

A demonstration of stable lithium-oxygen batteries based on high–donor number liquid electrolytes and an ionomer-protected anode.
Keywords: lithium-oxygen cells, solid-electrolyte interphases, Energy storage, redox mediators, high donor number electrolytes, lithium dendrites
Abstract
An electrochemical cell based on the reversible oxygen reduction reaction: 2Li+ + 2e− + O2 ↔ Li2O2, provides among the most energy dense platforms for portable electrical energy storage. Such Lithium-Oxygen (Li-O2) cells offer specific energies competitive with fossil fuels and are considered promising for electrified transportation. Multiple, fundamental challenges with the cathode, anode, and electrolyte have limited practical interest in Li-O2 cells because these problems lead to as many practical shortcomings, including poor rechargeability, high overpotentials, and specific energies well below theoretical expectations. We create and study in-situ formation of solid-electrolyte interphases (SEIs) based on bromide ionomers tethered to a Li anode that take advantage of three powerful processes for overcoming the most stubborn of these challenges. The ionomer SEIs are shown to protect the Li anode against parasitic reactions and also stabilize Li electrodeposition during cell recharge. Bromine species liberated during the anchoring reaction also function as redox mediators at the cathode, reducing the charge overpotential. Finally, the ionomer SEI forms a stable interphase with Li, which protects the metal in high Gutmann donor number liquid electrolytes. Such electrolytes have been reported to exhibit rare stability against nucleophilic attack by Li2O2 and other cathode reaction intermediates, but also react spontaneously with Li metal anodes. We conclude that rationally designed SEIs able to regulate transport of matter and ions at the electrolyte/anode interface provide a promising platform for addressing three major technical barriers to practical Li-O2 cells.
INTRODUCTION
The rechargeable lithium-oxygen (Li-O2) electrochemical cell is peerless among energy storage technologies for its high theoretical specific energy (3500 Wh/kg), which far exceeds that of current state-of-the-art Li-ion battery technology (1–4). Li-O2 cells are under intense study for applications in electrified transportation because they are viewed as the gateway to Li-air storage technology that is capable of offering competitive specific storage capacities to fossil fuels. A Li-O2 cell consists of a Li metal anode, an electrolyte that conducts Li+ ions, and uses O2 gas hosted in a porous carbon or metal support as the active material in the positive electrode (cathode). Ideally, the cell operates on the principle that Li2O2 is reversibly formed and decomposed in the cathode, with the net electrochemical reaction of 2(Li+ + e−) + O2 ↔ Li2O2 at an equilibrium potential of 2.96 V versus Li/Li+. However, in practice, the physicochemical processes in Li-O2 cells rarely, if ever, live up to these ideals. For example, the insoluble electrically insulating Li2O2 is difficult to oxidize, which leads to high charging overpotential (charge voltage, ~4.3 V) on the oxygen evolution reaction (OER) and limits the cell efficiency to ~60 to 70% (5–7). Additionally, decomposition reactions between the electrolyte and reactive oxygen species at the positive electrode and lithium metal at the negative electrode form undesirable products that further limit cell life and efficiency (8, 9). Finally, insoluble Li2O2 produced upon discharge accumulates in the cathode, eventually clogging pores in the cathode support, which simultaneously limits the Li-O2 cell discharge capacity and compromises rechargeability (10, 11). A grand challenge in the field concerns the development of materials and cell running protocols that can overcome all of these limitations without compromising the favorable attributes of the Li-O2 cell.
Several approaches to overcoming each of the challenges with the Li-O2 cell have been proposed, but a frustrating observation is that promising solutions to one problem often come at the expense of others or create new problems in some cases (1, 4). For example, significant theoretical and experimental efforts to lower the overpotential of the OER have resulted in the exploration of soluble redox mediators as electrolyte additives. Redox mediators are first oxidized electrochemically at a lower potential than Li2O2; the oxidized form of a soluble redox mediator can therefore diffuse to and oxidize otherwise electrochemically inaccessible Li2O2 deposited on the cathode surface, regenerating the mediator and allowing the Li-O2 battery to be recharged at a lower overpotential. Since their introduction by Chen et al. (12), multiple successful demonstrations of this concept have been reported using tetrathiafulvalene (12), (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (13), nitroxides (13, 14), lithium iodide (LiI) (15–17), tris[4-(diethylamino)phenyl]amine (18), iron phthalocyanine (19), and lithium bromide (LiBr) (20) as electrolyte additives. A drawback of this approach is that, with a few exceptions (13, 21), the redox mediator is free to diffuse throughout the cell and is reduced by Li metal in a parasitic process that depletes both the anode and the redox mediator. Likewise, efforts to improve the stability of electrolytes in the presence of the highly nucleophilic O2• species produced at the cathode and the Li metal anode (22, 23) have produced mixed results.
It is now known that electrolytes based on ethers, carbonates, ketones, and esters are all broken down at the cathode of a Li-O2 cell by the highly nucleophilic Li2O2 discharge product. At the anode, no liquid electrolyte that can survive long-term contact with metallic Li presently exists, and few form a stable solid-electrolyte interphase (SEI) with Li (24). Results from electrochemical mass spectrometry studies have shown that straight-chain alkyl amides N,N-dimethylformamide and N,N-dimethylacetamide (DMA) are unique among electrolyte solvents because of their stability against nucleophilic attack at the Li-O2 cathode (25, 26). Burke et al. (27) reported that high–donor number (DN) solvents (such as dimethyl sulfoxide in their case) induce a solution-mediated reaction pathway at the cathode by stabilizing LiO2 intermediates and the anion NO3−, which leads to higher cell discharge capacity (27–31). A possibly obvious drawback is that these high-DN electrolytes undergo a continuous chemical reaction with the Li anode, degrading the anode and electrolyte. LiNO3 salt additives have been investigated for their ability to form stable coatings on Li metal in certain electrolytes, which passivate the metal against attack even by electrolytes containing oxidizing sulfur species (32–35). In an important study, Walker et al. (36) showed that electrolytes that combine the beneficial effects of LiNO3 and DMA enable longer-term cycling of Li-O2 cells, underscoring the synergistic benefits of a high-DN electrolyte and anode protection in the Li-O2 cell.
An unprotected Li metal anode can fail as a result of other processes that are more catastrophic than those precipitated by uncontrolled chemical reactions with a liquid electrolyte (24, 37, 38). Electrodeposition of lithium metal during battery recharge is physically unstable toward the formation of rough/dendritic structures on the anode that ultimately grow to short-circuit the cell. The ohmic heat generated by this process can trigger thermal runaway of the cell in organic liquid electrolytes, leading to cell failure by fire and/or explosions (39–41). Furthermore, because rough electrodeposition increases the surface area of Li in contact with liquid electrolytes, physical instability of the Li anode exacerbates chemical instability at the anode/electrolyte interface. Three recent reviews provide a comprehensive assessment of the strengths and shortcomings of practiced strategies for stabilizing rechargeable lithium batteries against failure by dendrite-induced short circuits (24, 42, 43). An important conclusion is that because Li deposition is fundamentally unstable, fundamentally based approaches that take advantage of multiple physical processes are likely to be the most successful in guaranteeing long-term stability of rechargeable batteries that use metallic lithium as an anode.
Herein, we report on the stability of Li-O2 cells using liquid electrolytes containing an ionomer salt additive that spontaneously forms a multifunctional SEI at the anode. The additive and the in situ–formed SEI that it forms are deliberately designed to take advantage of three fundamentally based mechanisms for stabilizing electrochemical processes at the anode and cathode of the Li-O2 cell. First, consistent with predictions from recent continuum (44, 45) and density functional analyses of lithium deposition (46), we report that ionomer electrolyte additives that can ensure low diffusion barriers and high cation fluxes in the SEI at the anode are highly effective in stabilizing deposition of Li. We demonstrate the success of these additives by means of electrochemical analysis and postmortem imaging. Second, we show that if the ionomer additives are designed to form thin conformal coatings at the Li surface, it is possible to passivate the anode surface against chemical attack by high-DN (DN = 27.8) liquid electrolytes capable of stabilizing oxide intermediates on the cathode. Finally, we report that the same material that stabilizes Li deposition on the anode also functions as an effective redox mediator that lowers the overpotential for the OER reaction at the Li-O2 cathode.
RESULTS AND DISCUSSION
Understanding the anode protection mechanism
Characterization of the anode.
The electrolyte ionomer salt additive (2-bromoethanesulfonate lithium salt) investigated in the present study is illustrated in Fig. 1A. The material was chosen because of its ability to react with lithium to simultaneously anchor lithium ethanesulfonate at the anode/electrolyte interface and to generate partially soluble LiBr in the electrolyte. The specific ionomer chemistry selected for the study is motivated by four fundamental considerations. First, recent continuum theoretical analysis (44, 45) and experiments (47–49) indicate that tethering anions, such as sulfonates at the anode/electrolyte interface, lowers the potential at the interface during Li deposition and in so doing stabilizes the deposition. Second, joint density functional theoretical (JDFT) calculations (46) show that the energy barrier Ea for Li+ diffusion at a Li anode coated with LiBr salt (Ea,LiBr ≈ 0.03 eV) is much lower, by a factor of around 8, compared to Li2CO3 (Ea,Li2CO3 ≈ 0.24 eV), which forms naturally when aprotic solvents react with Li. This means that under isothermal conditions, stable deposition of Li in a given electrolyte can occur at deposition rates more than three orders of magnitude higher on a LiBr-coated Li anode than on an anode with a spontaneously formed Li2CO3-rich SEI. Third, the short hydrocarbon stem that connects the tethered sulfonate groups to Li should allow a dense hydrocarbon brush to form at the interface to protect the Li electrode from chemical attack by a high-DN electrolyte required for stability at the cathode. Finally, soluble LiBr undergoes electrochemical oxidation and reduction in an appropriate potential window to function as a soluble redox mediator.
Fig. 1. Artificial SEI concept and experimental verification of its proposed operating mechanism.
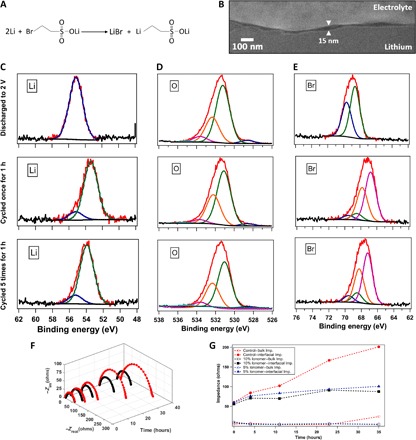
(A) Schematic for the reaction of lithium 2-bromoethanesulfonate with lithium metal forming LiBr and lithium-based organometallic. (B) SEM image of the interfacial layer between an intact electrolyte and a lithium electrode, revealed in a cross section produced by cryo-FIB milling. (C) Lithium 1s peak obtained from XPS of the lithium metal anode of a Li-O2 battery with the electrolyte ionomer [10% (by weight)] in 1 M LiNO3-DMA. (D) Oxygen 1s peak of the lithium anode. (E) Bromine 3d peak of the lithium anode. In (C) to (E), the first row shows the postmortem analysis after discharging until 2 V, the second row shows the result after cycling once with each half-cycle 5 hours long, and the third row shows the result after cycling five times with each half-cycle 1 hour long. (F) Three-dimensional diagram of Nyquist plots obtained by impedance measurements at different intervals of time using symmetric lithium cells, in which -Zim is the imaginary component of the impedance and -Zreal is the real component of the impedance. (G) Comparison of interfacial and bulk impedance values for ionomer-based and control electrolytes as a function of time. In (F) and (G), the red symbols denote results with the control electrolyte (1 M LiNO3-DMA), whereas the black and blue symbols represent batteries with 10 and 5% (by weight) ionomer additive, respectively, with the same electrolyte.
Cryo-focused ion beam (cryo-FIB) was used to characterize the morphology and thickness of the ionomer-enriched electrode/electrolyte interface with the liquid electrolyte intact but cryo-immobilized. In this technique, a symmetric lithium cell (with an ionomer-based electrolyte) was opened manually, and the sample was snap-frozen by immediately plunging it into slush nitrogen to preserve the electrolyte and to avoid air exposure. The sample was then transferred under vacuum into an FEI Strata 400 FIB fitted with a Quorum PP3010T Cryo-FIB/SEM Preparation System and maintained at −165°C for the duration of the experiment. To produce a cross section of the interface, we used the focused gallium ion beam to mill through the frozen electrolyte and into the electrode. This interface was then examined by scanning electron microscopy (SEM) and energy-dispersive x-ray (EDX) spectroscopy directly in the cryo-FIB. SEM images revealed an interfacial layer up to approximately 25 nm thick in most areas (see Fig. 1B). EDX analysis shows that the chemical composition of the layer is similar to that in the bulk electrolyte and that bromine species are distributed more or less uniformly throughout.
Additional insight into the nature of the interfacial region can be obtained by washing away the electrolyte and analyzing the ionomer layer that remains immobilized on the Li metal. For this purpose, we used EDX and high-resolution x-ray photoelectron spectroscopy (XPS) analytical measurements. The XPS measurements used monochromatic Al K-α x-rays (1489.6 eV) with a beam diameter of 1 mm to probe a surface layer on the electrodes approximately 15 to 25 nm thick, that is, comparable to the thickness of the interface revealed by cryo-FIB. Figure S1 reports the two-dimensional EDX results on a lithium anode that was thoroughly washed after cycling. Sulfur and bromine signals are evident everywhere on the surface of the material. XPS analysis was also performed using postmortem measurements on lithium anodes harvested from Li-O2 cells subjected to different running conditions. High-resolution scans for anodes retrieved after cycling or after a single discharge with the ionomer additive in the 1 M LiNO3-DMA electrolyte are reported in Fig. 1 (C to E). The corresponding results without the ionomer are shown in fig. S2. In Fig. 1C, it is apparent that after the first discharge, a Li 1s peak at 55.2 eV is observed on anodes with or without the ionomer present in the electrolyte. The peak may be attributed to the presence of LiOH, Li2O2, and Li2CO3 (50–57). A more prominent Li 1s peak is observed at 53.8 eV, accounting for about 85% of lithium, only in spectra of anodes cycled in the presence of the ionomer additive. This peak is indicative of the formation of a different SEI in electrolytes containing the ionomer; Li 1s peaks with comparable binding energy are reported for organometallics containing Li–C bonds (54.2 eV) (58, 59). This observation is consistent with the hypothesis that the ionomer reacts at the Li anode surface to form a lithium ethanesulfonate–rich SEI at the interface. Also, the fact that this binding energy is observed in the cycled anodes confirms that the SEI layer is stable and present even after repeated insertion and extraction of lithium ions into the underlying electrode.
Further evidence that the ionomer additive forms a stable SEI on Li can be deduced from the O 1s (Fig. 1D) and Br 3d (Fig. 1E) high-resolution scans. The O 1s peak at 532.2 eV comprises approximately 18% of the oxygen signal in cells without the ionomer additive, whether the anodes originate from cells that were subjected to a single discharge or were cycled. The 532.2-eV peak has been previously reported to originate from sulfonates (60), which accounts for 27 and 38%, respectively, of the oxygen signal when the anode is discharged once or cycled in the presence of the ionomer additive. The corresponding sulfur atomic contribution for the same materials can be computed from the wide survey scans (table S1) to be about 2% for the once discharged anode and about twice as high for the cycled anodes. The high-resolution scans of Br 3d reveal the formation of a single bond (a 3d5/2 and 3d3/2 doublet) with a Br 3d5/2 peak at 68.5 eV when the anode is discharged once in the presence of the ionomer. We attribute this peak to the formation of the Br–Li bond, which has been previously reported to occur at binding energies between 68.8 and 69.5 (53, 61). The same peak persists when the anode is cycled in the presence of the ionomer, but with a contribution of only around 15%. The reduced Li-Br species in the anodes of cycled cells is an indication of LiBr being solvated by the DMA electrolyte that can further participate in the redox mediation of oxygen cathode recharging. A more prominent Br 3d peak at 67.0 eV is observed only for the cycled anodes, likely originating from Br–C bonds [binding energies between 66.7 and 71.0 eV (60–65)] in the SEI originating from an untethered ionomer. The untethered ionomer in the electrolyte can help in the regeneration of the SEI layer in repeated cycling. Our results based on XPS analysis thus show that the ionomer-added electrolyte forms a SEI layer of lithium ethanesulfonate and LiBr, in accordance with the proposed reaction mechanism.
The effectiveness of ionomer-based SEI on Li was analyzed using impedance spectroscopy measurements on symmetric lithium cells. The results are shown in Fig. 1F with Nyquist-type plots at progressive time periods for control cells and those that contain 10% (by weight) ionomer additive. The Nyquist plots for electrolytes containing 5% ionomer additives are shown in fig. S3. The experimental data points are fitted with the circuit model illustrated in fig. S4 to deduce the bulk and interfacial resistances (Fig. 1G) as a function of time for the control electrolyte as well as with 10 and 5% (by weight) ionomer additive. It is seen that the bulk resistance for all cells remain essentially constant for approximately 20 hours, beyond which the bulk resistance of the control diverges (the increase is much larger; see fig. S5 for the results for the control cells after 48 and 56 hours). The time-dependent interfacial impedance provides an even more sensitive indicator of the stability of the anode-electrolyte interphase in a high-DN solvent. It is seen that the initial interfacial resistances for control and ionomer SEI–stabilized Li electrodes are approximately equal (~50 ohms). However, there is an exponential rise in the interfacial resistance of the control cell over time consistent with rapid reaction between Li and DMA. It is important to note that this reaction is observed although LiNO3 is present at large concentrations in the electrolyte. These results therefore challenge the view that LiNO3 provides an effective means of passivating Li metal anodes against reactive liquid electrolytes. In contrast, the results in Fig. 1G show that the interfacial resistance remains constant (see also fig. S4) when the ionomer-based SEI is present. It is seen that the stabilization with 10% ionomer additive is marginally better than the 5% case. Together, these findings demonstrate that a SEI based on bromide ionomers has a large stabilizing effect on Li anodes in DMA-based electrolyte solvents.
Lithium-electrolyte stability.
Figure 2 (A and B) reports on the quality of lithium ion deposition on stainless steel substrates mediated by control and ionomer-containing 1 M LiNO3-DMA electrolytes. For these experiments, cells were assembled with lithium as an anode and stainless steel as a virtual cathode. Lithium with a capacity of 10 mAh/cm2 was deposited at a rate of 1 mA/cm2 onto stainless steel, after which the cell was rested for a period of 10 hours and the voltage was monitored over time. Figure 2B shows that in case of a control electrolyte, Li deposition takes place at a higher voltage compared to the ionomer-containing electrolyte. Also, it can be observed that after the rest period, the voltage measured in the control cells immediately rises to approximately 0.5 V. This high open-circuit potential after Li deposition is a reflection of the complete decomposition of Li deposits on stainless steel due to corrosion by the electrolyte. It is again worth noting that despite using the Li-passivating salt LiNO3 at high concentrations in the electrolyte, the freshly deposited lithium reacts completely with the electrolyte solvent. Figure 2B also reports the corresponding voltage profiles observed in rested cells containing the ionomer as an electrolyte additive. It is seen that the cell voltage remains close to 0 V (versus Li/Li+), that is, near the open-circuit potential of a symmetric lithium cell, which means that the Li electrode is chemically stable in the reactive DMA electrolyte solvent.
Fig. 2. Stabilizing the lithium-electrolyte interface.
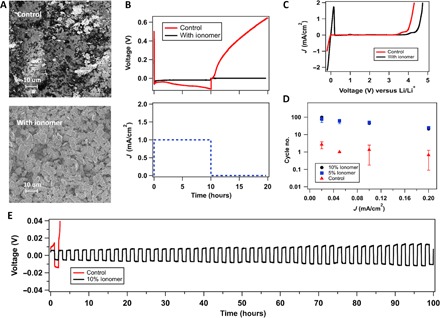
(A) SEM images of stainless steel (SS) electrode after depositing lithium (10 mAh/cm2) in a Li||SS cell with and without the ionomer additive using the same electrolyte of 1 M LiNO3-DMA. (B) Voltage profile of the Li||SS cell plotted over time. In this experiment, Li+ ions were deposited onto the stainless steel side at a current density of 1 mA/cm2 for 10 hours, after which the cell was kept at rest for an additional 10 hours, as shown in the current-versus-time curve. In the voltage-versus-time graph, the red line represents the profile of the control electrolyte (1 M LiNO3-DMA), whereas the black line is for the same electrolyte enriched with 10% (by weight) ionomer additive. The dashed blue line in the current-versus-time graph is the applied current for both cases. (C) Linear scan voltammetry showing current as a function of voltage versus Li/Li+, with Li as both working and reference electrode and SS being the counter electrode. (D) In a Li||SS cell, lithium with 10-mAh/cm2 capacity is deposited onto SS, and the battery was charged and discharged consecutively at various current densities. The cycle number associated with the divergence of voltage is plotted against the respective current densities. (E) Voltage profile for the strip-and-plate experiment under the abovementioned condition using a current density of 0.05 mA/cm2. In all figures, red indicates the control electrolyte (1 M LiNO3-DMA) and black represents the addition of 10% (by weight) ionomer additive, whereas blue denotes 5% (by weight) addition.
To further examine the morphology of Li deposits, we performed postmortem analysis, wherein the surface features of the electrodes were visualized under a SEM. Figure 2A shows the SEM image of the surface of stainless steel in the control and ionomer-based electrolytes. For the control, there are few patches of Li observed, and large sections of bare stainless steel are clearly visible. In contrast, in electrolytes containing the ionomer, the stainless steel surface is covered with a thick layer of lithium. It is also seen that Li electrodeposits formed in the latter electrolytes are evenly sized and spherical, even at a relatively high current density of 1 mA/cm2. This observation is consistent with previous reports of more compact electrodeposition of Li in electrolytes with halide salt–enriched SEIs and single-ion–conducting features (24, 42).
To fundamentally understand the basis of these observations, we characterized the electrochemical stability of the electrolytes by means of linear scan voltammetry in the range of −0.2 to 5 V versus Li/Li+, at a fixed scan rate of 1 mV/s. Figure 2C shows current as a function of voltage in a two-electrode setup of Li||stainless steel. It is seen that for the control (indicated by the red curve), the current diverges at a value around 4 V versus Li/Li+, whereas for electrolytes containing ionomer additives, the current diverges at a higher voltage, around 4.3 V versus Li/Li+. This improved stability is consistent with previous reports of electrolyte composites with tethered anions (49), wherein anions fixed at or near the electrode surface limit access to and chemical reaction of anions in an electrolyte with the negative electrode. Another important feature of the results can be seen at a potential close to 0 V versus Li/Li+. The significant current peak apparent at approximately −0.2 V versus Li/Li+ for both control and ionomer-containing electrolytes is a characteristic of lithium plating onto stainless steel. However, as the voltage is progressively increased, the corresponding Li stripping peak is not seen in the control cell but is readily apparent in cells with the ionomer-containing electrolyte. This behavior is indicative of the complete consumption of lithium deposits on stainless steel in the control cells and is consistent with previous results of SEM.
Figure 2 (D and E) reports results from so-called galvanostatic “plating-stripping” experiments. These experiments are used to evaluate the stability of Li electrodeposition and to assess the propensity of the material to electrodeposit as rough, dendritic structures. In contrast to previous studies (38), where thick (~0.75 mm) Li foil is used on both electrodes in plate-strip protocols, we performed these experiments using asymmetric Li/Li cells composed of one thick Li and one Li-lean (10 mAh/cm2 of Li deposited on stainless steel at 1 mA/cm2) electrode. The stability of the Li deposition reaction is normally assessed using three criteria: (i) magnitude of overpotential of lithium deposition, (ii) steep decrease of the cell voltage to zero with continuous charge-discharge, and (iii) a steady increase of the voltage over extended cycles of charge and discharge. In the first criterion, higher overpotential is indicative of formation of insulating products on the surface of the Li electrodes. It can be seen from Fig. 2E that at a fixed current density (0.05 mA/cm2), the voltage response for cells with ionomer-based SEI is low (approximately 6 mV), whereas the corresponding value for the control is much higher (approximately 150 mV). The second criterion is related to the short-circuiting of the cell when dendritic lithium that formed at one or both electrodes bridges the two electrodes. It is apparent that this phenomenon is not observed either in the control or for the ionomer SEI–stabilized electrodes. Thirdly, a rise in voltage over cycles represents an unstable SEI that grows continuously, eventually consuming the Li deposited on the stainless steel substrate. As seen in fig. S6, after only two cycles at both current densities studied, the control cell fails after a steep rise in voltage. This is quite different from what is observed for cells in which Li is stabilized by an ionomer SEI, which is stable for over 150 cycles. Figure 2D reports the number of cycles at which the cell voltage diverges as a function of current density (J). The ionomer-based SEI is seen to improve cell lifetime at a fixed current density by nearly two orders of magnitude. These results underscore the effectiveness of the ionomer-based SEI in stabilizing electrodeposition of Li in amide-based electrolytes, which were previously thought to be unfeasible for lithium metal batteries because of their high reactivity with and ready decomposition by Li.
Anode protection mechanism.
We hypothesize that the stability of the Li anode in DMA originates from two fundamental sources: (i) accumulation of LiBr salt at the Li/electrolyte interface, which facilitates Li-ion transport to the Li electrode during charging; and (ii) the existence of tethered sulfonate anions at the interface, which lowers the electric field at the electrode. Previous JDFT analysis revealed that the presence of lithium halides in the SEI of Li metal anode lowers the activation energy barrier by an order of magnitude or more for lateral Li diffusion at a Li/electrolyte interface, thereby increasing the tendency of Li to form smooth deposits (46). Comparing the surface diffusion barriers for various constituents of a typical SEI layer, Ozhabes et al. (46) found that Li2CO3, a common SEI constituent in carbonate electrolytes, has an energy barrier of 0.23 eV, whereas the barrier for a SEI composed of LiF is 0.17 eV. This difference has been argued previously to explain the much greater tendency of Li to form flat, compact deposits during battery recharge, as revealed by experiments in which weakly soluble LiF salts are enriched in the SEI by precipitating out of liquid electrolytes (37). The JDFT analysis shows that the activation energy barrier for Li-ion diffusion at a LiBr/Li interface is much lower (0.062 eV) and comparable to that of magnesium (46, 66), which is known in the literature to electrodeposit without formation of dendrites (67). Thus, the LiBr created during the formation of the SEI should provide an even more powerful (than LiF) stabilizing effect on Li deposition.
In addition to the presence of LiBr, the SEI created by the ionomer contains bound anionic groups in the form of lithium ethanesulfonate (Li-CH2CH2-SO3−). Thus, the electrolyte consists of a combination of free and tethered anions. In the past, researchers have realized the importance of single-ion–conducting electrolytes (42, 68), because these electrolytes prevent the formation of ion concentration regions within a cell, leading to stable ion transport even at a high charge rate. Recent linear stability analysis of electrodeposition by Tikekar et al. (24, 44, 45) showed that the stability of an electrolyte can be significantly enhanced by immobilizing only a small fraction (10%) of the anions. The design of our electrolyte, which is composed of a fraction of anions near the anodic surface, with LiNO3 as the free salt, is explicitly motivated by this theoretical framework. Thus, a modified SEI based on bromide ionomers tethered to the Li anode provides a powerful combination of processes that stabilize the anode against unstable electrodeposition.
Understanding the cathode stabilization mechanism
Characterizing cathode products.
Figure 3A shows a representative voltage profile for the galvanostatic discharge and charge for a Li-O2 cell with 1 M LiNO3 in an ionomer-enriched DMA electrolyte. Cutoff voltages of 2.2 and 4.3 V were used for the discharge and charge cycles, respectively, and both processes were performed at a fixed current density of 31.25 μA/cm2. Postmortem SEM analysis was used to study the evolution of discharge products on the cathode at three stages of discharge (D1, D2, and D3) and two stages of charge (C1 and C2). The SEM images show the reversible formation and decomposition of an insoluble solid product on the cathode. Complementary x-ray diffraction (XRD) analysis (Fig. 3B) shows that the cathode product is exclusively Li2O2 (no other products, such as LiOH, are observed). The SEM analysis shows that Li2O2 particles grow increasingly larger as the discharge progresses and nucleation sites for growth are filled, and the full discharge capacity of the cell is reached. Analysis of the particle sizes on discharge (see fig. S7) reveals that, at low current densities (for example, 15 μA/cm2), large Li2O2 particles (1 μm and higher) are formed. Comparing these results to those reported by Lau and Archer (10) for Li-O2 cells discharged in a 1 M LiTF in tetraethylene glycol dimethyl ether (TEGDME) (a low–donor number solvent), the Li2O2 particles formed in DMA are at least four times larger (see Fig. 3C). These findings are consistent with expectations for the high DN of DMA, which solvates Li+ cations and enables a solution-mediated mechanism, circumventing capacity limitations from the passivation layer formed at the cathode, which enables deep discharge (31). At higher current densities, the particle size at the voltage cutoff decreases drastically, consistent with the idea that kinetic diffusion limitations (27) set the maximum particle size. Upon charge, the SEM images (R1 and R2) show a cathode that closely resembles that of the pristine electrode before discharge. Redox mediation from lithium 2-bromoethanesulfonate is thought to aid in the electrochemical decomposition of the large, insulating Li2O2 particles formed on the cathode. Support for this hypothesis comes from the effectiveness of the recharge process as well as from the flat charge profile observed until the full capacity of the discharge is reached; the voltage ultimately begins to rise because of the set voltage limit of 4.3 V. Thus, Fig. 3 shows that a Li-O2 cell with 1 M LiNO3-DMA in an ionomer-based SEI on Li can reach a high capacity through LiO2 disproportionation, fully use the formed Li2O2 during the recharge, and cycles with features indicative of the presence of a redox mediator.
Fig. 3. Characterization and electrochemical analysis of oxygen cathode.
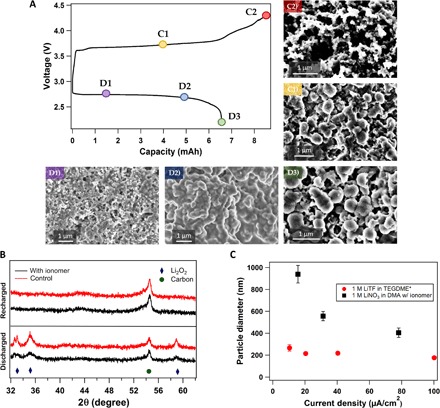
(A) Full charge-discharge cycle of a Li||O2 cell using ionomer-enriched 1 M LiNO3-DMA electrolyte operated at a current density of 31.25 μA/cm2. The different points on the voltage profile indicate various stages at which the same-type cells were stopped for ex situ analysis. The images below the voltage profile show the surface of a carbon cathode at the D1, D2, and D3 discharge phases. The size of the Li2O2 is seen to be increasing over the course of discharge. C1 and C2 show the stages of recharge; it is seen in C1 that the cathode is absent of Li2O2 particles. (B) XRD analysis showing various characteristic peaks for a fully discharged and a recharged Li||O2 battery. Here, diamonds denote Li2O2 peak and circles represent carbon. The red lines refer to the control electrolyte (1 M LiNO3-DMA), whereas black lines show the result for the same electrolyte with the ionomer additive. (C) The diameter of Li2O2 particles obtained by fully discharging a Li||O2 cell is plotted as a function of current density. Here, black indicates the electrolyte (1 M LiNO3-DMA) with the ionomer additive, whereas red represents data from Lau and Archer’s paper that used the electrolyte 1 M LiTF in TEGDME. *From Lau and Archer (10).
Cycling performance.
To evaluate the hypothesis that a high-DN electrolyte solvent and a redox mediator provide significant synergistic benefits for Li-O2 cells, we compare the voltage profiles for fully discharged cells without and with these attributes (see Fig. 4A). It is seen that the discharge capacity of Li-O2 cells with a 1 M LiNO3-DMA + ionomer electrolyte is noticeably higher (~6.5 mAh) than the discharge capacity of Li-O2 cells with a conventional 1 M lithium bis(trifluoromethane)sulfonimide (LiTFSI)–diglyme (~5.1 mAh) with the same cathode loading. This finding is consistent with the observation of large-sized lithium peroxide structures owing to the solution-mediated nucleation of peroxides. Comparison of the charge cycle shows that with the diglyme electrolyte, the voltage diverges to >4.2 V in ~3.5-mAh capacity, which is believed to be an indication of Li2CO3 formation and its effect on the charging process, whereas with the ionomer-based electrolyte, the voltage diverges at ~6.5 mAh (same as discharge). Figure 4B shows the cyclic voltammetry experiment for a Li-O2 cell in a two-electrode setup with lithium as both reference and counter electrode. The measurements were performed between 1.9 and 4.5 V (versus Li/Li+) at a scan rate of 1 mV/s, and the normalized current is plotted against voltage. The current peaks for the ionomer-based electrolyte are an order of magnitude higher than those for the control electrolyte. Thus, it can be inferred that there is higher electrochemical activity owing to the higher stability of the electrolyte and redox mediation due to the presence of LiBr. The peak seen at ~3.5 V can be attributed to a Br3−/Br− redox couple. The inset shows three cycles with ionomer-added electrolytes, where there is a slight shift of the current peaks to lower values.
Fig. 4. Galvanostatic cycling performance of lithium-oxygen electrochemical cell.
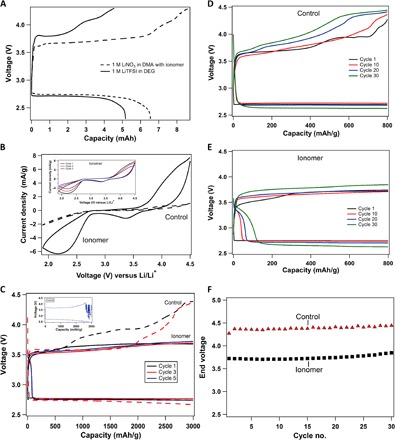
(A) Voltage profile for batteries fully discharged and recharged with 1 M LiNO3-DMA + ionomer electrolyte (shown with a solid black line) and a low–donor number electrolyte, 1 M LiTFSI-diglyme (shown with a dashed black line), at a current density of 31.25 μA/cm2. (B) Comparison of cycling voltammetry results for the control electrolyte (1 M LiNO3-DMA; shown with dashed lines) and the same electrolyte with the ionomer additive (shown with solid lines). The inset shows three cycles of cyclic voltammetry for the ionomer case. (C) Voltage profile of the Li||O2 battery with a cutoff capacity of 3000 mAh/g and a current density of 0.04 mA/cm2. The solid lines indicate ionomer-based electrolytes, whereas the control is shown with dashed lines. The inset shows the noisy profile of the fifth cycle with the control electrolyte. (D) Voltage profile with a capacity cutoff of 800 mAh/g and a current density of 0.08 mA/cm2 for a Li||O2 cell using the control electrolyte (1 M LiNO3-DMA). (E) Voltage-versus-capacity curve with the same cutoff of 800 mAh/g using the ionomer additive in the electrolyte. (F) End voltage of charging cycle for the control and the ionomer-added electrolyte is plotted as function of cycle number.
Discharge and charge profiles for cells having the electrolyte 1 M LiNO3-DMA with and without ionomers with a capacity cutoff of 3000 mAh/g and a current density of 0.04 mA/cm2 are shown in Fig. 4C. It is seen that both discharge and charge voltage curves tend to diverge to lower and higher values, respectively. Further, it can be seen from the inset of Fig. 4C that the voltage profile becomes extremely noisy in the fifth cycle of the control electrolyte, whereas that with the ionomer additive is stable. This instability without ionomers can be attributed to the degradation of the electrolyte by reaction with the unprotected lithium metal. One major benefit of cells cycled with ionomers is reduced overpotential during charge relative to that of the control cell, thus increasing cycling efficiency. This is studied in a Li-O2 battery with a lower capacity cutoff of 800 mAh/g at a current density of 0.08 mA/cm2 for the control electrolyte (Fig. 4D) and the ionomer-added electrolyte (Fig. 4E). As shown in Fig. 4E, the highest voltage on charge for cells with ionomers is approximately 3.7 V, close to the Br−/Br3− redox reaction at 3.48 V. Control cells with solely 1 M LiNO3-DMA reach voltages of around 4.45 V, as seen in Fig. 4D. This suggests a similar action to a redox mediator, in which Li2O2 is oxidized by Br3− to reform Br− in a cycle that lowers charge overpotential. The discharge and charge profiles remain similar over 30 cycles for cells with additive, whereas the charge profile in untreated cells increases more drastically. The distinct gentle slope of the initial portion of the discharge profile in cells with ionomers can be attributed to the presence of bromine species in the system. Figure 4F compares the end voltage of recharge with and without the ionomer additive. The ~1-V improvement in the round-trip efficiency not only saves loss of input energy but also ensures long-life cycling by preventing electrolyte decomposition (4).
Cathode stabilization mechanism.
At the cathode surface, LiBr is thought to participate in the redox mediation that promotes the OER reaction. In this process, the Li2O2 can be co-reduced with Br− to form O2 and Br3−. The potential for Br−→Br3− is 3.48 V; thus, the charging of a Li-O2 cell can be limited to this voltage. DMA’s ability to dissolve peroxides also aids in the effective electrolyte-side redox mediation. Support for the uniqueness of these ideas comes from recent experiments that demonstrate the efficacy of LiI and LiBr as redox mediators in Li-O2 cells based on glymes (20). In the absence of water in the electrolyte, LiI was reported to produce a gradual rise in the discharge voltage due to formation of iodine and similar products. LiBr was found to be ineffective in maintaining a steady charge voltage. In electrolytes with high water content and LiI, LiOH has been shown to be the primary discharge product, which has been reported to be thermodynamically impossible to undergo OER. Our results therefore clearly show that protecting the Li anode in a 1 M LiNO3-DMA electrolyte with a SEI based on bromide ionomer overcomes fundamental limitations of the anode, cathode, and electrolyte in previously studied systems and enables stable cycling of these cells.
CONCLUSIONS
In summary, we demonstrate that the addition of lithium 2-bromoethanesulfonate (ionomer) to 1 M LiNO3-DMA electrolytes produces a SEI at the lithium surface that stabilizes the anode in Li-O2 cells by at least two powerful processes. Compared to control cells with the ionomer SEI, Li-O2 cells based on lithium 2-bromoethanesulfonate exhibit flatter, more stable charge profiles and can withstand deeper cycling. Furthermore, we show that electrochemical charge-discharge processes in the cells coincide with the formation and decomposition of large Li2O2 particles as the principal OER product in the cathode. Analysis by linear scan voltammetry and “plate-strip” cycling analysis of the Li anode show that a SEI based on a lithium 2-bromoethanesulfonate ionomer on the anode provides chemical stability to Li against attack by DMA, as well as physical stability against rough, dendritic electrodeposition. Although we expect significant additional work from the “perfect” electrolyte for Li-O2 cells, by addressing fundamental issues that limit performance of the anode and cathode, we predict that multifunctional SEIs of the sort discussed in this study will emerge as critical to further progress.
MATERIALS AND METHODS
Li-O2 battery methods and materials
Cathode preparation.
A cathode slurry was prepared by mixing 180 mg of Super P carbon (TIMCAL), 20 mg of polyvinylidene fluoride (Sigma-Aldrich), and 2000 mg of N-methyl-2-pyrrolidone (Sigma-Aldrich) in a ball mill at 50 Hz for 1 hour. Toray TGP-H-030 carbon paper was coated with an 80-μm-thick layer of carbon slurry using a doctor blade. The resulting coated carbon paper was dried at 100°C overnight under vacuum and transferred into an argon-filled glove box [O2, <0.2 parts per million (ppm); H2O, <1.0 ppm; Innovative Technology] without exposure to air. Disks (15.9 mm in diameter) were punched and weighed from the carbon paper to yield individual carbon cathodes. The weight of the active carbon layer (not including the carbon paper) averaged 1.0 ± 0.1 mg.
Electrolyte preparation.
LiNO3 and LiTFSI were heated under vacuum overnight at 100°C to remove all traces of water and transferred directly into the glove box. DMA (Sigma-Aldrich) and bis(2-methoxyethyl) ether (diglyme; Sigma-Aldrich) solvents were dried over 3 Å molecular sieves (Sigma-Aldrich). Lithium 2-bromoethanesulfonate was obtained through ion exchange with sodium 2-bromoethanesulfonate (Sigma-Aldrich).
Coin cell assembly.
First, a 0.5-inch-diameter (12.7-mm-diameter) hole was punched in the top (cathode) side of each CR2032 case. Then, a stainless steel wire cloth disk [disk diameter, 0.75 inches (19 mm); wire diameter, 0.0055 inches (0.140 mm)] from McMaster-Carr was added, followed by a cathode disk, a 19-mm-diameter separator (either Whatman GF/D glass fiber or Celgard 3501), 100 μl of desired electrolyte, 0.5-inch-diameter lithium metal, a 15.5-mm-diameter stainless steel spacer disk, a stainless steel wave spring (MTI Corporation), and an anode cap of the CR2032 case. The assembly was crimped to a pressure of 14 MPa with a hydraulic coin cell crimple (BT Innovations).
Testing environment.
Cells were tested at a regulated pure O2 environment of 1.3 atm and allowed to equilibrate for 6 hours before electrochemical testing. Galvanostatic measurements were conducted using a Neware CT-3008 battery tester.
Cyclic voltammetry.
The cyclic voltammetry test was performed in a two-electrode setup of Li||air cathode. The batteries were cycled between 1.9 and 4.5 V at a scan rate of 1 mV/s several times.
Evaluating anode stability
Impedance spectroscopy.
Cells in the symmetric configuration were assembled in an Ar glove box. Measurements were carried out using a Solatron frequency analyzer at a frequency range of 10−3 to 107 Hz. The data were fitted into Nyquist-type plots using the equivalent circuit shown in fig. S2 with the software ZSimpWin. Impedance was conducted at room temperature at various time intervals.
Linear scan voltammetry.
Linear scan voltammetry was performed in a Li||stainless steel cell. The batteries were first swept to −0.2 V versus Li/Li+ and then they were swept in reverse direction until the voltage diverges.
Lithium versus stainless steel cycling.
For cycling tests, lithium versus stainless steel cells were prepared and were cycled at 0.01 mA/cm2 between 0 and 0.5 V 10 times to form a stable SEI layer. Then, different tests were carried out as previously described.
Characterization techniques
SEM and energy-dispersive analysis of x-rays.
Discharged cells were disassembled inside the glove box, and the cathodes were removed and transported to the SEM (Zeiss, LEO 1550 Field Emission SEM) within an airtight container. The cathodes were loaded onto the stage in the presence of a nitrogen stream. Images were taken with a single pass after focusing on a nearby region. Energy-dispersive analysis of x-ray (EDAX) measurements were performed by taking multiple counts on a small section of the sample.
X-ray diffraction.
Cathodes were mounted on a glass microscope slide inside an argon-filled glove box and coated with paraffin oil to protect them from air during the XRD measurements. Measurements were performed on a Scintag Theta-Theta x-ray diffractometer using Cu K-α radiation at λ = 1.5406Å and fitted with a two-dimensional detector. Frames were captured with an exposure time of 10 min, after which they were integrated along χ (the polar angle orthogonal to 2θ to yield an intensity-versus-2θ plot.
X-ray photoelectron spectroscopy.
XPS was conducted using Surface Science Instruments SSX-100 with an operating pressure of ~2 × 10−9 torr. Monochromatic Al K-α x-rays (1486.6 eV) with a beam diameter of 1 mm were used. Photoelectrons were collected at an emission angle of 55°. A hemispherical analyzer determined electron kinetic energy using a pass energy of 150 V for wide survey scans and 50 V for high-resolution scans. Samples were ion-etched using 4-kV Ar ions, which were rastered over an area of 2.25 mm × 4 mm with a total ion beam current of 2 mA, to remove adventitious carbon. Spectra were referenced to adventitious C 1s at 284.5 eV. CasaXPS software was used for XPS data analysis with Shelby backgrounds. Li 1s and O 1s were assigned to single peaks for each bond, whereas Br 3d was assigned to double peaks (3d5/2 and 3d3/2) for each bond with 1.05-eV separation. Residual SD was maintained close to 1.0 for the calculated fits. Samples were exposed to air only during the short transfer time to the XPS chamber (less than 5 s).
Supplementary Material
Acknowledgments
Funding: We are grateful to the Advanced Research Projects Agency-Energy (award DE-AR-0000750) for supporting this study. The study made use of the characterization facilities of the King Abdullah University of Science and Technology (KAUST)–Cornell University Center for Energy and Sustainability, which was supported by the KAUST through award number KUS-C1-018-02. Electron microscopy facilities at the Cornell Center for Materials Research, an NSF-supported Materials Research Science and Engineering Center through grant DMR-1120296, were also used for the study. Additional support for the FIB/SEM cryo-stage and transfer system was provided by the Kavli Institute at Cornell and the Energy Materials Center at Cornell and the U.S. Department of Energy, Energy Frontier Research Center, Basic Energy Sciences (DE-SC0001086). M.J.Z. and L.F.K. acknowledge support by the David and Lucile Packard Foundation. Author contributions: S.C., C.T.-C.W., and L.A.A. conceived the study, conducted the electrochemical measurements, and analyzed the resulting data. S.L. designed and built the electrochemical cell used for the study. W.I.A.S. performed XPS analysis, and Z.T., M.J.Z., and L.F.K. performed cryo-SEM and EDX analysis of the Li anode. All authors contributed to the writing of this article. Competing interests: L.A.A. is a founder and scientific advisor for NOHMs Technologies, a technology company focused on commercializing electrolytes and electrodes for lithium-sulfur and lithium-ion batteries. L.A.A. is a member of the Board of Directors of NOHMs Technologies. L.A.A. filed a patent through Cornell University to the U.S. Patent and Trademark Office related to this work, U.S. Provisional No. 62/436,248, on 19 December 2016. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1602809/DC1
fig. S1. Two-dimensional EDAX mapping of lithium-deposited stainless steel substrate with 1 M LiNO3-DMA electrolyte and 10% ionomer additive.
fig. S2. XPS results showing the binding energy of Li and O atoms with the control electrolyte (1 M LiNO3-DMA).
fig. S3. Nyquist plots of 1 M LiNO3-DMA enriched with 5% (by weight) ionomer additive, showing impedance for different storage times of the battery.
fig. S4. Equivalent circuit model to fit the Nyquist plot obtained from impedance spectroscopy measurement comprising bulk resistance, interfacial resistance parallel to a constant phase element, and a solid-state diffusion element.
fig. S5. Nyquist plots showing experimental as well as circuit model–fitted results of impedance measurements with symmetric cells for the control electrolyte and ionomer-added batteries after 48 and 56 hours of storage.
fig. S6. Stripping and plating of Li versus stainless steel cell after depositing lithium (10 mAh/cm2) onto stainless steel.
fig. S7. Size analysis of lithium peroxide particles after discharging a Li-O2 cell with 1 M LiNO3-DMA electrolyte and the ionomer additive at different current densities, as indicated in the box.
table S1. Atomic percentage of detected elements on lithium anodes.
REFERENCES AND NOTES
- 1.Bruce P. G., Freunberger S. A., Hardwick L. J., Tarascon J.-M., Li–O2 and Li–S batteries with high energy storage. Nat. Mater. 11, 19–29 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Luntz A. C., McCloskey B. D., Nonaqueous Li–air batteries: A status report. Chem. Rev. 114, 11721–11750 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Girishkumar G., McCloskey B., Luntz A. C., Swanson S., Wilcke W., Lithium-air battery: Promise and challenges. J. Phys. Chem. Lett. 1, 2193–2203 (2010). [Google Scholar]
- 4.Aurbach D., McCloskey B. D., Nazar L. F., Bruce P. G., Advances in understanding mechanisms underpinning lithium–air batteries. Nat. Energy 1, 16128 (2016). [Google Scholar]
- 5.Lu Y.-C., Gasteiger H. A., Parent M. C., Chiloyan V., Shao-Horn Y., The influence of catalysts on discharge and charge voltages of rechargeable Li–oxygen batteries. Electrochem. Solid State Lett. 13, A69–A72 (2010). [Google Scholar]
- 6.Mccloskey B. D., Valery A., Luntz A. C., Gowda S. R., Wallraff G. M., Garcia J. M., Mori T., Krupp L. E., Combining accurate O2 and Li2O2 assays to separate discharge and charge stability limitations in nonaqueous Li–O2 batteries. J. Phys. Chem. Lett. 4, 2989–2993 (2013). [DOI] [PubMed] [Google Scholar]
- 7.McCloskey B. D., Bethune D. S., Shelby R. M., Mori T., Scheffler R., Speidel A., Sherwood M., Luntz A. C., Limitations in rechargeability of Li-O2 batteries and possible origins. J. Phys. Chem. Lett. 3, 3043–3047 (2012). [DOI] [PubMed] [Google Scholar]
- 8.McCloskey B. D., Speidel A., Scheffler R., Miller D. C., Viswanathan V., Hummelshøj J. S., Nørskov J. K., Luntz A. C., Twin problems of interfacial carbonate formation in nonaqueous Li–O2 batteries. J. Phys. Chem. Lett. 3, 997–1001 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Ottakam Thotiyl M. M., Freunberger S. A., Peng Z., Chen Y., Liu Z., Bruce P. G., A stable cathode for the aprotic Li–O2 battery. Nat. Mater. 12, 1050–1056 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Lau S., Archer L. A., Nucleation and growth of lithium peroxide in the Li–O2 battery. Nano Lett. 15, 5995–6002 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Højberg J., McCloskey B. D., Hjelm J., Vegge T., Johansen K., Norby P., Luntz A. C., An electrochemical impedance spectroscopy investigation of the overpotentials in Li–O2 batteries. ACS Appl. Mater. Interfaces 7, 4039–4047 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Chen Y., Freunberger S. A., Peng Z., Fontaine O., Bruce P. G., Charging a Li–O2 battery using a redox mediator. Nat. Chem. 5, 489–494 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Lee D. J., Lee H., Kim Y.-J., Park J.-K., Kim H.-T., Sustainable redox mediation for lithium-oxygen batteries by a composite protective layer on the lithium-metal anode. Adv. Mater. 28, 857–863 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Bergner B. J., Hofmann C., Schürmann A., Schröder D., Peppler K., Schreiner P. R., Janek J., Understanding the fundamentals of redox mediators in Li–O2 batteries: A case study on nitroxides. Phys. Chem. Chem. Phys. 17, 31769–31779 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Liu T., Leskes M., Yu W., Moore A. J., Zhou L., Bayley P. M., Kim G., Grey C. P., Cycling Li-O2 batteries via LiOH formation and decomposition. Science 350, 530–533 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Kwak W.-J., Hirshberg D., Sharon D., Shin H.-J., Afri M., Park J.-B., Garsuch A., Chesneau F. F., Frimer A. A., Aurbach D., Sun Y.-K., Understanding the behavior of Li–oxygen cells containing LiI. J. Mater. Chem. A 3, 8855–8864 (2015). [Google Scholar]
- 17.Lim H.-D., Song H., Kim J., Gwon H., Bae Y., Park K.-Y., Hong J., Kim H., Kim T., Kim Y. H., Lepró X., Ovalle-Robles R., Baughman R. H., Kang K., Superior rechargeability and efficiency of lithium-oxygen batteries: Hierarchical air electrode architecture combined with a soluble catalyst. Angew. Chem. Int. Ed. 53, 3926–3931 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Kundu D., Black R., Adams B., Nazar L. F., A highly active low voltage redox mediator for enhanced rechargeability of lithium–oxygen batteries. ACS Cent. Sci. 1, 510–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun D., Shen Y., Zhang W., Yu L., Yi Z., Yin W., Wang D., Huang Y., Wang J., Wang D., Goodenough J. B., A solution-phase bifunctional catalyst for lithium–oxygen batteries. J. Am. Chem. Soc. 136, 8941–8946 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Kwak W.-J., Hirshberg D., Sharon D., Afri M., Frimer A. A., Jung H.-G., Aurbach D., Sun Y.-K., Li–O2 cells with LiBr as an electrolyte and redox mediator. Energy Environ. Sci. 9, 2334–2345 (2016). [Google Scholar]
- 21.Zhang T., Liao K., He P., Zhou H., A self-defense redox mediator for efficient lithium–O2 batteries. Energy Environ. Sci. 9, 1024–1030 (2016). [Google Scholar]
- 22.Veith G. M., Nanda J., Delmau L. H., Dudney N. J., Influence of lithium salts on the discharge chemistry of Li–air cells. J. Phys. Chem. Lett. 3, 1242–1247 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Black R., Oh S. H., Lee J.-H., Yim T., Adams B., Nazar L. F., Screening for superoxide reactivity in Li-O2 batteries: Effect on Li2O2/LiOH crystallization. J. Am. Chem. Soc. 134, 2902–2905 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Tikekar M. D., Choudhury S., Tu Z., Archer L. A., Design principles for electrolytes and interfaces for stable lithium-metal batteries. Nat. Energy 1, 16114 (2016). [Google Scholar]
- 25.Freunberger S. A., Chen Y., Peng Z., Griffin J. M., Hardwick L. J., Bardé F., Novák P., Bruce P. G., Reactions in the rechargeable lithium–O2 battery with alkyl carbonate electrolytes. J. Am. Chem. Soc. 133, 8040–8047 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Bryantsev V. S., Uddin J., Giordani V., Walker W., Addison D., Chase G. V., The identification of stable solvents for nonaqueous rechargeable Li-air batteries. J. Electrochem. Soc. 160, A160–A171 (2013). [Google Scholar]
- 27.Burke C. M., Pande V., Khetan A., Viswanathan V., Mccloskey B. D., Enhancing electrochemical intermediate solvation through electrolyte anion selection to increase nonaqueous Li–O2 battery capacity. Proc. Natl. Acad. Sci. U.S.A. 112, 9293–9298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson L., Li C., Liu Z., Chen Y., Freunberger S. A., Ashok P. C., Praveen B. B., Dholakia K., Tarascon J.-M., Bruce P. G., The role of LiO2 solubility in O2 reduction in aprotic solvents and its consequences for Li–O2 batteries. Nat. Chem. 6, 1091–1099 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Khetan A., Luntz A., Viswanathan V., Trade-offs in capacity and rechargeability in nonaqueous Li–O2 batteries: Solution-driven growth versus nucleophilic stability. J. Phys. Chem. Lett. 6, 1254–1259 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Knudsen K. B., Vegge T., McCloskey B. D., Hjelm J., An electrochemical impedance spectroscopy study on the effects of the surface- and solution-based mechanisms in Li-O2 cells. J. Electrochem. Soc. 163, A2065–A2071 (2016). [Google Scholar]
- 31.Aetukuri N. B., McCloskey B. D., García J. M., Krupp L. E., Viswanathan V., Luntz A. C., Solvating additives drive solution-mediated electrochemistry and enhance toroid growth in non-aqueous Li–O2 batteries. Nat. Chem. 7, 50–56 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Rosenman A., Elazari R., Salitra G., Markevich E., Aurbach D., Garsuch A., The effect of interactions and reduction products of LiNO3, the anti-shuttle agent, in Li-S battery systems. J. Electrochem. Soc. 162, A470–A473 (2015). [Google Scholar]
- 33.Zhang S. S. Role of LiNO3 in rechargeable lithium/sulfur battery. Electrochim. Acta 70, 344–348 (2012). [Google Scholar]
- 34.Liang X., Wen Z., Liu Y., Wu M., Jin J., Zhang H., Wu X., Improved cycling performances of lithium sulfur batteries with LiNO3-modified electrolyte. J. Power Sources 196, 9839–9843 (2011). [Google Scholar]
- 35.Aurbach D., Pollak E., Elazari R., Salitra G., Kelley C. S., Affinito J., On the surface chemical aspects of very high energy density, rechargeable Li–sulfur batteries. J. Electrochem. Soc. 156, A694–A702 (2009). [Google Scholar]
- 36.Walker W., Giordani V., Uddin J., Bryantsev V. S., Chase G. V., Addison D., A rechargeable Li–O2 battery using a lithium nitrate/N,N-dimethylacetamide electrolyte. J. Am. Chem. Soc. 135, 2076–2079 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Choudhury S., Archer L. A.. Lithium fluoride additives for stable cycling of lithium batteries at high current densities. Adv. Electron. Mater. 2, 1500246 (2016). [Google Scholar]
- 38.Choudhury S., Mangal R., Agrawal A., Archer L. A.. A highly reversible room-temperature lithium metal battery based on crosslinked hairy nanoparticles. Nat. Commun. 6, 10101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agrawal A., Choudhury S., Archer L. A., A highly conductive, non-flammable polymer–nanoparticle hybrid electrolyte. RSC Adv. 5, 20800–20809 (2015). [Google Scholar]
- 40.Tarascon J.-M., Armand M., Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Kashiwagi T., Du F., Douglas J. F., Winey K. I., Harris R. H. Jr, Shields J. R., Nanoparticle networks reduce the flammability of polymer nanocomposites. Nat. Mater. 4, 928–933 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Tu Z., Nath P., Lu Y., Tikekar M. D., Archer L. A., Nanostructured electrolytes for stable lithium electrodeposition in secondary batteries. Acc. Chem. Res. 48, 2947–2956 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Cheng X.-B., Zhang R., Zhao C.-Z., Wei F., Zhang J.-G., Zhang Q., A review of solid electrolyte interphases on lithium metal anode. Adv. Sci. 3, 1500213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tikekar M. D., Archer L. A., Koch D. L., Stability analysis of electrodeposition across a structured electrolyte with immobilized anions. J. Electrochem. Soc. 161, A847–A855 (2014). [Google Scholar]
- 45.Tikekar M. D., Archer L. A., Koch D. L., Stabilizing electrodeposition in elastic solid electrolytes containing immobilized anions. Sci. Adv. 2, e1600320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozhabes Y., Gunceler D., Arias T. A., Stability and surface diffusion at lithium-electrolyte interphases with connections to dendrite suppression. arXiv:1504.05799 (2015). [Google Scholar]
- 47.Lu Y., Tikekar M., Mohanty R., Hendrickson K., Ma L., Archer L. A., Stable cycling of lithium metal batteries using high transference number electrolytes. Adv. Energy Mater. 5, 1402073 (2015). [Google Scholar]
- 48.Schaefer J. L., Yanga D. A., Archer L. A., High lithium transference number electrolytes via creation of 3-dimensional, charged, nanoporous networks from dense functionalized nanoparticle composites. Chem. Mater. 25, 834–839 (2013). [Google Scholar]
- 49.Bouchet R., Maria S., Meziane R., Aboulaich A., Lienafa L., Bonnet J.-P., Phan T. N. T., Bertin D., Gigmes D., Devaux D., Denoyel R., Armand M., Single-ion BAB triblock copolymers as highly efficient electrolytes for lithium-metal batteries. Nat. Mater. 12, 452–457 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Yao K. P. C., Kwabi D. G., Quinlan R. A., Mansour A. N., Grimaud A., Lee Y.-L., Lu Y.-C., Shao-Horn Y., Thermal stability of Li2O2 and Li2O for Li-air batteries: In situ XRD and XPS studies. J. Electrochem. Soc. 160, A824–A831 (2013). [Google Scholar]
- 51.Lu Y.-C., Crumlin E. J., Veith G. M., Harding J. R., Mutoro E., Baggetto L., Dudney N. J., Liu Z., Shao-Horn Y., In situ ambient pressure X-ray photoelectron spectroscopy studies of lithium-oxygen redox reactions. Sci. Rep. 2, 715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kundu D., Black R., Berg E. J., Nazar L. F., A highly active nanostructured metallic oxide cathode for aprotic Li–O2 batteries. Energy Environ. Sci. 8, 1292–1298 (2015). [Google Scholar]
- 53.J. F. Moulder, W. Stickle, P. Sobol, K. Bomben, Handbook of X-ray Photoelectron Spectroscopy (Perkin-Elmer Corp., 1992). [Google Scholar]
- 54.Zhang Z., Lu J., Assary R. S., Du P., Wang H.-H., Sun Y.-K., Qin Y., Lau K. C., Greeley J., Redfern P. C., Iddir H., Curtiss L. A., Amine K., Increased stability toward oxygen reduction products for lithium-air batteries with oligoether-functionalized silane electrolytes. J. Phys. Chem. C 115, 25535–25542 (2011). [Google Scholar]
- 55.Lu J., Jung H.-J., Lau K. C., Zhang Z., Schlueter J. A., Du P., Assary R. S., Greeley J., Ferguson G. A., Wang H.-H., Hassoun J., Iddir H., Zhou J., Zuin L., Hu Y., Sun Y.-K., Scrosati B., Curtiss L. A., Amine K., Magnetism in lithium-oxygen discharge product. ChemSusChem 6, 1196–1202 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Basile A., Bhatt A. I., O’Mullane A. P., Stabilizing lithium metal using ionic liquids for long-lived batteries. Nat. Commun. 7, ncomms11794 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hausbrand R., Cherkashinin G., Ehrenberg H., Gröting M., Albe K., Hess C., Jaegermann W., Fundamental degradation mechanisms of layered oxide Li-ion battery cathode materials: Methodology, insights and novel approaches. Mater. Sci. Eng. B 192, 3–25 (2015). [Google Scholar]
- 58.Xiong S., Xie K., Diao Y., Hong X., Properties of surface film on lithium anode with LiNO3 as lithium salt in electrolyte solution for lithium–sulfur batteries. Electrochim. Acta 83, 78–86 (2012). [Google Scholar]
- 59.Wu Y., Fang S., Jiang Y., Effects of nitrogen on the carbon anode of a lithium secondary battery. Solid State Ionics 120, 117–123 (1999). [Google Scholar]
- 60.Desai S. M., Solanky S. S., Mandale A. B., Rathore K., Singh R. P., Controlled grafting of N-isopropyl acrylamide brushes onto self-standing isotactic polypropylene thin films: Surface initiated atom transfer radical polymerization. Polymer. 44, 7645–7649 (2003). [Google Scholar]
- 61.NIST X-ray Photoelectron Spectroscopy Database, Version 4.1 (National Institute of Standards Technology, 2012).
- 62.Ferrighi L., Píš I., Nguyen T. H., Cattelan M., Nappini S., Basagni A., Parravicini M., Papagni A., Sedona F., Magnano E., Bondino F., Di Valentin C., Agnoli S., Control of the intermolecular coupling of dibromotetracene on Cu(110) by the sequential activation of C–Br and C–H bonds. Chem. A Eur. J. 21, 5826–5834 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Basagni A., Ferrighi L., Cattelan M., Nicolas L., Handrup K., Vaghi L., Papagni A., Sedona F., Di Valentin C., Agnoli S., Sambi M., On-surface photo-dissociation of C–Br bonds: Towards room temperature Ullmann coupling. Chem. Commun. 51, 12593–12596 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Gutzler R., Cardenas L., Lipton-Duffin J., El Garah M., Dinca L. E., Szakacs C. E., Fu C., Gallagher M., Vondráček M., Rybachuk M., Perepichka D. F., Rosei F., Ullmann-type coupling of brominated tetrathienoanthracene on copper and silver. Nanoscale 6, 2660–2668 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Di Giovannantonio M., El Garah M., Lipton-Duffin J., Meunier V., Cardenas L., Fagot Revurat Y., Cossaro A., Verdini A., Perepichka D. F., Rosei F., Contini G., Insight into organometallic intermediate and its evolution to covalent bonding in surface-confined ullmann polymerization. ACS Nano 7, 8190–8198 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Jäckle M., Groß A., Microscopic properties of lithium, sodium, and magnesium battery anode materials related to possible dendrite growth. J. Chem. Phys. 141, 174710 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Ha S.-Y., Lee Y.-W., Woo S. W., Koo B., Kim J.-S., Cho J., Lee K. T., Choi N.-S., Magnesium(II) Bis(trifluoromethane sulfonyl) imide-based electrolytes with wide electrochemical windows for rechargeable magnesium batteries. ACS Appl. Mater. Interfaces 6, 4063–4073 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Feng S., Shi D., Liu F., Zheng L., Nie J., Feng W., Huang X., Armand M., Zhou Z., Single lithium-ion conducting polymer electrolytes based on poly[(4-styrenesulfonyl)(trifluoromethanesulfonyl)imide] anions. Electrochim. Acta 93, 254–263 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1602809/DC1
fig. S1. Two-dimensional EDAX mapping of lithium-deposited stainless steel substrate with 1 M LiNO3-DMA electrolyte and 10% ionomer additive.
fig. S2. XPS results showing the binding energy of Li and O atoms with the control electrolyte (1 M LiNO3-DMA).
fig. S3. Nyquist plots of 1 M LiNO3-DMA enriched with 5% (by weight) ionomer additive, showing impedance for different storage times of the battery.
fig. S4. Equivalent circuit model to fit the Nyquist plot obtained from impedance spectroscopy measurement comprising bulk resistance, interfacial resistance parallel to a constant phase element, and a solid-state diffusion element.
fig. S5. Nyquist plots showing experimental as well as circuit model–fitted results of impedance measurements with symmetric cells for the control electrolyte and ionomer-added batteries after 48 and 56 hours of storage.
fig. S6. Stripping and plating of Li versus stainless steel cell after depositing lithium (10 mAh/cm2) onto stainless steel.
fig. S7. Size analysis of lithium peroxide particles after discharging a Li-O2 cell with 1 M LiNO3-DMA electrolyte and the ionomer additive at different current densities, as indicated in the box.
table S1. Atomic percentage of detected elements on lithium anodes.