


Abstract
We propose a method for analyzing the magnitude and direction of curvature within nucleic acids, based on the curvilinear helical axis calculated by Curves+. The method is applied to analyzing curvature within minicircles constructed with varying degrees of over- or under-twisting. Using the molecular dynamics trajectories of three different minicircles, we are able to quantify how curvature varies locally both in space and in time. We also analyze how curvature influences the local environment of the minicircles, notably via increased heterogeneity in the ionic distributions surrounding the double helix. The approach we propose has been integrated into Curves+ and the utilities Canal (time trajectory analysis) and Canion (environmental analysis) and can be used to study a wide variety of static and dynamic structural data on nucleic acids.
INTRODUCTION
Measuring DNA curvature has never been particularly easy. The simplest approaches reduce the problem to the deformation of individual base pair steps, generally linking curvature to a combination of the helical parameters roll and tilt and taking into account the helical twist between successive steps (1). However, as pointed out earlier (2), many different combinations of the inter-base pair helical parameters may correspond to the same curvature and, similarly, regular non-zero values of these parameters do not imply a curved helical axis (e.g. within regular A-DNA). An alternative approach is to try to define regular helical regions within a structure, determine their linear axes and then describe an overall bend as the angle formed between these axes (2), although clearly this is not always possible. Another attempt used the projection of the base pair normals into a plane perpendicular to the average, linear helical axis (a method that is not easy to interpret since it assumes that the normals are aligned with the helical axis; an assumption that is only valid for conformations close to a canonical B-DNA) (3). None of these methods is really satisfactory. Curvature is often the result of subtle deformations involving several base pairs, which rarely align in a plane and thus are not simply additive. A good example of this is the curvature induced by A-tracts (runs of several AT pairs with the purines in one strand). The difficulty of defining curvature in these cases, even after high-resolution crystal structures became available, led to a very long debate in the literature, where base pair step interpretations competed with more global views involving ‘junctions’ between the A-tracts and the flanking DNA segments (4–7). Since this time, the problem of understanding and quantifying curvature has remained, notably because of a rapidly increasing database of experimental and computationally derived structural information on deformed DNA. Such deformation may be caused in many ways: by the base sequence alone, by bound ligands, by bound proteins or protein complexes or by topological constraints, as in looped or circular DNA.
In developing the DNA conformational analysis program Curves (8,9), and its more recent incarnation Curves+ (10), we aimed at defining not only helical, backbone and groove parameters, but also a curvilinear helical axis that would help to resolve some of the questions raised above. Although we believe that such a helical axis has been a very useful guide to interpreting DNA curvature in a visual sense, it did not provide quantitative information on curvature. In our case, an overall bend was defined using the angle between the vectors forming the ends of the curvilinear helical axis. Calculating this angle for an axis extending to the terminal base pairs was not advisable since these base pairs often undergo significant deformations themselves. However, deciding which base pairs to ignore in any given case was not an easy choice to make. Similarly, local curvature could be defined using the angle between the helical axis vectors at successive base pair levels, but this angle is not easy to interpret and Curves+ provided no accompanying directional information. Indeed, while we have mainly discussed measuring the magnitude of curvature above, it is equally important to known its direction, and thus, whether successive local curvatures contribute to a significant overall bend.
If we accept that a curvilinear helical axis can serve for measuring the magnitude of curvature, measuring its direction requires defining a reference system. If curvature always occurred in a plane then this plane could be used. However, given the fact that most helical axes do not lie in a plane, or follow any simple three-dimensional (3D) shape, we need some reference based on DNA itself. The most obvious choice seems to be the base pairs, since they can be represented with a well-defined reference axis system (10,11). This allows the direction of curvature to be defined, at least at the level of each base pair. The importance of defining the direction of curvature became clear in our early studies of sequence-induced curvature. This led us to use the term ‘register’ to define the local curvature direction with respect to a given base pair (12). With intrinsically-bent DNA, or DNA where bending is induced, for example by circularization, register enables us to define which face of DNA is oriented toward the direction of curvature and, in the case of molecular simulations, whether register is fixed or variable with time.
In the present work, we propose a method for defining the local magnitude of curvature (hereafter termed simply curvature) and register, based on the curvilinear helical axis calculated by Curves+. It can be applied to both static DNA structures and to molecular dynamics (MD) trajectories. In addition, the availability of quantitative data on curvature, coupled with our recent work on describing ions or molecules surrounding DNA (13,14), enables us to go further and analyze how curvature affects the environment of DNA.
We have chosen to illustrate this approach using MD simulations of DNA minicircles, since we can easily control the overall curvature of minicircles via over- or under-twisting, by changing the number of base pairs or by changing the linking number (15–17). However, the same analysis can equally be applied to linear DNA, helical RNA's or multi-stranded nucleic acids. We remark in passing that the Curves+ helical axis derived for minicircles, as well as an alternative axis definition, have previously been used for estimating writhe, but this work did not consider the magnitude or direction of local curvature (18).
MATERIALS AND METHODS
Minicircle construction
The present analysis is based on the study of three different DNA minicircles. The first ‘relaxed’ minicircle, termed RE_94, contains 94 base pairs (bp) and has a linking number Lk = 9 (i.e. nine helical turns), implying an average helical twist of 34.5° (in the absence of writhe), close to the optimal value for B-DNA (however, see remarks below). This sequence was taken from the study of Cloutier and Widom where it was found to have a high ring closure probability (19). In the second ‘unwound’ minicircle, UN_94, the sequence is the same as that of RE_94 (i.e. 94 bp), but Lk has been reduced to 8, implying a full turn of undertwisting. Lastly, in the third ‘overwound’ minicircle, OV_89, the sequence has been shortened by 5 bp with respect to RE_94, again with Lk = 9, adding roughly half a turn of overtwisting.
In order to determine the actual degree of under- or over-twisting to be expected during MD simulations of these minicircles, it is possible to introduce a fractional linking number Lk_0 for the same sequence in a cyclized, but torsionally relaxed state. Then each minicircle with covalently closed backbones has an integer linking number Lk and a relative linking number, or superhelical density, σ = (Lk - Lk_0)/Lk_0. For a given DNA fragment it is possible to estimate Lk_0 by summing all of the sequence-dependent average base pair step twists for a linear DNA of the same sequence simulated using the same force field and solvent/ion environment that we employ (see below). This approach neglects contributions to Lk_0 from any intrinsic bends in the helical axis of the average shape of the linear fragment, but any associated error will be small if the average helical axis is close to being straight. Here, the sequence-dependent twists were obtained from the μABC dataset (20) by summing up the twists of the central base pair steps of the corresponding tetranucleotide fragments. The results are shown in Table 1, where we can see that RE_94 is actually slightly overwound (σ = 0.055), UN_94 is underwound by roughly the same magnitude (σ = −0.063) and OV_89 is significantly overwound (σ = 0.113). These values result from an average reduction of twist of 1.6° compared to the currently accepted sequence-averaged value of 34.3° (i.e. 10.5 bp/turn) for DNA in solution. This difference can be partially attributed to the force field and partially to the specific sequence used for these minicircles.
Table 1. Minicircles studied: showing the linking number Lk, the unconstrained Lk0, the superhelical density σ and the corresponding base sequence.
| Minicircle | Lk | Lk0 | σ | Sequence (5΄-3΄) |
|---|---|---|---|---|
| RE_94 | 9 | 8.53 | 0.055 | GGCCGGGTCGTAGCAAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCT ACCGCGTTTTAACCGCCAATAGGATTACTTACTAGTCTCTAC |
| UN_94 | 8 | 8.53 | −0.063 | GGCCGGGTCGTAGCAAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCT ACCGCGTTTTAACCGCCAATAGGATTACTTACTAGTCTCTAC |
| OV_89 | 9 | 8.09 | 0.113 | GGCCGGGTCGTAGCAAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCT ACCGCGTTTTAACCGCCAATAGGATTACTTACTAGTC |
The initial construction of each minicircle was made using the internal coordinate nucleic acid modeling program JUMNA, using its superhelical symmetry and ring closure options (12,21). The minicircles were energy minimized in JUMNA using the Amber parm99/BSC0 force field (22–24) combined with a simple continuum solvent/counterion representation, before being transferred to Amber to carry out MD simulations.
Molecular dynamics (MD) simulations
Simulations were performed using the Amber 12 program suite (22,25) with the BSC0 modifications (24) to the parm99 parameter set (22,23). Each minicircle was placed in a truncated octahedral cell and solvated with SPC/E water (26) creating a solvent layer at least 10 Å thick. The minicircle charge was neutralized with an appropriate number of potassium cations and then sufficient K+Cl− ion pairs were added to achieve a bulk ionic strength of 0.15 M, using the Dang force field parameters (27). All ions were initially placed randomly, but at least 5 Å from the solute and at least 3.5 Å from one another. The resultant system size varied from 131,409 to 164,196 atoms. After heating and equilibrating the systems, using a protocol described earlier (28,29), simulations were carried out for at least 300 ns using periodic boundary conditions. Electrostatic interactions were treated with the particle mesh Ewald method (30) with a 9 Å real-space cutoff. The length of chemical bonds involving hydrogen was restrained using SHAKE (31), allowing the use of a 2 fs time step. An NPT ensemble was generated using the Berendsen algorithm (32) with a 5 ps coupling constant to keep the temperature at 300 K and the pressure at 1 atm. During each simulation, the solute was kept in the center of the simulation cell by removing any motion of its center of mass every 5000 steps (33). Conformational snapshots were saved every ps for analysis. The average conformation for any given minicircle simulation (subsequently used to define the average helical axis) was obtained by averaging the superposed Cartesian coordinates of the MD snapshots.
Measuring local curvature and register
Our approach to measuring curvature along a nucleic acid fragment (whether or not it belongs to a minicircle) is to locally fit a circle to the curvilinear helical axis obtained from a Curves+ analysis (10). The Curves+ axis is defined by a set of orthogonal reference frames Ai centered at points Ui for each base pair i. In order to obtain the curvature at a given base pair step i → i + 1, we start by fitting a circle to the axis points Ui - 1, Ui, Ui+ 1 and a second circle to the points Ui, Ui+ 1, Ui+ 2. The radii ri and ri +1 of these circles (termed osculating circles) define the curvature ci and ci +1 at levels i and i + 1 respectively (equal to the inverse of the corresponding radii). We then define the curvature at step i → i + 1 as geometrical average (ci.ci+ 1)1/2. Using this procedure, we associate curvature with base pair steps rather than base pairs. This seems appropriate since curvature largely arises from the perturbation of the stacking between successive base pairs. This procedure also decreases the impact of abrupt changes in the curvature of the helical axis to some extent, but, as shown below, curvature nevertheless can vary rapidly along a nucleic acid fragment. It should be added that the curvature obtained by fitting a circle to three points depends on the separation of the points. This dependence is very small for weakly deformed minicircles, or for helical DNA, such as that wound around the histone core of nucleosomes. For more strongly deformed helical axes, the dependence is more important, but a separation of single base pair steps is small enough to lead to accurate results even in very extreme cases. As an example, consider an ellipse with a circumference equal to an 89 bp minicircle and a ratio of three between the major and minor axes. This shape has a range of radii of curvature from 7 to 206 Å (beyond the range of radii seen in the most deformed minicircle studied here). Our approach yields these values to within 0.3 Å at the highest curvature and 0.2 Å at the lowest. Increasing the separation of the points would naturally decrease this accuracy.
In order to make the values of curvature more meaningful in nucleic acid terms, we use a scaling factor of 40 Å, making curvature a dimensionless quantity (hereafter termed Ci). This choice makes the curvature of DNA on the nucleosome approximately equal to 1.0 (to be precise, analyzing the 147 bp of DNA from the high-resolution nucleosome structure 1KX5 (34), leads to Ci = 1.05) and it makes easier to interpret the degree of curvature in other cases.
We now need to define the direction of curvature at a given base pair step i → i + 1 with respect to the orientation of the DNA base pairs. We have previously termed this variable rotational register, or, more simply, register (12). This variable is important because it determines which portion of the double helix (groove or backbone) faces inward or outward with respect to the local curvature and, in the case of MD simulations, whether this orientation is static or dynamic.
The register, Gi, at base pair step i → i + 1 is calculated by first generating the corresponding mid-step axis reference frame 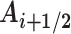 by a half-rotation of frame Ai around the screw axis linking the frames Ai and Ai+ 1 (see the Supplementary Data concerning Curves+ for more details (10)). We then calculate the mean of unit radial vectors of the osculating circles determined at positions Ui and Ui+ 1 and project this vector into the plane perpendicular to the local helical axis
by a half-rotation of frame Ai around the screw axis linking the frames Ai and Ai+ 1 (see the Supplementary Data concerning Curves+ for more details (10)). We then calculate the mean of unit radial vectors of the osculating circles determined at positions Ui and Ui+ 1 and project this vector into the plane perpendicular to the local helical axis 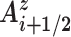 . We term this vector Gi + 1/2. Finally, we calculate the angle between the vectors Gi + 1/2 and
. We term this vector Gi + 1/2. Finally, we calculate the angle between the vectors Gi + 1/2 and 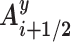 , positive angles being defined as a clockwise rotation around
, positive angles being defined as a clockwise rotation around 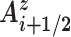 (
(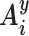 is derived from the BL vector describing the long axis of base pair i, pointing toward the strand whose 5΄-3΄ direction is aligned with
is derived from the BL vector describing the long axis of base pair i, pointing toward the strand whose 5΄-3΄ direction is aligned with 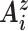 (10)). In this way, a register G = 90° implies that the minor groove is on the inside face of the local curvature, while G = −90° places the major groove on the inside.
(10)). In this way, a register G = 90° implies that the minor groove is on the inside face of the local curvature, while G = −90° places the major groove on the inside.
Because instantaneous values of curvature and register both fluctuate rapidly as a function of time during MD trajectories, time-averaging was carried out using a sliding window with a default width of 1 ns. The user can however change the width of the time window, or remove time-averaging completely using options in Canal.
Analyzing ion distributions
Ion distributions around a nucleic acid fragment are analyzed using curvilinear helicoidal coordinates as described in our earlier publications (13,14). These coordinates R, D and A, define the position of any given ion with respect to the Curves+ helical axis, where R is the radial distance from the axis (in Å), D is the distance along the axis (measured in units of base pair steps, starting from 1.0 at the first base pair level) and A is the angle of the ion with respect to the corresponding Ay vector described in the previous section. This angle is measured in degrees and the sign of the rotation is as defined above. In the case of an MD trajectory, Curves+ calculates the RDA coordinates of every ion in each snapshot with respect to the instantaneous helical axis. This data is passed to the Canion utility that can then calculate the molarity and the ion population in any chosen region surrounding the nucleic acid. Molarities and populations can be analyzed as time series, or as averages over a given time interval, using a single average helical axis derived from the simulation (13). This approach provides a precise picture of the ionic environment by avoiding ‘blurring’ the distribution of ions close to the nucleic acid due to their coupling with the thermal fluctuations of the nucleic acid (notably, bending, twisting and stretching).
Using the definition of register described above, it now becomes possible to distinguish ions in terms of their location on the ‘inside’ or ‘outside’ of a curved nucleic acid fragment. For an ion with coordinates R, D, A, this is achieved by calculating the difference between the angular position of the ion, A and the register GD at the location D. Ions lying ‘inside’ the local direction of curvature are those for which |A-GD| < 90°, while those for which |A-GD| ≥ 90° are classed as ‘outside’. Note that the register vector defining GD at any position within a given base pair step can be obtained from the corresponding Gi using the screw transform discussed in the previous section.
Canion has been modified to allow the user to optionally limit any analysis to either the inside or the outside face of a chosen nucleic acid fragment. This distinction is only relevant in zones where the curvature C is significant. We have therefore introduced a cutoff below which inside/outside distinctions are ignored. The default is set to C = 0.4, but this can be modified by the user. In order to check the choice of volumes in which ion densities will be calculated, Canion now also includes an option to visualize the volumes (by generating a Gaussian cube density matrix that can be read into in a graphic program such as Chimera (35,36)). Supplementary Figure S1 shows examples of this output.
RESULTS AND DISCUSSION
Local curvature and register during MD simulations
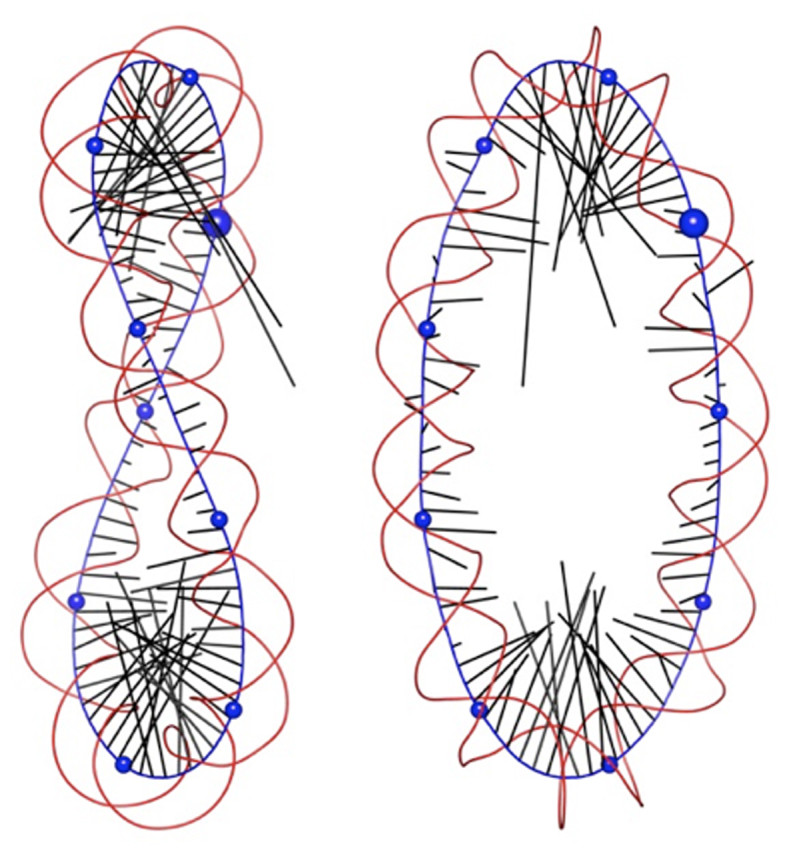
We begin by discussing the slightly overwound 94 bp minicircle (RE_94) and, in particular, the conformation of the average structure of this minicircle derived from the MD simulation. A Dreiding representation of the average structure is shown in Figure 1. The Curves+ analysis of this structure provides the helical axis and the curvature and register for each base pair step. This data is illustrated in Figure 2, where the radial vectors G (black) attached to the helical axis (blue), point in the direction of the local curvature and have lengths that are proportional to the magnitude of the curvature. The red curves represent the phosphodiester backbones. The data on curvature and register is provided in the line plots shown in Figure 3.
Figure 1.

Average structures of the RE_94 (top left), UN_94 (top right) and OV_89 (bottom) minicircles, shown in a wire representation, surrounded by transparent surface envelopes. Each minicircle is oriented with base pair 1 toward the top of the figure, with the numbering increasing in a clockwise sense. Key features are the strongly unwound segment in UN_94 C29-A33 (indicated by the arrow) and the out-of-plane writhing of the overwound OV_89.
Figure 2.

Two orthogonal views of data derived from a Curves+ analysis of the average conformation of the RE_94 minicircle, showing the helical axis (blue), phosphodiester backbones spline curves (red) and curvature vectors (black) for each base pair step indicating the direction and the magnitude of curvature. Blue spheres are placed on the helical axis every 10 bp, starting at base pair step 1 (larger sphere) and increasing in a clockwise sense.
Figure 3.

Curvature (top) and register (bottom) within the RE_94 minicircle. Black lines correspond to the analysis of the average MD structure and red lines to the time averages from the MD simulation.
Although this minicircle is somewhat overwound (σ = 0.055, see Table 1), the average helical axis of RE_94 is close to being a planar circle. However, looking closely we see that the curvature along the minicircle is not constant, reflecting the fact that it is easier to bend DNA toward the minor or major grooves than toward the backbones. This is visible in Figure 2, where shortest radial vectors (i.e. the lowest curvature) occur when a phosphodiester backbone is located inside the helical axis, whereas the longest vectors correspond to inward pointing grooves. The preference for bending toward the grooves of the double helix also explains why the direction of curvature oscillates above and below the plane of the minicircle (see the right-hand image in Figure 2), as the result of a balance between conserving the overall bending direction (the plane of the minicircle) and the ease of bending toward the grooves.
In Figure 3, the bending direction (lower plot) is described by the local register, namely the direction of@ curvature with respect to the local orientation of the DNA base pairs (where 90° and −90° correspond respectively to bending toward the minor and major grooves, while 0° and ±180° correspond to bending toward the backbones). For a planar minicircle, the right-handed rotation of the double helix every 10–11 bp, implies that register will decrease more or less uniformly by 360° over the same interval. This behavior can be seen in Figure 3, where register (lower plot) is kept in the range −180° → 180° and the plot is interrupted for visual reasons each time the register jumps from one extreme value to the other. In an idealized (uniformly twisting) case the nine segments of decreasing register (corresponding to nine helical turns within RE_94) would be diagonal lines with identical slopes proportional to the twist. The fluctuations visible in the figure reflect the oscillations in bending direction discussed above, modulated by specific base sequence effects.
So far, the discussion of the conformation of RE_94 has been based on its MD average structure, however we can also obtain information by time averaging the appropriate structural parameters from the MD time series. The results for curvature and register are shown in Figure 3 as red lines. We can see that, for register, the structure and time averages correspond very well (with a correlation coefficient of 0.997). For the curvature, although the two measures vary in a similar way (correlation coefficient 0.944), the time-averaged curvature is higher than that of the average structure. The difference between the two curves is also variable, being generally small for positions of high curvature and large for positions of low curvature. This is simply because low curvature regions generally do not have well-defined directions of curvature. Thus, averaging the magnitudes of curvature for all the snapshots will result in a larger value than that obtained from the average structure that was obtained by 3D superposition of helical axes that locally bend in different directions.
This behavior is illustrated by the data in Supplementary Figure S2 that shows register time series for steps with different degrees of curvature. (Note that the 1 ns sliding window time-averaging described in the ‘Materials and Methods’ section has been applied to these time series to smooth the data). The figure shows the results for three steps belonging to RE_94. Steps 5 and 11, which are associated with high curvature in the average structure (<C> = 1.43 and <C> = 1.13, corresponding to bending into the minor and major grooves respectively, black lines in the figure), indeed show little variation in register during the simulation. In contrast, step 2 is associated with low curvature (<C> = 0.41 and bending toward the phosphodiester backbone, red line) and shows significant changes in bending direction.
While discussing register, we have implicitly assumed that the minicircle is not rotating significantly around its helical axis during the simulation, since this would cause the register of all steps to vary with time. The results shown in Supplementary Figure S2 confirm that this assumption is justified for RE_94, but also show that rotational stability can only be tested reliably by looking at the register of steps with high curvature.
We now pass to the first of our modified minicircles, UN_94, that still has 94 bp, but that has been unwound by one helical turn (Lk = 8) leading to a superhelical density of −0.063. As seen for RE_94, Supplementary Figure S3 confirms that UN_94 is rotationally stable throughout the simulation. However, in contrast to RE_94, this minicircle shows considerable irregularity (see Figure 1;Supplementary Figures S4 and 5). The curvature, whether measured from the average structure, or by time-averaging from the MD trajectory, shows a dominant region of strong curvature between base pairs 26 and 35 (Supplementary Figure S5, upper plot). The data in Figure 4 shows that the unwinding has been strongly concentrated in very few steps, notably C29pT30, T31pA32 and A32pA33 (see also Figure 1). These three steps account for 134° (i.e. 37%) of the total unwinding between RE_94 and UN_94. We remark that all the strongly untwisted steps belong to the pyrimidine-purine or purine-purine families that we have previously shown to be capable of adopting low twist states (20) (although not nearly as extreme as the values seen in this constrained situation). This region roughly corresponds to what has been defined earlier as a DNA ‘wrinkle’ (17), although a strict alternation of normal and low twists does not apply here (the twists for the segment G28-A33 being 23.8°, −3.8°, 32.5°, −18.6° −6.5°, 33.9°). Since bending in the direction of roll (i.e. perpendicular to the long axis of the base pairs) is easier than through tilt, strong local unwinding helps to align the roll directions of successive base pair steps and explains the concentration of curvature that we observe in this region. The local conformational perturbation in minicircles of this size and this degree of negative supercoiling is in line with earlier simulations (17,37) and with experimental studies of their sensitivity to single stranded nucleases (38).
Figure 4.

Change of twist between RE_94 and UN_94. The unwinding is strongly concentrated at steps 29, 31 and 32 within the sequence fragment CGC29TT31A32AA. The total decrease in twist for these three steps is 134°.
We lastly consider the shortened minicircle OV_89. By removing 5 bp from this minicircle (without changing the linking number) we have created an extra half turn of overwinding. First, note that, in contrast to RE_94 and UN_94, roughly 100 ns are necessary for this minicircle to reach a stable rotational state (see Supplementary Figure S6). As shown in Figure 1, and in more detail in Figure 5, this minicircle is significantly writhed and is beginning to approach a figure of eight conformation (to better understand this 3D shape see Supplementary Movie S1). The axis of OV_89 however remains smooth and no kinks occur. The average curvature plot in Supplementary Figure S7 shows that this conformation leads to two strongly curved regions, each involving roughly three helical turns of DNA (covering the base pairs 65 → 89 + 1 → 2 and 19 → 50 respectively). Note that these two bends are centered on base pairs 34 and 78 and are thus almost exactly opposite one another within the 89 bp minicircle. This would be expected from mechanical studies of the buckling of a twisted ring (see for example, (39)), where the lowest energy modes involve the formation of an ellipse and then a figure of eight. Both these structures have two diametrically opposing regions of high curvature (in the case of OV_89, covering roughly 1.5 helical turns) that allow the bending to be strongly reduced in the remainder of the ring. Warped, approximately elliptical conformations have also been observed using stereo cryo-EM studies of small minicircles (40,41). For OV_89, this inhomogeneity of curvature leads to values covering a range of 3.63 (0.21 ≤ C ≤ 3.84), in striking contrast to the range of 1.09 observed for RE_94 (measured from the corresponding average structures).
Figure 5.
Two orthogonal views of data derived from a Curves+ analysis of the average conformation of the OV_89 minicircle, showing the helical axis (blue), phosphodiester backbones spline curves (red) and curvature vectors (black) for each base pair step indicating the direction and the magnitude of curvature. Blue spheres are placed on the helical axis every 10 bp, starting at base pair step 1 (larger sphere) and increasing in a clockwise sense.
Ion distributions around minicircles
The measurement of curvature in terms of magnitude and direction enables us to add a new dimension to the analysis of ion distributions around DNA. In particular, we can now define the ‘inside’ and ‘outside’ of a minicircle based on the local register, as described in the ‘Materials and Methods’ section. We have performed this analysis for our three minicircles, dividing the space around the double helix in three ways. The first two ways have already been used in our earlier work: (i) within the grooves, R ≤ 10.25 Å (where 10.25 Å is the average distance of the backbone phosphorus atoms from the helical axis) hereafter denoted ‘<P’, or beyond the grooves 10.25 < R ≤ 18 Å, denoted ‘>P’; (ii) within the angular range of the minor groove (33° < A ≤ 147°, denoted ‘Min’) or within that of the major groove (A ≤ 33° or A > 147°, denoted ‘Maj’). We now add the distinction ‘IN’ and ‘OUT’ based respectively on the regions within, or beyond, ±90° of the local direction of curvature (see the ‘Materials and Methods’ section for details). The total potassium cation populations in each region for each of the three minicircles are listed in Supplementary Table S1 and the corresponding volumes of the regions analyzed (excluding the volume occupied by the atoms of the DNA minicircles) are given in Supplementary Table S2. In each case, we have started analyzing the trajectories only after the first 100 ns to allow for equilibration of the ion distribution and to allow for the slow rotational relaxation of OV_89. Although we will discuss some values of both populations and volumes, this data can be usefully condensed into the average molarities that are shown in in Table 2. We remark that the bulk molarity of the potassium cations (allowing for the extra ions added to achieve charge neutrality) lies between 0.34 M and 0.37 M for the three minicircle simulations.
Table 2. Potassium ion molarities inside (IN) and outside (OUT) the minicircles, showing values within the grooves (<P, R ≤ 10.25 Å) and beyond the grooves (>P, 10.25 Å < R ≤ 18 Å) for the minor groove (Min), the major groove (Maj) or for both combined (All).
| Minicircle | All/IN | All/OUT | Min/IN | Min/OUT | Maj/IN | Maj/OUT |
|---|---|---|---|---|---|---|
| RE_94 < P | 1.70 | 0.90 | 2.17 | 0.98 | 1.48 | 0.80 |
| RE_94 > P | 1.04 | 0.78 | 1.21 | 0.93 | 0.96 | 0.72 |
| UN_94 < P | 1.29 | 0.99 | 1.71 | 0.99 | 1.12 | 0.99 |
| UN_94 > P | 1.03 | 0.78 | 1.22 | 0.88 | 0.94 | 0.74 |
| OV_89 < P | 1.39 | 0.56 | 1.97 | 0.78 | 1.14 | 0.47 |
| OV_89 > P | 0.98 | 0.44 | 1.10 | 0.53 | 0.93 | 0.40 |
We begin by again considering the almost planar and circular case RE_94. The most striking effects of minicircle curvature are seen within the DNA grooves (<P). As shown in Supplementary Table S1, there are 45% more ions inside (IN) than outside (OUT) (27.4 versus 18.9), although, due to curvature, the total IN groove volume is 23% less than the OUT volume (obviously the two volumes would be identical in a straight DNA segment). This implies an even bigger difference in molarity, the IN grooves average being 89% higher than that of the OUT grooves (Table 2). The concentration of ions inside the minicircle clearly reflects the closer inter-phosphate distances on the inside of the minicircle. The Curves+ groove analysis shows that, on average, curvature makes the minor groove inside the minicircle 3 Å narrower than that outside, and the major groove width 4 Å narrower (see Figure 6).
Figure 6.

Minor groove widths (lower curve) and major groove widths (upper curve) along the RE_94 minicircle. Missing data in the upper curve correspond to very wide major grooves where no width measurement can be obtained (see (10)).
If we look beyond the grooves (>P), the IN molarities again dominate, being 33% higher, despite the IN volume being 31% smaller in that region (see Supplementary Table S2). Returning to distributions within the grooves (R ≤ 10.25 Å), we can also see that the most important increase in ion population due to curvature concerns the minor groove. This is reflected in the IN molarity that is 2.2 times higher than the OUT molarity for the minor groove, while for the major groove the ratio is only 1.7 (Table 2). This can be explained by noting that the Debye length is roughly 8 Å at the present ionic strength. As shown in Figure 6, this implies that electrostatic damping will reduce the superposition of negative phosphate potentials across the major groove significantly more than those across the minor groove (although one must be cautious in applying a bulk property such as the Debye length at the atomic scale). It should be pointed out that there are nevertheless more ions in the IN major groove (16.4 versus 10.9 in the minor groove), reflecting the fact that its volume is 2.2 times larger than that of the IN minor groove (Supplementary Table S2).
Another way to understand the effect of curvature on the ion distribution is provided by 3D plots of ion density, as shown for RE_94 in Figure 7. Here, the colored volumes are 2 M isomolarity surfaces for K+ ions. Those colored blue correspond to ions classed IN, while those colored red are classed OUT. This image clearly shows how the ion distribution has adjusted to compensate for the increased phosphate charge density inside the minicircle, while still strongly accumulating within the major and minor grooves (as also seen earlier in the analysis of linear DNA fragments (13,14)).
Figure 7.

Isomolarity surfaces at 2 M for potassium ions surrounding the RE_94 minicircle. Blue: ion density inside. Red: ion density outside. For clarity, the minicircle is represented only by its helical axis and its backbone spline curves (black lines). Black spheres are placed on the on the helical axis every 10 bp, starting at base pair step 1 (larger sphere, top) and increasing in a clockwise sense; note that some of these spheres are hidden by the isomolarity surfaces.
For the strongly underwound UN_94 minicircle, the results in Table 2 show that the impact of curvature is apparently smaller than in RE_94 since the IN and OUT ion molarities within the grooves are now 1.29 M versus 0.99 M (an increase of only 30%). As shown by the isomolarity plot in Figure 8, this result is partially explained by the localized unwinding that occurs between base pairs 29 and 33, where irregularities in the local curvature perturb the regions classed as inside and outside. The same effect occurs near points of low curvature around positions 7 and 53. This case illustrates the importance of viewing the axis curvature in 3D before drawing conclusions based on values averaged over the full minicircle.
Figure 8.

Isomolarity surfaces at 2 M for potassium ions surrounding the UN_94 (top) and OV_89 (bottom). Blue: ion density inside. Red: ion density outside. For clarity, the minicircles are represented only by their helical axis and backbone spline curves (black lines). Base pair number 1 is at the top of each image and numbering increases in a clockwise sense.
For the overwound OV_89 case, as discussed above, strong curvature is concentrated in two opposing regions, separated by almost straight arms. Supplementary Table S1 shows that this minicircle has the highest proportion of IN versus OUT ions within the grooves (a 77% increase) and, a correspondingly higher molarity (1.39 M IN versus 0.56 M OUT, an increase of 148%). This result is mainly due to the strongly curved segments as strikingly shown by the isomolarity plot in Figure 8 (qualitatively similar results have been observed in other minicircle simulations (17,42)). Within these segments (defined as in the previous section) the ion population inside dominates that outside by 259% and 97% for the bends centered on base pairs 34 and 78 respectively.
Considering all three minicircles together, we can summarize the impact of curvature on the ionic environment by noting that potassium ion molarities are always higher inside the minicircle than outside, whether we consider the regions within the grooves (R ≤ 10.25 Å) or beyond the grooves (10.25 < R ≤ 18 Å). Within the grooves, it is also always the case that curvature modulates the minor groove molarities more than those of the major groove.
CONCLUSIONS
We have developed a new method for quantifying the magnitude and the direction of curvature of nucleic acids, based on the curvilinear helical axis provided by Curves+. Application of this method to analyzing the MD trajectories of three DNA minicircles provides information that was not previously accessible. First, it is shown that apparently smooth planar curvature conceals underlying local variations of curvature that are caused by the mechanical properties of individual base pair steps and a strong preference for bending in the direction of the major and minor grooves. These effects cause the direction of bending to oscillate above and below an apparent plane of curvature. Second, by studying the time dependence of the local direction of curvature ‘register’ (but only for significantly curved regions), it is possible to see whether a minicircle rotates around its own helical axis or, if not, which stable orientation it adopts and how quickly this orientation is achieved. Third, in the case of under- or overwound minicircles, it is possible to analyze heterogeneous curvature and to determine how this is coupled to changes in other structural parameters. Last, it is possible to analyze the links between curvature and the environment of DNA, notably the significant impact curvature has on the distribution of cations around the double helix. In the cases studied here, curvature concentrates potassium cations toward the interior of the minicircle, this effect being strongest in the minor groove.
This method is now a part of the freely available Curves+ software (and the accompanying utilities Canal and Canion, aimed at analyzing MD trajectories) that can be found at http://curvesplus.bsc.es/.
Although we have concentrated here on applying our new technique to circularized DNA in this work, the method is equally applicable to linear DNA and it should be of interest for analyzing the more subtle curvatures induced in DNA either by its own base sequence, or by external factors. In the latter case, it should hopefully contribute to a better understanding of the impact of deformation of the double helix on its solvent/salt environment and on the formation of complexes with proteins or small ligands.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to acknowledge GENCI for a generous allocation of computer time on the CINES supercomputer OCCIGEN.
Footnotes
Present address: Marco Pasi, Centre for Biomolecular Sciences and School of Pharmacy, University of Nottingham, Nottingham NG7 2RD, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Sidaction [A125-1-02333]; ANR Project CHROME [ANR-12-BSV5-0017-01]; Swiss National Science Foundation (including provision for computational resources) [200020_163324 to J.H.M., in part]. Funding for open access charge: Sidaction [A125-1-02333].
Conflict of interest statement. None declared.
REFERENCES
- 1. Goodsell D.S., Dickerson R.E.. Bending and curvature calculations in B-DNA. Nucleic Acids Res. 1994; 22:5497–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu X.J., Olson W.K.. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008; 3:1213–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dickerson R.E., Goodsell D.S., Neidle S.. “...the tyranny of the lattice...”. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:3579–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bolshoy A., McNamara P., Harrington R.E., Trifonov E.N.. Curved DNA without A-A: experimental estimation of all 16 DNA wedge angles. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:2312–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crothers D.M. DNA curvature and deformation in protein-DNA complexes: a step in the right direction. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:15163–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barbic A., Zimmer D.P., Crothers D.M.. Structural origins of adenine-tract bending. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:2369–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhurkin V.B., Tolstorukov M.Y., Fei X., Colasanti A.V., Olson W.K.. Ohyama T. Sequence-dependent variability of B-DNA: an update on bending and curvature. DNA Conformation and Transcription. 2005; Springer; 18–34. [Google Scholar]
- 8. Lavery R., Sklenar H.. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J. Biomol. Struct. Dyn. 1988; 6:63–91. [DOI] [PubMed] [Google Scholar]
- 9. Lavery R., Sklenar H.. Defining the structure of irregular nucleic acids: conventions and principles. J. Biomol. Struct. Dyn. 1989; 6:655–667. [DOI] [PubMed] [Google Scholar]
- 10. Lavery R., Moakher M., Maddocks J.H., Petkeviciute D., Zakrzewska K.. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009; 37:5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olson W.K., Bansal M., Burley S.K., Dickerson R.E., Gerstein M., Harvey S.C., Heinemann U., Lu X.J., Neidle S. et al. . A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 2001; 313:229–237. [DOI] [PubMed] [Google Scholar]
- 12. Sanghani S.R., Zakrzewska K., Harvey S.C., Lavery R.. Molecular modelling of (A4T4NN)n and (T4A4NN)n: sequence elements responsible for curvature. Nucleic Acids Res. 1996; 24:1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lavery R., Maddocks J.H., Pasi M., Zakrzewska K.. Analyzing ion distributions around DNA. Nucleic Acids Res. 2014; 42:8138–8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pasi M., Maddocks J.H., Lavery R.. Analyzing ion distributions around DNA: sequence-dependence of potassium ion distributions from microsecond molecular dynamics. Nucleic Acids Res. 2015; 43:2413–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lankas F., Lavery R., Maddocks J.H.. Kinking occurs during molecular dynamics simulations of small DNA minicircles. Structure. 2006; 14:1527–1534. [DOI] [PubMed] [Google Scholar]
- 16. Harris S.A., Laughton C.A., Liverpool T.B.. Mapping the phase diagram of the writhe of DNA nanocircles using atomistic molecular dynamics simulations. Nucleic Acids Res. 2008; 36:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mitchell J.S., Laughton C.A., Harris S.A.. Atomistic simulations reveal bubbles, kinks and wrinkles in supercoiled DNA. Nucleic Acids Res. 2011; 39:3928–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sutthibutpong T., Harris S.A., Noy A.. Comparison of molecular contours for measuring writhe in atomistic supercoiled DNA. J. Chem. Theory Comput. 2015; 11:2768–2775. [DOI] [PubMed] [Google Scholar]
- 19. Cloutier T.E., Widom J.. Spontaneous sharp bending of double-stranded DNA. Mol. Cell. 2004; 14:355–362. [DOI] [PubMed] [Google Scholar]
- 20. Pasi M., Maddocks J.H., Beveridge D., Bishop T.C., Case D.A., Cheatham T., Dans P.D., Jayaram B., Lankas F. et al. . μABC: a systematic microsecond molecular dynamics study of tetranucleotide sequence effects in B-DNA. Nucleic Acids Res. 2014; 42:12272–12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lavery R., Zakrzewska K., Sklenar H.. JUMNA (Junction Minimization of Nucleic-Acids). Comput. Phys. Commun. 1995; 91:135–158. [Google Scholar]
- 22. Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R., Merz K.M., Onufriev A., Simmerling C., Wang B., Woods R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheatham T.E.3., Cieplak P., Kollman P.A.. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999; 16:845–862. [DOI] [PubMed] [Google Scholar]
- 24. Pérez A., Marchán I., Svozil D., Sponer J., Cheatham T.E., Laughton C.A., Orozco M.. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pearlman D.A., Case D.A., Caldwell J.W., Ross W.S., Cheatham T.E., DeBolt S., Ferguson D., Seibel G., Kollman P.. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995; 91:1–41. [Google Scholar]
- 26. Berendsen H.J.C., Grigera J.R., Straatsma T.P.. The missing term in effective pair potentials. J. Phys. Chem. 1987; 91:6269–6271. [Google Scholar]
- 27. Dang L.X. Mechanism and thermodynamics of ion selectivity in aqueous-solutions of 18-crown-6 ether—a molecular dynamics study. J. Am. Chem. Soc. 1995; 117:6954–6960. [Google Scholar]
- 28. Beveridge D.L., Barreiro G., Byun K.S., Case D.A., Cheatham T.E. III, Dixit S.B., Giudice E., Lankas F., Lavery R. et al. . Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. I. Research design and results on d(CpG) steps. Biophys. J. 2004; 87:3799–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dixit S.B., Beveridge D.L., Case D.A., Cheatham T.E. III, Giudice E., Lankas F., Lavery R., Maddocks J.H., Osman R. et al. . Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophys. J. 2005; 89:3721–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G.. A smooth particle mesh Ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 31. Ryckaert J.P., Ciccotti G., Berendsen H.J.C.. Numerical-integration of cartesian equations of motion of a system with constraints—molecular-dynamics of N-alkanes. J. Comput. Phys. 1977; 23:327–341. [Google Scholar]
- 32. Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R.. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984; 81:3684–3690. [Google Scholar]
- 33. Harvey S.C., Tan R.K.Z., Cheatham T.E. III. The flying ice cube: velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 1998; 19:726–740. [Google Scholar]
- 34. Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J.. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002; 319:1097–1113. [DOI] [PubMed] [Google Scholar]
- 35. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 36. Goddard T.D., Huang C.C., Ferrin T.E.. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007; 157:281–287. [DOI] [PubMed] [Google Scholar]
- 37. Sutthibutpong T., Matek C., Benham C., Slade G.G., Noy A., Laughton C., K Doye J.P., Louis A.A., Harris S.A.. Long-range correlations in the mechanics of small DNA circles under topological stress revealed by multi-scale simulation. Nucleic Acids Res. 2016; 44:9121–9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Du Q., Kotlyar A., Vologodskii A.. Kinking the double helix by bending deformation. Nucleic Acids Res. 2008; 36:1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Benham C.J. Onset of writhing in circular elastic polymers. Phys. Rev. A Gen. Phys. 1989; 39:2582–2586. [DOI] [PubMed] [Google Scholar]
- 40. Amzallag A., Vaillant C., Jacob M., Unser M., Bednar J., Kahn J.D., Dubochet J., Stasiak A., Maddocks J.H.. 3D reconstruction and comparison of shapes of DNA minicircles observed by cryo-electron microscopy. Nucleic Acids Res. 2006; 34:e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Demurtas D., Amzallag A., Rawdon E.J., Maddocks J.H., Dubochet J., Stasiak A.. Bending modes of DNA directly addressed by cryo-electron microscopy of DNA minicircles. Nucleic Acids Res. 2009; 37:2882–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noy A., Sutthibutpong T., A Harris S.. Protein/DNA interactions in complex DNA topologies: expect the unexpected. Biophys. Rev. 2016; 8:233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.