture
Human aortic endothelial cells (HAECs, Cambrex, Walkersville, MD) were cultured in EGM-2 medium with 5% fetal bovine serum (Cambrex) and used between passages 5 and 9. All cells were maintained at 37°C with 5% CO2. All cells were seeded in plates such that they would reach 90–95% confluence on the day of the experiment.
Measurement of adhesion molecules, cytokines and chemokines protein expressions
HAEC at 1.0×104 cells per well were plated into a 96-well plate the night before. HAEC were pre-treated for 1 hr with AGI-1067 or control (0.1% DMSO) then co-treatment with 2 μg/ml LPS. Cell surface expression of VCAM-1 and E-selectin were determined by FACS as described previously 7, 44. The mRNA expression of IL-6 and MCP-1 were determined by qRT-PCR normalized with GAPDH.
Immunoprecipitation and Western Blots
HAECs were pre-treated for 1 hr with AGI-1067 or control (0.1% DMSO) before co-treatment with 2 μg/ml LPS for the given time points. Whole cell extracts were collected. Samples were subjected to electrophoresis on SDS-PAGE gels and then transferred to a nitrocellulose membrane. Blots were probed with primary antibodies in Odyssey Blocking Buffer (BB; Licor; Lincoln, NE) + 0.1% Tween 20 (BBT) overnight at 4°C or for 2 hr at room temperature. Membranes were washed and incubated for 1 hr with IRdye-labeled secondary antibodies in BBT. Images were obtained using the Odyssey Infrared Imaging System (Licor; Lincoln, NE). For immunoprecipitation to analyze protein interaction in vivo, 400 μg of cell lysate supernatant were precleared by incubating with 5 μg of normal rabbit serum plus protein A/G-agarose beads on rotator at 4 °C overnight. The lysates were then incubated with 5 μg of the first protein-specific antiserum for 2 h with 50 μl of protein A/G-agarose beads. The immune complexes were collected after each immunoprecipitation by centrifugation at 14,000 × g for 10 min followed by four washes with lysis buffer. The immune complexes were subjected to SDS-PAGE followed by immunoblot (Immobilon P, Millipore, Milford, MA) with the second protein (e.g. ASK1)-specific antibody (H300, Santa Cruz Biotechnology).
Statistical Analysis
Values were expressed as the means ± standard errors of at least three independent experiments. The data were analyzed using the two sample t-test assuming equal variances and values were considered significantly different at the 95% confidence level.
RESULTS
AGI inhibits LPS/TLR4-induced EC adhesion molecules and chemokines
VCAM-1 and E-selectin are up regulated by a variety of inflammatory stimuli and mediate the attachment of leukocytes to the endothelium 7, 45. We evaluated the ability of AGI-1067 to inhibit the surface expression of VCAM-1 and E-selectin induced by the pro-inflammatory ligands LPS. AGI-1067 inhibits the LPS-induced expression of VCAM-1 and E-selectin in a concentration-dependent manner with IC50 values of approximately 4.5 μM for both adhesion molecules (Figure 1A). IL-6, IL-8 and MCP-1 are pro-inflammatory cytokines associated with the pathogenesis of atherosclerosis46. Pretreatment of HAEC with AGI-1067 inhibits LPS-induced gene expression of IL-6, IL-8 and MCP-1 in a concentration-dependent manner with IC50 values of 3.4 μM, 3.8 μM and 4.0 μM, respectively (Figure 2B).
Figure 1. AGI inhibits LPS/TLR4-induced EC adhesion molecules and chemokines.

A. HAEC were pre-treated for 1 hr with AGI-1067 or control (0.1% DMSO) before co-treatment with (A) 2 μg/ml LPS for 4 hr. VCAM-1 and E-selectin surface expressions were determined by FACS. Surface expression (mean fluorescence intensity: MFI) are presented as the mean ± SEM. The data are from at least 3 pooled independent experiments with N = 5, * P <0.05.
B. AGI-1067 inhibits TLR4-induced release of cytokines and chemokines. HAEC were pre-treated for 1 hr with AGI-1067 or control (0.1% DMSO) before co-treatment with (A) 2 μg/ml LPS for 2 hr. The gene expression of IL-6 and MCP-1 was determined by qRT-PCR normalized with GAPDH. The data are from at least 3 pooled independent experiments with N ≥ 5, * P <0.05.
Figure 2. AGI inhibits LPS/TLR4-induced activation of MAPK and transcriptional factors.

A–B. HAEC were pre-treated with AGI-1067 at 10 μM or control (0.1% DMSO) for 1 hour prior to co-treatment with 2 μg/ml LPS for indicated times. Whole-cell lysates were used to determine the levels of total and phospho-p38 or total and phospho-JNK1/2 by Western blot analysis. Relative p38 (p-p38:p38) and JNK (p-JNK1/2:JNK1/2) were quantified. The figures are representative data from at least three separate experiments. * P <0.05.
C–D. HAEC were pre-treated with AGI-1067 at various doses (0.1% DMSO) for 1 hour prior to co-treatment with 2 μg/ml LPS for 30 min. Whole-cell lysates were used to determine the levels of total and phospho-p38 or total and phospho-JNK1/2 by Western blot analysis. Relative p38 (p-p38:p38) and JNK (p-JNK1/2:JNK1/2) were quantified. The figures are representative data from at least three separate experiments. * P <0.05.
E–F. AGI-1067 inhibits TLR4-induced activation of MAPK dependent transcription factors ATF2 and c-JUN. HAEC were pre-treated with AGI-1067 at 10 μM or control (0.1% DMSO) for 1 hour prior to co-treatment with 2 μg/ml LPS for indicated times. Whole-cell lysates were used to determine the levels of phospho-ATF2 or phospho-cJun by Western blot analysis. Relative phosphorylations of ATF2 (p-ATF2:ATF2) and c-Jun (p-cJun:cJun) were quantified. The figures are representative data from at least three separate experiments. * P <0.05.
AGI inhibits LPS/TLR4-induced activation of MAPK and transcriptional factors
The MAPK pathways regulate the expression of many inflammatory genes including cell surface adhesion molecules, cytokines and chemokines47. Treatment of HAEC with LPS activates p38 and JNK1/2. Pretreatment with AGI-1067 at 10 μM inhibits LPS-induced p38 and JNK activation (Figure 2A with quantifications in 2B). Furthermore, inhibition of both p38 and JNK by AGI-1067 occurred in a concentration-dependent manner with an IC50 value under 5 μM (Figure 2C with quantifications in 2D).
Activated JNK and p38 lead to the phosphorylation of activating protein 1 (AP-1) transcription factors c-Jun and ATF248. AP-1 transcription factors regulate many inflammatory genes49. Treatment of HAEC with LPS induces time-dependent phosphorylation of c-Jun and ATF2 within 1 hour of stimulation (Figure 2E with quantifications in 2F). AGI-1067 at 10 μM inhibits LPS-induced c-JUN and ATF2 phosphorylation.
AGI inhibits LPS/TLR4-induced ASK1 activation by specifically dissociating Trx1 but not 14-3-3 from ASK1
ASK1, a redox regulated MAP3K, is an up stream regulator of p38 and JNK1/226. As expected, treatment of HAEC with LPS activated ASK1 activity as indicated by ASK1 autophosphorylation at Thr845 (a marker of ASK1 activity). AGI-1067, at 10 μM, inhibits LPS-induced ASK1 phosphorylation at Thr845 (Figure 3A with quantifications in 3B). Trx1 and 14-3-3 bind to ASK1 and inhibit ASK1 activity.
Figure 3. AGI inhibits LPS/TLR4-induced ASK1 activation by specifically dissociating Trx1 but not 14-3-3 from ASK1.

A–B. HAEC were pre-treated with AGI-1067 at 10 μM or control (0.1% DMSO) for 1 hour prior to co-treatment with 2 μg/ml LPS for indicated times. Whole-cell lysates were used to determine the levels of total and phospho-ASK1 by Western blot analysis. Relative phosphorylations of ASK1 (p-ASK1:ASK1) were quantified. The The figures are representative data from at least three separate experiments. * P <0.05.
C–D. AGI-1067 prevent LPS-induced dissociation of Trx1, but not 14-3-3, from ASK. HAEC were pre-treated with AGI-1067 at 10 μM or control (0.1% DMSO) for 1 hour prior to co-treatment with 2 μg/ml LPS for indicated times. Cell lysates were immunoprecipitated with anti-Trx1 or anti-14-3-3 antibody followed by Western blot with anti-ASK1 or respective antibodies as indicated. LPS induces release of ASK1 from both Trx1 and 14-3-3. However, AGI1067 blocks the release of ASK1 from Trx1, but not from 14-3-3, consistent with the anti-oxidant effects of AGI-1067. Relative Trx1-and 14-3-3-bound ASK1 (pulldown ASK1:input ASK1) were quantified. TThe figures are representative data from at least three separate experiments. * P <0.05.
Cytokines such as TNF-α activate ASK1 in part by releasing both Trx1 and 14-3-3 from ASK1. We hypothesize that AGI-1067 may prevent the release of Trx1 or 14-3-3 to inhibit activation of ASK1. As shown in figure 3C with quantifications in 3D, association of ASK1 with Trx1 and 14-3-3 was detected in untreated HAECs. LPS treatment reduced the interaction of ASK1 with both Trx1 and 14-3-3 in a time dependent manner. Treatment with AGI-1067 prevented the LPS-induced dissociation of ASK1 from thioredoxin, but had no effects on dissociation of 14-3-3 from ASK1. These data are consistent with the anti-oxidant effects of AGI-1067, suggesting that AGI-1067 may prevent oxidation of Trx1 and as a result prevent dissociation of Trx1 with ASK1 and block ASK1 activation (Figure 4 for a model).
Figure 4. A model for the effect of AGI-1067 on LPS/TLR4-induced ASK1 activation and inflammation.
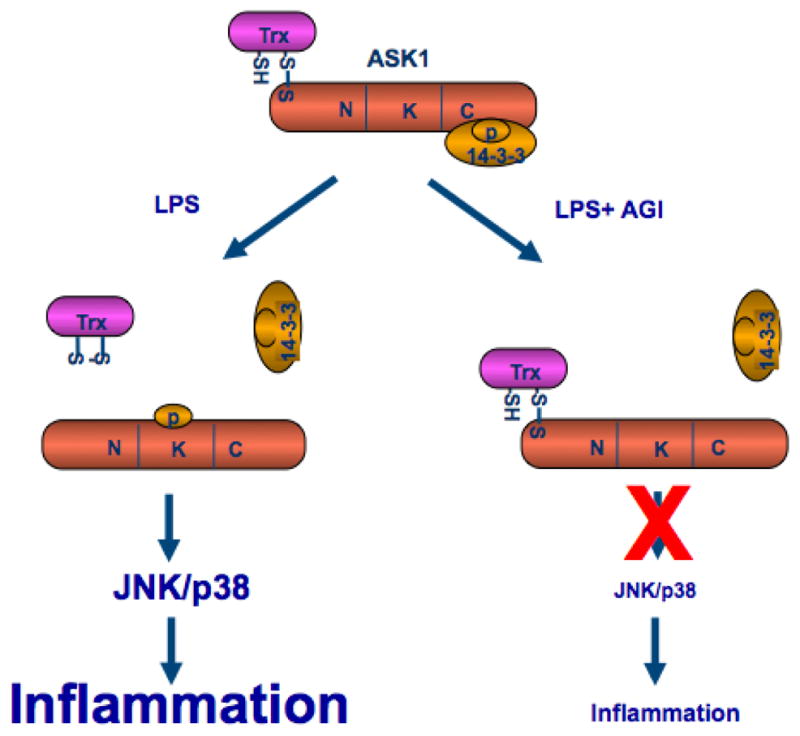
AGI-1067 prevents the LPS/TLR4-induced dissociation ASK1 from its inhibitor Trx1, but not from 14-3-3, leading to inhibition of inflamamtion (see Text for details).
DISCUSSION
Inflammation is a key initial step in vascular diseases such as atherosclerosis. Inflammation is complicated by the fact that multiple cytokines are involved in the process and each cytokine has pleiotropic functions. One commonality of proinflammatory stimuli is that they activate NF-κB and ASK1-JNK/p38 two parallel signaling cascades, leading to induction of inflammatory molecules. Many anti-inflammatory drugs have been selected to target NF-κB pathway50. However, it has been realized that NF-κB triggers survival signal whereas ASK1 induces apoptotic signal in many cell types. Thus, inhibition of ASK1 without disruption of NF-κB survival signal provides a valid approach for anti-inflammatory therapy. This is supported by the findings that proatherosclerotic stimuli (e.g., TNF, IL-1, LPS, oxidized lipids and oxidative stress) activate ASK1 while atheroprotective laminar flow inhibits ASK1 activation without effects on NF-κB34. Our current data suggest that AGI-1067 belongs to a new category of potential anti-inflammatory chemicals as it inhibits ASK1 activation without effects on NF-κB.
The clinical significance of our work is that AGI-1067 inhibits ASK1-JNK/p38-dependent gene expression of porinflamamtory molecules. It has been well documented that both JNK and p38 MAP kinases are downstream of ASK1 and play an important role in the regulation of inflammatory gene expression. Inhibition of p38 activity down-regulates pro-inflammatory gene expressions such as TNF-α, TNF-β, MCP-1, IL-1β, IL-6, iNOS, COX and adhesion molecules51–53. While the JNK pathway regulates many inflammatory genes including TNF-α, IL-2, E-selectin, VCAM-1 and MMPs such as collagease-1 44, 47, 54, 55. Downstream of the MAPKs are the members of the AP-1 family of transcription factors. The AP-1 transcription factors are heterodimers between members of the Jun family and the Fos family. AP-1 binding sites are present in many endothelial cell pro-inflammatory genes including VCAM-1, E-selectin, tissue factor and MCP-1 49. Adenovirus-mediated over-expression of c-Jun and c-Fos in human endothelial cells induces the expression of adhesion molecules and release of chemokines. Additionally, in vivo transfection of dominant negative c-Jun into rats inhibits intimal thickening after balloon injury by preventing vascular smooth muscle cell proliferation56. ATF2, another transcription factor activated by p38 and JNK, also forms heterodimers with members of the AP-1 family57. ATF2 knock-out mice show decreased LPS-induced inflammation including reduced expression of E-selectin, P-selectin, VCAM-1, IL-6 and KC 58. ATF2 knock-out mice show decreased LPS-induced inflammation including reduced expression of E-selectin, P-selectin, VCAM-1, IL-6 and KC 58. Consistent with the inhibition of p38 and JNK1/2, AGI-1067 inhibits the phosphorylation of c-Jun and ATF2 which could explain the reduction in LPS-induced pro-inflammatory gene expression in AGI-1067-treated HAEC.
AGI-1067 inhibits IL-6 and MCP-1, which are pro-inflammatory cytokines linked to the pathogenesis of atherosclerosis. IL-6 mRNA is found in atherosclerotic lesions and elevated IL-6 plasma concentrations are associated with an increased risk of myocardial infarction59, 60. MCP-1, another chemokine present in atherosclerotic lesions, recruits monocytes and plays a role in their adhesion to and infiltration through the endothelium. Additionally, over-expression of MCP-1 in atherosclerotic prone ApoE−/− mice accelerates the disease state, while MCP-1 deficiency reduces the susceptibility of the mice to develop atherosclerosis 61, 62. Our results demonstrate that AGI-1067 inhibits endothelial cell TLR4-mediated activation of p38 and JNK MAP kinases and expression of cytokines and chemokines. These data suggest that modulation of the endothelial intracellular redox state by AGI-1067 inhibits the redox-sensitive ASK1-p38/JNK pathways and expression of pro-inflammatory genes. These findings may provide a mechanistic framework for understanding the anti-inflammatory and anti-atherosclerotic properties of AGI-1067. LPS also activates the interferon (IFN) regulatory factor (IRF)-3 pathway that participates in the transcriptional induction of IFN-α, IFN-β, and a subset of IFN-stimulated genes (ISGs) as a result of viral infection. Interestingly, it has been reported that LPS activates IRF3 in an ASK1-p38-dependent manner30. We are investigating if AGI-1067 inhibits the IRF3-mediated innate immune responses.
One important finding of our study is that AGI-1067 compound prevents the LPS/TLR4-induced dissociation ASK1 from its inhibitor Trx1, but not from 14-3-3, leading to inhibition of inflamamtion (Figure 4 for a model). The mecahnism for ASK1 activation has been extensively investigated. The reduced form of Trx1 directly associates with ASK1 in the N-terminal domain of ASK1, inhibiting its kinase activity 63 and blocking activation of ASK1 by TNF43, 63. Inflammatory signals activate ASK1 in part by oxidizing Trx to release it from ASK143, 63. We have previously shown that a single Cys residue in the catalytic site of Trx1 (C32 or C35) and C250 of ASK1 are critical for the formation of the complex. Furthermore, Trx-C32S and Trx-C35S constitutively bind to ASK1, even in the presence of hydrogen peroxide in vitro or of TNF in vivo, most likely because they cannot be oxidized to form a disulfide bond between the two catalytic cysteines leading to its release from ASK143, 64. These results suggest that the binding of Trx1 to ASK1 is critical for the regulation of ASK1 activity. Our current data demonstrate that AGI-1067 retains Trx1-ASK1 complex to lock ASK1 in an inactive state even in the presence of inflammatory stimuli LPS. Therefore, AGI-1067 could be an effective therapeutics to treat inflammatory diseases.
Our results further provides novel insights into the mechanism by which ASK1 activity is regulated by its two inhibitors Trx1 and 14-3-3. Previously, we have shown that apoptotic stimuli such as TNF together with protein synthesis inhibitor cycloheximide (CHX) specifically induce dissociation of ASK1 from both Trx and 14-3-3 leading to EC apoptosis 10, 35–37, 43, 64. TNF induced reactive oixygen species (ROS) likely contribuyes to Trx1 release by oxidizing Trx1. TNF-activated AIP1 recruits PP2A, a phosphatase that dephosphorylates the 14-3-3 binding site pSer-967 on ASK1, to facilitate the 14-3-3 release10, 35–37, 43, 64. Therefore, Trx1 and 14-3-3 are dissoicated at different steps of ASK1 activation through distinct mechanisms. Interestingly, our recent finding suggests that an anti-cancer prodrug Laromustine derivative methyl isocyanate dissociates ASK1 from Trx1 but not from 14-3-3 and leads to the non-apoptotic death of EC65. These data indicate that 14-3-3-bound ASK1 is partially active with a unique function. Our current study demonstrates another scenario, i.e, Trx1-bound (14-3-3 free) ASK1 is sufficient to block LPS/TLR4-induced ASK1 autoactivation, and downstream JNK/p38 signaling as well as gene expression of inflammatory molecules. It will be interesting to determine if other inflammatory stimuli have similar effects on Trx1-ASK1 and 14-3-3-ASK1 complexes. Of note, laminar flow, the most potent physiological anti-atehrosclerotic factor, can prevent the release of both inhibitors Trx1 and 14-3-3 from ASK1. Impotantly, our data may provide insights why AGI-1067 failed to meet primary endpoint in late-stage (Phase III) trial as an anti-atherosclerotic drug, but did achieve other predefined endpoints (http://www.firstwordpharma.com/node/111149). Therefore, it needs to screen more potent anti-atherosclerotic drugs that can prevent both Trx1 and 14-3-3 release from ASK1. On the other hand, AGI-1067 may have benificenry effects for other inflammatory diseases such as myocardial infarction and retinopathy as we demonstrated with ASK1 inhibitor GS-44421738 and other drugs66.
Acknowledgments
Source of funding: This work was supported by NIH grants R01 HL085789 and R01 HL109420 to WM.
Footnotes
Disclosure: None declared.
References
- 1.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 2.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 3.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–44. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 4.Hartvigsen K, Chou MY, Hansen LF, Shaw PX, Tsimikas S, Binder CJ, Witztum JL. The role of innate immunity in atherogenesis. J Lipid Res. 2009;50(Suppl):S388–93. doi: 10.1194/jlr.R800100-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hermansson A, Ketelhuth DF, Strodthoff D, Wurm M, Hansson EM, Nicoletti A, Paulsson-Berne G, Hansson GK. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. 2010;207:1081–93. doi: 10.1084/jem.20092243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Q, Qin L, Dai S, Zhang H, Pasula S, Zhou H, Chen H, Min W. AIP1 suppresses atherosclerosis by limiting hyperlipidemia-induced inflammation and vascular endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2013;33:795–804. doi: 10.1161/ATVBAHA.113.301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin L, Huang Q, Zhang H, Liu R, Tellides G, Min W, Yu L. SOCS1 prevents graft arteriosclerosis by preserving endothelial cell function. J Am Coll Cardiol. 2014;63:21–9. doi: 10.1016/j.jacc.2013.08.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–55. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–15. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 10.Pober JS, Min W, Bradley JR. Mechanisms of Endothelial Dysfunction, Injury, and Death. Annu Rev Pathol. 2009;4:71–95. doi: 10.1146/annurev.pathol.4.110807.092155. [DOI] [PubMed] [Google Scholar]
- 11.Wan T, Liu T, Zhang H, Tang S, Min W. AIP1 functions as Arf6-GAP to negatively regulate TLR4 signaling. J Biol Chem. 2010;285:3750–7. doi: 10.1074/jbc.M109.069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blasi F, Denti F, Erba M, Cosentini R, Raccanelli R, Rinaldi A, Fagetti L, Esposito G, Ruberti U, Allegra L. Detection of Chlamydia pneumoniae but not Helicobacter pylori in atherosclerotic plaques of aortic aneurysms. J Clin Microbiol. 1996;34:2766–9. doi: 10.1128/jcm.34.11.2766-2769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiu B, Viira E, Tucker W, Fong IW. Chlamydia pneumoniae, cytomegalovirus, and herpes simplex virus in atherosclerosis of the carotid artery. Circulation. 1997;96:2144–8. doi: 10.1161/01.cir.96.7.2144. [DOI] [PubMed] [Google Scholar]
- 14.Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, Muggeo M, Xu Q, Wick G, Poewe W, Willeit J. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation. 2001;103:1064–70. doi: 10.1161/01.cir.103.8.1064. [DOI] [PubMed] [Google Scholar]
- 15.Kuo CC, Shor A, Campbell LA, Fukushi H, Patton DL, Grayston JT. Demonstration of Chlamydia pneumoniae in atherosclerotic lesions of coronary arteries. J Infect Dis. 1993;167:841–9. doi: 10.1093/infdis/167.4.841. [DOI] [PubMed] [Google Scholar]
- 16.Shor A, Kuo CC, Patton DL. Detection of Chlamydia pneumoniae in coronary arterial fatty streaks and atheromatous plaques. S Afr Med J. 1992;82:158–61. [PubMed] [Google Scholar]
- 17.Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, Willeit J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J Am Coll Cardiol. 1999;34:1975–81. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 18.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–61. [PubMed] [Google Scholar]
- 19.Engelmann MG, Redl CV, Nikol S. Recurrent perivascular inflammation induced by lipopolysaccharide (endotoxin) results in the formation of atheromatous lesions in vivo. Lab Invest. 2004;84:425–32. doi: 10.1038/labinvest.3700065. [DOI] [PubMed] [Google Scholar]
- 20.Hollestelle SC, De Vries MR, Van Keulen JK, Schoneveld AH, Vink A, Strijder CF, Van Middelaar BJ, Pasterkamp G, Quax PH, De Kleijn DP. Toll-like receptor 4 is involved in outward arterial remodeling. Circulation. 2004;109:393–8. doi: 10.1161/01.CIR.0000109140.51366.72. [DOI] [PubMed] [Google Scholar]
- 21.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kol A, Sukhova GK, Lichtman AH, Libby P. Chlamydial heat shock protein 60 localizes in human atheroma and regulates macrophage tumor necrosis factor-alpha and matrix metalloproteinase expression. Circulation. 1998;98:300–7. doi: 10.1161/01.cir.98.4.300. [DOI] [PubMed] [Google Scholar]
- 23.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1213–9. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- 24.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–33. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 25.Walton KA, Hsieh X, Gharavi N, Wang S, Wang G, Yeh M, Cole AL, Berliner JA. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8. A role for Toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2003;278:29661–6. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- 26.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–4. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 27.Hattori K, Naguro I, Runchel C, Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell communication and signaling : CCS. 2009;7:9. doi: 10.1186/1478-811X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–8. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–92. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 30.Chiang E, Dang O, Anderson K, Matsuzawa A, Ichijo H, David M. Cutting edge: apoptosis-regulating signal kinase 1 is required for reactive oxygen species-mediated activation of IFN regulatory factor 3 by lipopolysaccharide. J Immunol. 2006;176:5720–4. doi: 10.4049/jimmunol.176.10.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song JJ, Lee YJ. Dissociation of Akt1 from its negative regulator JIP1 is mediated through the ASK1-MEK-JNK signal transduction pathway during metabolic oxidative stress: a negative feedback loop. J Cell Biol. 2005;170:61–72. doi: 10.1083/jcb.200502070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Chen J, Fu H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc Natl Acad Sci U S A. 1999;96:8511–5. doi: 10.1073/pnas.96.15.8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Yin G, Surapisitchat J, Berk BC, Min W. Laminar flow inhibits TNF-induced ASK1 activation by preventing dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2001;107:917–23. doi: 10.1172/JCI11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang R, He X, Liu W, Lu M, Hsieh JT, Min W. AIP1 mediates TNF-alpha-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2003;111:1933–43. doi: 10.1172/JCI17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Zhang H, Lin Y, Li J, Pober JS, Min W. RIP1-mediated AIP1 phosphorylation at a 14-3-3-binding site is critical for tumor necrosis factor-induced ASK1-JNK/p38 activation. J Biol Chem. 2007;282:14788–96. doi: 10.1074/jbc.M701148200. [DOI] [PubMed] [Google Scholar]
- 37.Min W, Lin Y, Tang S, Yu L, Zhang H, Wan T, Luhn T, Fu H, Chen H. AIP1 recruits phosphatase PP2A to ASK1 in tumor necrosis factor-induced ASK1-JNK activation. Circ Res. 2008;102:840–8. doi: 10.1161/CIRCRESAHA.107.168153. [DOI] [PubMed] [Google Scholar]
- 38.Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W. Thioredoxin-2 Inhibits Mitochondrial ROS Generation and ASK1 Activity to Maintain Cardiac Function. Circulation. 2015 doi: 10.1161/CIRCULATIONAHA.114.012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sundell CL, Somers PK, Meng CQ, Hoong LK, Suen KL, Hill RR, Landers LK, Chapman A, Butteiger D, Jones M, Edwards D, Daugherty A, Wasserman MA, Alexander RW, Medford RM, Saxena U. AGI-1067: a multifunctional phenolic antioxidant, lipid modulator, anti-inflammatory and antiatherosclerotic agent. J Pharmacol Exp Ther. 2003;305:1116–23. doi: 10.1124/jpet.102.048132. [DOI] [PubMed] [Google Scholar]
- 40.Kunsch C, Luchoomun J, Grey JY, Olliff LK, Saint LB, Arrendale RF, Wasserman MA, Saxena U, Medford RM. Selective inhibition of endothelial and monocyte redox-sensitive genes by AGI-1067: a novel antioxidant and anti-inflammatory agent. J Pharmacol Exp Ther. 2004;308:820–9. doi: 10.1124/jpet.103.059733. [DOI] [PubMed] [Google Scholar]
- 41.Luyendyk JP, Piper JD, Tencati M, Reddy KV, Holscher T, Zhang R, Luchoomun J, Chen X, Min W, Kunsch C, Mackman N. A Novel Class of Antioxidants Inhibit LPS Induction of Tissue Factor by Selective Inhibition of the Activation of ASK1 and MAP Kinases. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/ATVBAHA.107.143552. [DOI] [PubMed] [Google Scholar]
- 42.Meng CQ, Somers PK, Rachita CL, Holt LA, Hoong LK, Zheng XS, Simpson JE, Hill RR, Olliff LK, Kunsch C, Sundell CL, Parthasarathy S, Saxena U, Sikorski JA, Wasserman MA. Novel phenolic antioxidants as multifunctional inhibitors of inducible VCAM-1 expression for use in atherosclerosis. Bioorg Med Chem Lett. 2002;12:2545–8. doi: 10.1016/s0960-894x(02)00516-4. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Cir Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- 44.Min W, Pober JS. TNF initiates E-selectin transcription in human endothelial cells through parallel TRAF-NF-kappa B and TRAF-RAC/CDC42-JNK-c-Jun/ATF2 pathways. J Immunol. 1997;159:3508–18. [PubMed] [Google Scholar]
- 45.Carlos T, Kovach N, Schwartz B, Rosa M, Newman B, Wayner E, Benjamin C, Osborn L, Lobb R, Harlan J. Human monocytes bind to two cytokine-induced adhesive ligands on cultured human endothelial cells: endothelial-leukocyte adhesion molecule-1 and vascular cell adhesion molecule-1. Blood. 1991;77:2266–71. [PubMed] [Google Scholar]
- 46.Ito T, Ikeda U. Inflammatory cytokines and cardiovascular disease. Curr Drug Targets Inflamm Allergy. 2003;2:257–65. doi: 10.2174/1568010033484106. [DOI] [PubMed] [Google Scholar]
- 47.Berk BC, Abe JI, Min W, Surapisitchat J, Yan C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann N Y Acad Sci. 2001:93–109. doi: 10.1111/j.1749-6632.2001.tb03932.x. 94721653590. discussion 109–11. [DOI] [PubMed] [Google Scholar]
- 48.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–6. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 49.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. Faseb J. 1995;9:899–909. [PubMed] [Google Scholar]
- 50.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pietersma A, Tilly BC, Gaestel M, de Jong N, Lee JC, Koster JF, Sluiter W. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem Biophys Res Commun. 1997;230:44–8. doi: 10.1006/bbrc.1996.5886. [DOI] [PubMed] [Google Scholar]
- 52.Rovin BH, Wilmer WA, Danne M, Dickerson JA, Dixon CL, Lu L. The mitogen-activated protein kinase p38 is necesssary for interleukin 1beta-induced monocyte chemoattractant protein 1 expression by human mesangial cells. Cytokine. 1999;11:118–26. doi: 10.1006/cyto.1998.0409. [DOI] [PubMed] [Google Scholar]
- 53.Xu W, Yan M, Lu L, Sun L, Theze J, Zheng Z, Liu X. The p38 MAPK pathway is involved in the IL-2 induction of TNF-beta gene via the EBS element. Biochem Biophys Res Commun. 2001;289:979–86. doi: 10.1006/bbrc.2001.6069. [DOI] [PubMed] [Google Scholar]
- 54.Conze D, Krahl T, Kennedy N, Weiss L, Lumsden J, Hess P, Flavell RA, Le Gros G, Davis RJ, Rincon M. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activation. J Exp Med. 2002;195:811–23. doi: 10.1084/jem.20011508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–65. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 56.Yasumoto H, Kim S, Zhan Y, Miyazaki H, Hoshiga M, Kaneda Y, Morishita R, Iwao H. Dominant negative c-jun gene transfer inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia in rats. Gene Ther. 2001;8:1682–9. doi: 10.1038/sj.gt.3301590. [DOI] [PubMed] [Google Scholar]
- 57.Gupta S, Campbell D, D’Erijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–93. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 58.Reimold AM, Kim J, Finberg R, Glimcher LH. Decreased immediate inflammatory gene induction in activating transcription factor-2 mutant mice. Int Immunol. 2001;13:241–8. doi: 10.1093/intimm/13.2.241. [DOI] [PubMed] [Google Scholar]
- 59.Seino Y, Ikeda U, Ikeda M, Yamamoto K, Misawa Y, Hasegawa T, Kano S, Shimada K. Interleukin 6 gene transcripts are expressed in human atherosclerotic lesions. Cytokine. 1994;6:87–91. doi: 10.1016/1043-4666(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 60.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–72. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 61.Aiello RJ, Bourassa PA, Lindsey S, Weng W, Natoli E, Rollins BJ, Milos PM. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–25. doi: 10.1161/01.atv.19.6.1518. [DOI] [PubMed] [Google Scholar]
- 62.Gosling J, Slaymaker S, Gu L, Tseng S, Zlot CH, Young SG, Rollins BJ, Charo IF. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest. 1999;103:773–8. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J. 1998;17:2596–606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res. 2004;94:1483–91. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]
- 65.Ji W, Yang M, Praggastis A, Li Y, He Y, Ghazvinian R, Cincotta D, Rice K, Min W. Carbamoylating activity associated with the activation of the antitumor agent laromustine inhibits angiogenesis by inducing ASK1-dependent endothelial cell death. PloS One. 2014;9:e103224. doi: 10.1371/journal.pone.0103224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang X, Zhou H, Ding Y, Li J, Yang C, Luo Y, Li S, Sun G, Liao X, Min W. TMP Prevents Retinal Neovascularization and Imparts Neuroprotection in an Oxygen-Induced Retinopathy Model. Invest Ophthalmol Vis Sci. 2012;53:2157–69. doi: 10.1167/iovs.11-9315. [DOI] [PMC free article] [PubMed] [Google Scholar]