


Abstract
Specific nucleoprotein complexes are formed strictly to prevent over-initiation of DNA replication. An example of those is the so-called handcuff complex, in which two plasmid molecules are coupled together with plasmid-encoded replication initiation protein (Rep). In this work, we elucidate the mechanism of the handcuff complex disruption. In vitro tests, including dissociation progress analysis, demonstrate that the dimeric variants of plasmid RK2 replication initiation protein TrfA are involved in assembling the plasmid handcuff complex which, as we found, reveals high stability. Particular proteases, namely Lon and ClpAP, disrupt the handcuff by degrading TrfA, thus affecting plasmid stability. Moreover, our data demonstrate that TrfA monomers are able to dissociate handcuffed plasmid molecules. Those monomers displace TrfA molecules, which are involved in handcuff formation, and through interaction with the uncoupled plasmid replication origins they re-initiate DNA synthesis. We discuss the relevance of both Rep monomers and host proteases for plasmid maintenance under vegetative and stress conditions.
INTRODUCTION
Pairing of two DNA molecules or specific sites on a DNA molecule has been demonstrated to play a regulatory role in various processes including replication, transcription, recombination or conjugation. Disassembling of paired DNA molecules is critical for the release of inhibited activity. Handcuff complex pairs two plasmid origins in a manner which causes steric hindrance to their function. This regulatory mechanism involves specific nucleoprotein complex formed on direct repeats (iterons) located within the plasmid replication origin. It controls the initiation of DNA replication and therefore the copy number and stable maintenance of plasmid. Handcuffing prevents a new round of replication initiation by inhibition of origin melting, which is important for plasmid copy number control. Handcuff prevents metabolic overburdening of the cell by maintaining plasmid copies at a minimum level. Although the importance of this nucleoprotein complex has been identified, the structure and the mechanisms involved in assembling and resolving of the handcuffed plasmids are not fully elucidated.
Plasmid Rep proteins exist in a monomer-dimer equilibrium, which determines the efficiency of initiator protein in replication (1–3). Both forms, monomers and dimers, of the Rep protein are functional but their function is different. Monomers bind to the iterons at the origin, promoting replication initiation (4–7). Dimers, on the other hand, in some plasmid replicons can indirectly affect the frequency of replication initiation, via an auto-repression process, through the interaction with the inverted repeats, located within the sequence of rep promoter (5,8–10). The role of Rep dimers in mediating the origin pairing was proposed for the π replication protein of the R6K plasmid (11,12). It had been shown that the π dimers have a greater affinity to participate in handcuffing than π monomers, albeit the contribution of Rep monomers was not excluded. A model for Rep-mediated handcuffing indicating a direct interaction between two arrays of Rep monomers bound to iterons on two plasmid molecules was also suggested and such mechanism was proposed for the RepA protein from the pPS10 plasmid (13,14). It was also proposed that dimers of the Rep protein can bridge two monomers which bind iteron arrays. Such a model was conceived for handcuffing in F and RK2 plasmids (15,16). Although all of the above mentioned handcuffing models are different, they all assume the control of the plasmid replication re-initiation for stable retention of plasmid in host cells and maintaining a fixed plasmid copy number. The stability of handcuff complex has not been investigated to date, however, it is anticipated that the handcuff must be somehow resolved when the concentration of plasmid molecule decreases in bacterial cell (3).
RK2 is a 60-kb broad-host-range IncP1 plasmid that is stably maintained in a wide range of Gram-negative bacteria. Its replication and maintenance at defined copy number of four to seven copies per chromosome in Escherichia coli (17,18) depends on a specific origin of replication, oriV and a plasmid-encoded trans-acting replication initiator, TrfA protein. Both elements are sufficient for the initiation of RK2 plasmid replication and they regulate the frequency of plasmid RK2 replication initiation by creating handcuff complexes (16,19). Indeed, the handcuffing-defective initiator mutants were found to have abnormally high plasmid copy numbers (16). TrfA protein exists in the cell mainly as a dimer (20,21), but the active form of the protein are monomers, which can bind to the iterons at the RK2 origin and contribute to the initiation of DNA replication (6,21). Dimers of TrfA protein are activated to the monomeric form by chaperone proteins (22–24). The level of the active TrfA protein form is also regulated by the action of the cellular proteases. The ClpXP and ClpYQ degrade only the dimeric form of TrfA, while for the Lon and ClpAP proteases the oligomeric state of TrfA protein is not crucial (23,25). The TrfA proteolysis by ClpAP and Lon is stimulated by DNA (25).
According to the structural prediction (23), the C-terminal, major part of TrfA protein (residues 195–382), has two Winged–Helix (WH) domains, a feature typical of the known DNA-binding proteins, involved in plasmid replication initiation (26–31), as well as of the Archaeal and Eukaryotic replication initiators. Specific mutations within this region, which affect the stability of TrfA dimer, were described. Substitutions of amino acids: G254D, S267L and G254D/S267L result in predominantly monomeric form of the protein (16), while the S257F mutation, within the dimerization interface, causes stabilization of the TrfA dimers (16,23). Monomeric TrfA variants support mini-RK2 maintenance in E. coli at a copy number much higher than normal (16,32). The TrfA initiator is a protein with multiple functions. Its main activity is the formation of initial complex by binding to the five iteron sequences (6), which generates a localized strand destabilization of DNA unwinding element (DUE). It was shown (33), that TrfA can form a nucleoprotein complex with single-stranded DNA at the DUE, and that the complex is specific to the one of the two single strands. TrfA is also essential for the formation of a fully active replisome at the replication origin of the RK2 plasmid (34). All of the above mentioned TrfA functions provide positive regulation of the RK2 plasmid replication, but TrfA protein, through the ability to handcuff two plasmid molecules, takes part also in negative regulation by preventing RK2 plasmid replication initiation. To date, it is not clear whether it's the dimers or monomers that are involved in the RK2 handcuff complex formation. The available data indicates that the wild-type TrfA protein participates in the handcuff structure formation and shows that the monomeric TrfA form is defective in RK2 plasmid handcuffing. However, the involvement of both TrfA dimers and monomers in the handcuff structure has also been postulated (16,19,35). The lack of auto-repression in the RK2 plasmid seems to make the RK2 handcuffing a crucial mechanism which controls the plasmid copy number. Since RK2 has no other negative mechanisms of regulation of replication initiation, it is a good research model to explore handcuffing. In this work, we investigate the stability of the handcuff complex and the mechanisms for disassembling (uncuffing) the handcuffed plasmid molecules. We found that both proteases, through degradation of Rep dimers involved in handcuff complex, and Rep monomers by displacing those Rep dimers, are able to disrupt the handcuff. Our results describe new regulatory factors which trigger the re-initiation of DNA replication.
MATERIALS AND METHODS
Bacterial strains, plasmids and oligonucleotides
Bacterial strains and plasmids used in this work are listed in Supplementary Table S1. Escherichia coli cells were transformed with plasmids by standard procedure (36). Oligonucleotides used in this work are listed in Supplementary Table S2. Oligonucleotides used for amplification of 460 bp DNA fragment that contains oriV sequence were: IT1 and IT2. Oligonucleotides used for amplification of 465 bp no-iteron DNA fragment: NIT1 and NIT2. Biotinylated DNA fragments used in the analysis of handcuff formation and dissociation in experiments utilizing NeutrAvidin™ coated plates (assay described below) were obtained by hybridization of two complementary single-stranded oligonucleotides (Thermo Scientific). 5΄-end biotinylated double stranded DNA fragment, containing RK2 iterons sequence was ITB1. 5΄-end biotinylated double stranded no-iteron DNA fragment was NITB1. Oligonucleotides used for the RK2 copy number determination were: trfA1 and trfA2 for amplification of 268 bp fragment of trfA gene of pTJS42 plasmid. The clpP-1 and clpP-2 oligonucleotides were used for clpP gene amplification, which was used for pClpP plasmid construction (Supplementary Data).
Protein purification
In the experiments presented in this study highly purified proteins (95% purity or higher) were utilized. The TrfA preparations were the N-terminally histidine-tagged 33 kDa version of the protein. Purification of the His-tagged TrfA variants (including TrfA wt, TrfA G254D/S267L and TrfA S257F) was performed essentially as described previously (6). Published protocols were applied to the purification of ClpA (37), ClpP (38), ClpX (23) and Lon (39) proteins. The proteolytic activity of Clp and Lon proteases was measured using the previously described method (38), with α-casein as a substrate for ClpAP and Lon, and λO protein for ClpXP. The unit of the activity was defined as the degradation of 1 μg of substrate protein per hour.
In vitro NeutrAvidin™ coated plate assay (NA assay)
The 384-well black plates coated with NeutrAvidin™ (Pierce, Thermo Scientific) were used to observe the formation and disruption of handcuff complexes. The 500 nM solution of 120 bp biotin-labeled dsDNA which contains iterons was incubated for 1 h at 32°C in the NeutrAvidin™-coated plate, which immobilized biotinylated DNA with NeutrAvidin™. Addition of 40 nM solution of 460 bp AlexaFluor 555-labeled dsDNA-oriV and 570 nM solutions of the TrfA protein variants (TrfA wt, TrfA G254D/S267L or TrfA S257F), and incubation for an additional hour at 32°C, were performed for the handcuff complexes assembly. Incubations were conducted in 50 μl LAB buffer (40 mM Tris–HCl pH 7.8, 40 mM potassium glutamate, 10 mM MgCl2, 1 mM CaCl2, 10 mM DTT, 10 μg/ml BSA, 5 mM ATP). The unbound DNA and TrfA protein were removed by three washes with 100 μl of LAB buffer. The reaction was stopped by addition of 50 μl of 2% SDS solution. Handcuff complex formation was determined by measuring the fluorescence signal in Beckman Coulter DXT 880 Multimode Detector. The stability of RK2 handcuff complex was tested in continuous buffer rinsing (LAB buffer at 1 ml/min flow). The fluorescence signals of particular samples were analyzed after 0, 5, 15, 30, 45 and 60 min of buffer rinsing. To test the disruption of handcuff complexes, selected chaperones, proteases or monomeric TrfA, were added to the reaction mixture after the handcuff complex assembly.
Ligase assay
The modified ligase assay (40) was used to examine the handcuffing of DNA fragments, containing RK2 origin, by TrfA and for the analysis of disruption of the created handcuff complexes. A 489 bp PvuII-fragments from the pBK20 plasmid were used in the experiments. The ligation reactions were performed in LAB buffer. Handcuff complexes were formed by incubation of 7 pM of DNA fragments with 650 nM of TrfA protein for 30 min at 32°C. Subsequently, 5 units of T4 DNA ligase (Fermentas) were added, and samples were incubated at 22°C for 1.5 h. The reactions were stopped by ethanol precipitation of DNA and the precipitates were then resuspended in 30 μl of TE buffer. Fifteen microliters of each sample was electrophoresed on 1% agarose gel in 1× TBE for 1.5 h at 10 V/cm in the presence of ethidium bromide. The presence of multimeric plasmid DNA indicated formation of handcuff complex. Gels were quantified by densitometry using ImageLab™ Software (BioRad). Percentage of DNA multimers (all bands above free DNA) was estimated for each lane as the intensity of multimers compared with the total intensity of DNA. To analyze the disruption of handcuff complexes, 250 nM solutions of chaperones, proteases or TrfA G254D/S267L were added to the reaction mixture after the handcuff complex preformation, and were further incubated for 30 min at 32°C. Next, ligation, precipitation and electrophoresis of the samples were performed. For the analysis of the handcuff disruption, the ligated plasmid DNA multimers, before and after incubations with chaperones, proteases or TrfA G254D/S267L, were analyzed.
Transformation frequency
pTJS42 and pUC19 plasmids were purified by means of two CsCl buoyant density gradient centrifugations and 100 ng of the obtained DNA was used for the E. coli cells’ transformation according to standard Bio-Rad electroporation protocol. The cells were prepared for electroporation as described previously (41,42). Electroporations were performed in 2 mm cuvettes (Bio-Rad) and the parameters were 3000 V, 25 mF, 200 V for all E. coli strains. The transformation frequency was calculated as colony-forming unit (CFU) per μg of plasmid DNA.
In vivo plasmid stability assays
Cultures of E. coli C600 cells and derivatives containing insertion deactivations in lon and clp gens, carrying pTJS42 plasmid (Supplementary Table S1), were diluted from overnight cultures into R medium (20 g/l trypton, 10 g/l yeast extract, 5 g/l NaCl, 0.2% glycerol, 0.05 M KPi pH 7.2) containing appropriate antibiotics and incubated at 37°C to OD600 0.5. Cultures were then diluted into medium without antibiotics, to allow growth of mini-RK2-free segregants, and maintained in exponential growth for 120 generations by sequential dilutions. The samples collected during the experiment were plated on agar plates without antibiotics and, from each plating, ≥400 colonies were patched onto agar with and without antibiotics to score retention or loss of plasmids. Plasmids pBADLon or pClpP were used for supplementation of E. coli lon and clp mutants (Supplementary Table S1). Stability assays were performed in R medium, supplemented with tetracycline and ampicillin, for pBADLon, or kanamycin, for pClpP plasmids, at 37°C to OD600 0.5. To induce lon or clpP expression, the medium was supplemented with 0.02% arabinose or 0.1 mM IPTG, respectively. The percentage of plasmid loss per generation was calculated using the formula  , as described previously (43), where n is the number of generations elapsed, Fi is the fraction of cells containing plasmid at the initial time point and Ft is the fraction of cells containing plasmid at the final time point.
, as described previously (43), where n is the number of generations elapsed, Fi is the fraction of cells containing plasmid at the initial time point and Ft is the fraction of cells containing plasmid at the final time point.
Plasmids foci number determination
To obtain the images of plasmid RK2 mini-derivatives foci, modified fluorescent repressor-operator system (FROS) method was used (44). A mini-RK2 pCVI replicon containing an array of tetO repeats was visualized in E. coli cells which express a TetR-EYFP fusion protein. E. coli strains were grown overnight at 37°C, diluted 1:100 in fresh medium and grown to OD600 0.3. The expression of TetR-EYFP from pLAK53 plasmid was induced for 60 min with 0.4% l-arabinose. The expression of lon or clpP in protease-deficient strains was induced from pBADLon or pClpP plasmids. To induce the E. coli C600 cells filamentation, cephalexin (10 μg/ml) was added to the culture, 1–2 h prior to microscopy. Samples were collected every 20 min and prepared for microscopic observation. Cells were stained with FM4-64 dye (N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino) phenyl) hexatrienyl) pyridinium dibromide) (0.5 mg/ml), to visualize cell membrane, immobilized on poly-l-lysine-treated microscope coverslips and observed using an OLYMPUS BX51 fluorescence microscope. Microscope images were obtained using an F-View-II CCD camera. Measurements and image analysis were conducted with AnalySIS software. During each experiment minimum 200 cells were measured. All measured cells contained plasmid foci. The obtained result was the average result for specific bacterial population. The average number of plasmid foci was calculated for the whole cell or for 1 μm3 of the bacterial cell. The bacterial volume was calculated using the formula  , where
, where  is the radius of the cell, calculated as a
is the radius of the cell, calculated as a  of the cell diameter, and h is the average length of the plasmid containing bacterial cell.
of the cell diameter, and h is the average length of the plasmid containing bacterial cell.
Plasmid copy number determination
Plasmid copy number in E. coli C600 and its derivatives was determined by quantitative PCR (qPCR). Cells were grown at 37°C in LB medium, counted using a Petroff–Hauser counting chamber and then the amount of culture equivalent to 120 cells was harvested and total DNA was extracted (45). A 0.5 μl sample out of 100 μl of isolated total DNA solution was used as a template for quantitative PCR. PCR was performed in the LightCycler 2.0 Carousel-based System (Roche) with oligonucleotide primers trf1 and trf2, using the LightCycler FastStart DNA MasterSYBR Green I Kit (Roche) according to the manufacturer's specifications. During 45 quantification cycles, primer annealing was set to 58°C for 7 s, followed by 15 s extension. Amplification products were 268 bp for the trfA gene fragment from RK2 plasmid. Cycle threshold (Ct) values were determined after automatic adjustment of the baseline and fluorescence threshold using Light Cycle Software version 4.05 (Roche). External standard curve was obtained by amplifying serial dilution (from 1 × 106 to 1 × 102 copies/μl) of plasmid pTJS42 as a template. Amplification efficiencies were 92%. R2 values for the standard curve were 0.9945. Plasmid copy numbers (PCNs) per cell were calculated using the following equation:
 |
where the copy number of trfA sequence was calculated from the standard curve. PCN was calculated for DNA samples isolated from three independent bacterial cultures, and for each sample three independent reactions were prepared.
In vitro replication assay (FII)
In vitro replication was performed using an E. coli C600 and its protease-deficient strains crude extracts, active for oriV replication. The extract was prepared with the use of ammonium sulfate fractionation (0.28 g per 1 ml of cell lysate) as described previously (35). Reaction mixtures contained 250 ng supercoiled pTJS42 DNA; 40 mM Hepes⋅KOH (pH 8.0); 11 mM magnesium acetate; 2 mM ATP; 500 μM (each) GTP, CTP, and UTP; 50 μM (each) dNTPs; 150 cpm/pmol [methyl-3H]-dTTP; 80 mM creatine phosphate; 5% (wt/vol) PEG; 50 mM KCl; 16 μg/ml creatine kinase; 80 μg/ml BSA, and 500 ng of wild-type TrfA. Mixtures were assembled on ice, followed by incubation at 32°C for 1.5 h, which was stopped by addition of 0.1 M sodium pyrophosphate and 10% (wt/vol) TCA. Samples were filtrated onto Whatman GF/C glass microfiber filters, and total nucleotide incorporation (pM) was measured by liquid scintillation counting.
RESULTS
TrfA dimers are involved in assembling RK2 handcuff complex
Since different models, postulating Rep monomers or dimers contribution to the handcuff complex formation, were proposed (12,13,15,16,46,47), we tested if RK2 handcuff complex is formed by TrfA monomers or dimers. We performed the NA (Figure 1A) and ligase assays (Figure 1C) with the use of three TrfA protein variants: the wild-type TrfA, monomeric variant TrfA G254D/S267L and TrfA S257F (16,23). Wild-type TrfA is purified mainly as a dimer (6,23), TrfA G254D/S267L (16,19,23) is a monomeric variant which is a ‘hyperactive’ form of TrfA, causing run-away replication effect in replication assays (16,19) and TrfA S257F is isolated as a dimer and was described as constitutively dimeric (16,23). It has been previously shown that all analyzed TrfA variants, TrfA wt, TrfA G254D/S267L and TrfA S257F, bind iteron-containing DNA in vitro (16,48), however the binding of TrfA S257F is significantly weaker (23). The elevated fluorescence signal in the NA assay and high level of DNA multimers in ligase assay indicated handcuff formation by wild-type TrfA and TrfA S257F, in both types of experiments (Figure 1B and D, lanes 1 and 2). In contrast, the activity of monomeric protein TrfA G254D/S267L was barely detectable in coupling of the iteron-containing DNA (Figure 1B and D, lanes 3). We observed that the fluorescence signal in the NA assay and the level of DNA multimers in ligase assay were comparable to those recorded for negative control, in which no protein was used (Figure 1B and D, lanes 4). The obtained results indicate that the handcuff complexes of the RK2 plasmid are formed through the activity of dimers of the TrfA protein and TrfA monomers alone are not able to handcuff plasmid molecules.
Figure 1.

RK2 plasmid handcuff complex is formed by wild-type TrfA. Schematic representation of the experimental procedure is presented in panel (A) NA assay, and (C) ligation assay. The formation of handcuff complexes was analyzed using the following proteins: wild-type TrfA, monomeric form TrfA G254D/S267L and dimeric form TrfA S257F, as described under Materials and methods. The results of performed experiments are presented in panel (B) NA assay, and (D) ligase assay. On agarose gel free DNA and DNA multimers are marked F and M respectively. A quantified densitometry with using ChemiDoc MP System and ImageLab™ Software (BioRad) were applied for DNA multimers amount estimation. The error bars are derived from three different experiments.
RK2 handcuff is a stable nucleoprotein complex
Although it was speculated that the handcuffing is released by spontaneous dissociation when the plasmid concentration in bacterial cell decreases (47), no experimental data concerning the stability of handcuff is available to date. Therefore, we decided to analyze more closely the handcuff complex dynamics and make an attempt to resolve the mechanism of the uncuffing process in the RK2 plasmid. We tested the stability of the RK2 handcuff complex using the NA system with continuous buffer rinsing. The NA system is based on measuring the signal of the fluorescently labeled DNA. First, we assembled the handcuff complexes, as described under Materials and methods, using the NA assay. Next, we performed continuous rinsing with LAB buffer at 1 ml/min flow (Figure 2A). We stopped the flow at 0, 5, 15, 30, 45 and 60 min of rinsing and measured the fluorescence. We did not observe significant differences in the level of fluorescence at any of the particular time points (Figure 2B). The obtained data indicates that the handcuff complex is highly stable and excludes the possibility of spontaneous dissociation of the once formed handcuff complex in vitro.
Figure 2.

RK2 iteron plasmid forms stable handcuff structures. The scheme of the experimental procedure is presented in panel (A). Stability of the RK2 handcuff structures was tested in the NA assay (B), on pre-assembled handcuff structures, as described under Materials and methods. Stability test was performed in continuous buffer rinsing. The fluorescence signals of particular samples, were analyzed after 0, 5, 15, 30, 45 and 60 min of buffer rinsing.
RK2 plasmid uncuffing is mediated by Lon and ClpAP proteases
Since the obtained results indicated high stability of the handcuff complexes, we assumed that the spontaneous dissociation of the RK2 handcuff complex is very improbable. Therefore, we speculated that there must exist a mechanism which actively disrupts the once formed handcuff complex. To unravel how the re-initiation process occurs after handcuffing, we tested the influence of chaperones and proteases, on the preformed handcuff complexes. We assumed that both chaperones and proteases could potentially disrupt the handcuff nucleoprotein complex. Hence, we first assembled the RK2 handcuff complex with the use of linear DNA containing sequence of five iterons, and wild-type TrfA protein, then we treated the complexes with ClpX or ClpA chaperones (Figure 3A and C). We found out, by usage of FI* assay (Supplementary Data and Supplementary Figure S1), that ClpA, similar to ClpX chaperone (22), cause TrfA conformational activation. Chaperons are able to convert TrfA dimers to replicationally proficient monomers. In the absence of chaperones, dimers of TrfA protein dominate and thereby promote handcuffing, and high fluorescent signal in the NA assay (Figure 3B, lane 1) or high quantities of DNA multimers in the ligase assay (Figure 3D, lane 1). If the handcuff complexes had been disrupted, we should have observed a decrease in both the fluorescence signal in the NA assay and the number of oligomeric complexes in the ligase assay. Instead, we observed high fluorescence signal in the NA assay (Figure 3B, lanes 2 and 4) and the presence of multimeric complexes in the ligase assay (Figure 3D, lanes 2 and 4), exactly as it was observed in the control experiment, where no chaperones were added. Those results indicate that the tested ClpX and ClpA chaperones do not affect the already assembled RK2 handcuff complex.
Figure 3.

Uncuffing of the RK2 handcuff structures is mediated by Lon and ClpAP proteases. The scheme of the experimental procedure is presented in panel (A) NA assay, and (C) ligation assay. Uncuffing of the RK2 handcuff structures was tested on pre-assembled handcuff structures as described in Materials and methods. After handcuffing reaction the influence of several chaperones (ClpX and ClpA) and proteases (ClpXP, ClpAP and Lon) was analyzed by the NA (B) and ligase (D) assays. On agarose gel free DNA and DNA multimers are marked F and M, respectively. Positive control (lane 1, B and D) reactions with wild-type TrfA protein without the use of chaperones or proteases. Stability of TrfA protein, during analysis of handcuff structure disruption by chaperones and proteases in ligase assays, was tested by SDS-PAGE electrophoresis (E). A quantified densitometry with using ChemiDoc MP System and ImageLab™ Software (BioRad) were applied for TrfA protein and DNA multimers amount estimation. The error bars are derived from three different experiments.
To explore whether the presence of bacterial proteases can disrupt the handcuff complex we performed experiments with ClpXP, ClpAP and Lon proteins (Figure 3). All tested proteases recognize and can degrade the TrfA protein (25). In our experiments the influence of proteases on the handcuff complexes was observed only in the reactions where ClpAP or Lon proteins were present. We observed a substantial decrease in the fluorescence signal in the NA assay (Figure 3B, lanes 3 and 6) and a reduction of the multimeric DNA forms in ligase assay (Figure 3D, lanes 3 and 6). No such effects were observed during the experiments performed in the presence of ClpXP protease (Figure 3B and D, lane 5). In our tests, we also analyzed the stability of TrfA protein particles involved in the handcuff complex formation. Both ClpAP and Lon caused degradation of TrfA dimers in the handcuff complexes (Figure 3E, lanes 3 and 6).
The effect of particular proteases on the formation of handcuff complexes was also observed in the replication reaction in crude extracts, which were obtained from E. coli C600 or its protease deficient derivatives: lon(−), clpP(−) and lon(−)clpP(−). We applied essentially the same method for the detection of the handcuff effects on RK2 replication as it was described by Kittel and Helinski (35). We used the wild-type form of TrfA protein, the RK2 mini-replicon - pTJS42 - and plasmid pBK20 which is a pUC18 derivative containing RK2 iterons but no other oriV structural elements required for the origin activity. Addition of the pBK20 extra-iterons to the replication reaction with the wild-type C600 crude extract resulted in a 60% reduction of the DNA synthesis (Supplementary Figure S2). The observed DNA synthesis level was 40%, when compared to the reaction without extra iterons. This decrease of the DNA synthesis was a result of the handcuff being formed with pTJS42 and pBK20 plasmids, as it was impossible to restore it by the addition of excess of TrfA protein (35). The same reactions, performed in crude extracts from the protease-deficient strains, resulted in even greater, up to 90%, decrease of DNA synthesis. The DNA synthesis in clpP(−) extract was 30% and about 10% in both lon(−) and lon(−)clpP(−) extracts comparing to DNA synthesis observed with no pBK20 (Supplementary Figure S2). The most pronounced inhibitory effect in case of the reactions in lon(−) and lon(−)clpP(−) crude extracts indicates that the lack of Lon protease results in higher stability of handcuff complexes, as they are not disrupted.
RK2 plasmid is unstable in Lon-deficient E. coli strains
Based on our in vitro tests we concluded that the equilibrium between the handcuffed and the released forms of RK2 plasmid is probably affected by both Lon and ClpAP proteases. Therefore, we performed experiments in Lon- and ClpP-deficient strains, to estimate the in vivo effects of protease absence. We tested the transformation frequency with the pTJS42 plasmid and performed the stability assays on this RK2 minireplicon both in the wild-type and protease-deficient E. coli strains. The observed transformation frequency was more than one order of magnitude lower in lon(−) and lon(−)clpP(−) strain when compared to the wild-type strain (Supplementary Table S3). A reduction of the transformation frequency was also observed in the E. coli clpP(−) strain, however, when considering the reduction observed with the control pUC19 DNA, it was not relevant. The results of the stability assays are presented in Figure 4A and B and the analysis of plasmid loss per generation is shown in Supplementary Table S4. In comparison to the wild-type strain, the rate of plasmid loss was approximately 2-fold higher in the clpP(−) strain and fivefold higher in the lon(−) strain. The analysis of plasmid stability in the double clpP(−)lon(−) mutant showed a significant increase of plasmid loss, which occurred over ten times more frequently than in the wild-type strain. We also tested the stability of mini-RK2 derivatives in cells where the lack of Lon and ClpP proteases was complemented with the genes present on plasmid DNA. During the experiment the cells were grown in ampicillin- or kanamycin-containing medium, to maintain the pBADLon or pClpP plasmid, in lon(−) or clpP(−) strains, respectively. The RK2 minireplicon loss per generation in the lon(−) strain with supplementation of Lon protease was restored to the level comparable to the wild-type strain. Synthesis of ClpP protein in the clpP(−) strain had no significant effect on the pTJS42 plasmid stability. The obtained results demonstrate that the deficiency of specific proteases in bacterial cells has a negative effect on the plasmid maintenance.
Figure 4.
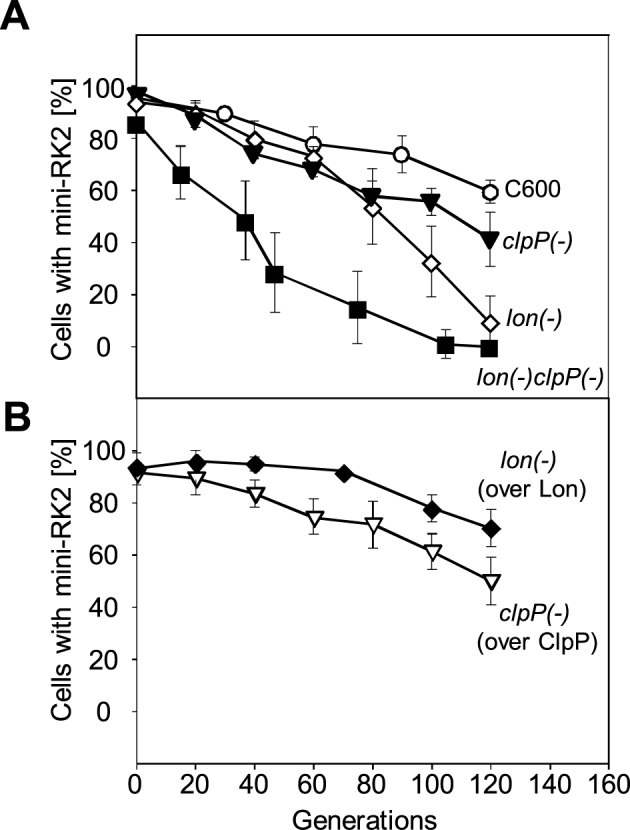
The mutation in Lon protease in E. coli strains affects the miniRK2 plasmid stability. Stability assays were performed as described under Materials and methods. The graphs show the dependence of the number of bacterial cells carrying plasmid pTJS42 on the number of generations after abolishment of the antibiotic selection pressure. Panel (A) shows the stability of mini-RK2 plasmid in E. coli C600 and its protease-deficient derivatives. Panel (B) shows the results of mini-RK2 plasmid stability experiments in Lon and ClpP supplemented strains. Cultures were grown for about 120 generations. The given results are means from three independent repeats of each experiment.
Protease deficiency results in bacterial cells having less plasmid foci containing more plasmids copies in one focus
Several reports show that RK2 plasmid is present in bacterial cell at five to eight copies per chromosome (32,49,50). Furthermore, the published data indicates that the number of plasmid foci per cell does not correspond to the number of plasmid copies per cell, indicating that each focus consists of more than one plasmid and that the RK2 plasmid molecules are grouped into a few clusters (44,51). To test if proteases might affect the formation of the plasmid clusters we used FROS system (see Materials and methods), allowing the localization of mini-RK2 in the E. coli strains - C600 and its protease-deficient derivatives lon(−), clpP(−) and lon(−)clpP(−). No substantial differences in the plasmid foci numbers were observed in the analyzed strains when we calculated them per single cell. Since E. coli Lon mutant strains form filaments (52,53), we also calculated the number of plasmid foci relative to cell volume. For the comparison between strains we determined the average volume (see Materials and methods) for the analyzed derivatives and after quantifying the number of EYFP foci, we calculated the number of foci per 1 μm3 of cell (Supplementary Figure S3) in order to determine the density of plasmid foci in cytosol. When comparing to the wild-type strain, the number of foci per 1 μm3 of the cell cytosol was two and half times lower in lon(−) and lon(−)clpP(−) strains (Supplementary Figure S3). No significant differences were observed in case of the clpP(−) strain. To verify whether the decreased number of mini-RK2 foci per 1 μm3 of the cell is not due to the cells’ filamentation itself, we performed an experiment with the wild-type E. coli C600 strain treated with cephalexin. Cephalexin is an inhibitor of cell division, which has no influence on chromosomal and plasmid DNA replication (44,54,55). We observed a very high number of plasmid foci in cephalexin treated cells calculated per single cell, however, the average number of the plasmid foci per 1 μm3 of the cephalexin-treated cells was comparable to the wild-type strain and over two times higher than this observed for Lon mutant strains (Supplementary Figure S3). This indicates that the filamentation has no influence on the reduction of the plasmid foci number/cellular concentration in the tested E. coli mutant strains. This reduction is most likely caused by Lon protease deficiency.
We also verified the number of mini-RK2 foci in lon(−) and clpP(−) strains complemented with the Lon or ClpP proteins, respectively. The proteases were complemented by using plasmids containing protease gene under control of an inducible promotor (see Materials and methods). The average number of foci per 1 μm3 of Lon-supplemented lon(−) cell was comparable to the wild-type strain. We also observed lack of filamentation and reduction of the average length of the Lon-supplemented lon(−) cells to the length similar to the one of the wild-type strain cells (Supplementary Figure S3). Supplementation by ClpP protein in clpP(−) strain had no significant effect on the average number of foci per 1 μm3 of ClpP-supplemented cells.
To determine the plasmid copy number of RK2 forming a single focus, in the wild-type C600 strain and its protease-deficient derivatives, we performed qPCR PCN analysis (see Materials and methods). The obtained results showed that in the wild-type C600 strain there was an average number of 5.8 copies of mini-RK2 plasmid per bacterial cell, while clpP(−), lon(−) and lon(−)clpP(−) strains contained approximately 4.7, 10.6 and 14.3 copies of the plasmid, respectively (Supplementary Figure S3). Based on our microscopy analysis (Supplementary Figure S3), the average number of foci per bacterial cell in the analyzed strains was 2.6 for the wild-type C600, 1.8 for clpP(−), 3.3 for lon(−) and 3.4 for lon(−)clpP(−) strains. The estimated number of plasmid molecules per focus was 2.3 in the wild-type C600 and 2.6 in clpP(−). We observed an increase in the number of plasmid molecules forming individual foci in lon(−) - 3.2, and lon(−)clpP(−) strain - 4.2 (Supplementary Figure S3). Supplementation of Lon protease in the lon(−) strain reduced the mini-RK2 to 2.6 per focus. We analyzed the mini-RK2 plasmid copy number in the C600 cephalexin-treated cells. The number of plasmid molecules per focus was 2.8, which is comparable to the wild-type strain. The obtained results indicate that the proteases affect the plasmid foci formation in bacterial cells. Protease-deficient bacterial cells have less plasmid foci per cell volume and more plasmid copy in one focus.
Handcuffed RK2 plasmid molecules are uncuffed by TrfA monomers
Our experiments showed that dimeric but not monomeric variants of TrfA take part in assembling handcuff complex and that the particular proteases are able to liberate handcuffed plasmid molecules by degrading TrfA involved in handcuff structure (Figures 1 and 3). Then we asked whether there is any effect of the TrfA monomers on the already formed handcuff. To address this issue we performed both the NA and the ligase assays, in which, after the handcuff complexes were formed with the wild-type TrfA, we titrated the reactions with increasing concentrations of the monomeric TrfA G254D/S267L protein (Figure 5A and C). Titration of the RK2 handcuff complexes with the monomeric TrfA showed the destructive effect of this protein on the already formed handcuff complex. In reactions titrated with the TrfA G254D/S267L protein, a reduction of fluorescent signal in the NA assay and a decreased number of DNA multimers in ligase assay was observed (Figure 5B and D). We also performed experiments with the wild-type TrfA protein activated by the ClpX chaperone (Supplementary Figure S4). The ClpX-dependent activation converts the wild-type TrfA dimers into monomers (22). The data obtained in the NA experiment with the ClpX activated wild-type TrfA was similar to this obtained in the assays with TrfA G254D/S267L. It must be pointed out that ClpX alone is not able to disrupt handcuff structure (Figure 3).
Figure 5.

Monomeric form of TrfA protein uncuffs the RK2 handcuff structures. Schemes of the experimental procedure are presented in panel (A) NA assay, and (C) ligation assay. Uncuffing of the RK2 handcuff structures was tested on pre-assembled handcuff structures as described under Materials and methods. After handcuffing reaction the influence of monomeric TrfA form was analyzed in NA (B) and ligase (D) assays. On agarose gel free DNA and DNA multimers are marked F and M, respectively. A quantified densitometry with using ChemiDoc MP System and ImageLab™ Software (BioRad) were applied for DNA multimers amount estimation. The error bars are derived from three different experiments.
Since TrfA monomer is able to disrupt handcuff complexes of plasmid RK2, we further asked how efficient this reaction is. Experiments which define the dynamics of the handcuff disruption by monomeric TrfA were performed (Supplementary Figure S5). After the assembly of the RK2 handcuff complex with the wild-type TrfA, we treated it with TrfA G254D/S267L (at 3 μM concentration), thereby inducing handcuff disruption (Supplementary Figure S5B), and then we analyzed the disruption of the handcuff complex at defined time points. After 5 min, we observed 40% reduction of the fluorescence signal in the NA assay. In 45 min, there was almost 80% reduction of fluorescence.
The TrfA monomers displace wild-type TrfA molecules and uncuff plasmid molecules
To analyze more thoroughly the process of RK2 handcuff disruption, an experiment, which allowed to explore the role of the monomeric TrfA form, was performed. We applied two versions of the NA assay. In the first assay, to assemble the handcuff complex, we used a fluorescently labeled wild-type TrfA protein, apart from the fluorescently labeled DNA (Figure 6A). We titrated the obtained complex with increasing amounts of TrfA G254D/S267L. After stopping the reaction, the decrease in the fluorescence signal from both DNA and the wild-type TrfA protein was observed (Figure 6B). It indicates that the monomeric TrfA variant released the wild-type TrfA from the handcuff complex and dissociated the coupled DNA fragments. In the second assay, we assembled the handcuff complex using fluorescently labeled DNA and an unlabeled wild-type TrfA (Figure 6C). Such complexes were titrated with increasing concentration of fluorescently labeled TrfA G254D/S267L. In this experiment, we observed a decrease in the fluorescence signal derived from the DNA (Figure 6D, white bars), and an increase in the fluorescence signal derived from TrfA G254D/S267L (Figure 6D, dark gray bars), which indicated that the handcuff was disrupted and the TrfA monomers bound the uncoupled DNA fragments. The obtained data shows that during the RK2 handcuff disruption the wild-type TrfA is displaced by the monomeric form of the initiator protein.
Figure 6.

Monomeric form of the TrfA protein replaces the wild-type form from the plasmid DNA iterons. Schemes of the experimental procedure are presented in panel (A) handcuff formation with the fluorescently-labeled wild-type TrfA AlexaFluor 488, and (C) handcuff formation with the wild-type TrfA. After handcuffing reaction the influence of monomeric TrfA form was analyzed: (B) handcuff structures formed with TrfA AlexaFluor 488, were titrated with the monomeric form of TrfA protein (0.3 and 3 μM) and (D) handcuff structures formed with the wild-type TrfA protein were titrated with fluorescently-labeled monomeric form TrfA G254D/S267L AlexaFluor 488 (0.3 and 3 μM).
The TrfA molecules are stably associated within the handcuff complex
We performed experiments to determine the TrfA turnover in the handcuff in which we titrated the pre-assembled handcuff complexes with the wild-type TrfA protein. When the handcuff complex was formed with the involvement of the AlexaFluor 488-labeled wild-type TrfA, it was titrated with the unlabeled form of the wild-type TrfA (Figure 7A), no change in the fluorescence signal was observed (Figure 7B, dark gray bars). We did not observe any change in the level of fluorescence derived from the AlexaFluor 555-labeled DNA (Figure 7B, white bars) as well. We performed an experiment in which we assembled the handcuff complex composed of the unlabeled wild-type TrfA and titrated it with the AlexaFluor 488-labeled wild-type TrfA (Figure 7C). In this reaction, we also did not observe an increase in the fluorescence signal (Figure 7D, dark gray bars). In the same experiment the fluorescence from the handcuffed AlexaFluor 555-labeled DNA fragment remained unchanged (Figure 7D, white bars). The presented experiments did not reveal any turnover of wild-type TrfA within the handcuff, hence we conclude that TrfA molecules are stable elements of the handcuff complex.
Figure 7.

The TrfA dimers are stably associated within the handcuff complex. Schemes of the experimental procedure are presented in panel (A) handcuff formation with fluorescently-labeled wild-type TrfA AlexaFluor 488, and (C) handcuff formation with wild-type TrfA. After handcuffing reaction the dynamics of such structure was analyzed: (B) handcuff structures formed with TrfA AlexaFluor 488, were titrated with wild-type TrfA protein (0.3 and 3 μM) and (D) handcuff structures formed with the wild-type TrfA protein were titrated with fluorescently-labeled wild-type TrfA AlexaFluor 488 (0.3 and 3 μM).
The disruption of handcuff by the TrfA monomers results in plasmid replication initiation
It was intriguing to test whether the activity of the monomeric TrfA in the disruption of the handcuff complexes and displacement of the wild-type protein from the handcuff (see sections above and Figure 6) would restore the plasmid DNA replication of the released plasmid molecules. To address this issue, we performed replication experiments with the use of crude extracts obtained from the wild-type E. coli C600 strain or its protease-deficient derivatives. The formation of handcuff complexes was generated by the addition of the plasmid pBK20 containing extra-iterons, as described above (see Supplementary Figure S2). We used the 2:1 molar ratio of the pBK20:pTJS42 mini-replicons which resulted in substantial inhibition of DNA synthesis (Supplementary Figure S2). In the extract from the wild-type E. coli, the DNA synthesis was reduced to 40% of that observed without pBK20. The most pronounced inhibition was observed in extracts prepared from the protease-deficient E. coli strains. Similarly, as in the experiment presented in Supplementary Figure S2, the DNA synthesis was reduced to 25%, 10% and 8% in clpP(−), lon(−) and lon(−)clpP(−) extracts, respectively, when comparing to reactions where the handcuff was not created (Supplementary Figure S2A and B). Much lower inhibition in the extract from the wild-type strain is most likely caused by the Lon and ClpAP proteases activity which disrupts the handcuff plasmid complexes. After preformation of the handcuff complexes, we titrated the mixtures with the monomeric variant TrfA G254D/S267L or the wild-type form of the TrfA protein (Figure 8A and B). No increase in the DNA synthesis level was observed in the reactions with wild-type TrfA, regardless of the applied concentration (Figure 8B). Even a significant excess of the wild-type TrfA did not restore the pTJS42 DNA replication, indicating that the plasmid molecules are permanently handcuffed. In contrast, when the mixtures containing the preformed handcuff complexes were titrated with the monomeric TrfA G254D/S267L, a substantial increase in DNA synthesis was observed (Figure 8A). The increase was more or less similar regardless of the type of extract used. Those results indicate that the monomeric TrfA G254D/S267L disrupts the handcuff complex, which results in re-establishment of replication initiation and DNA synthesis of plasmid RK2 mini-replicon.
Figure 8.
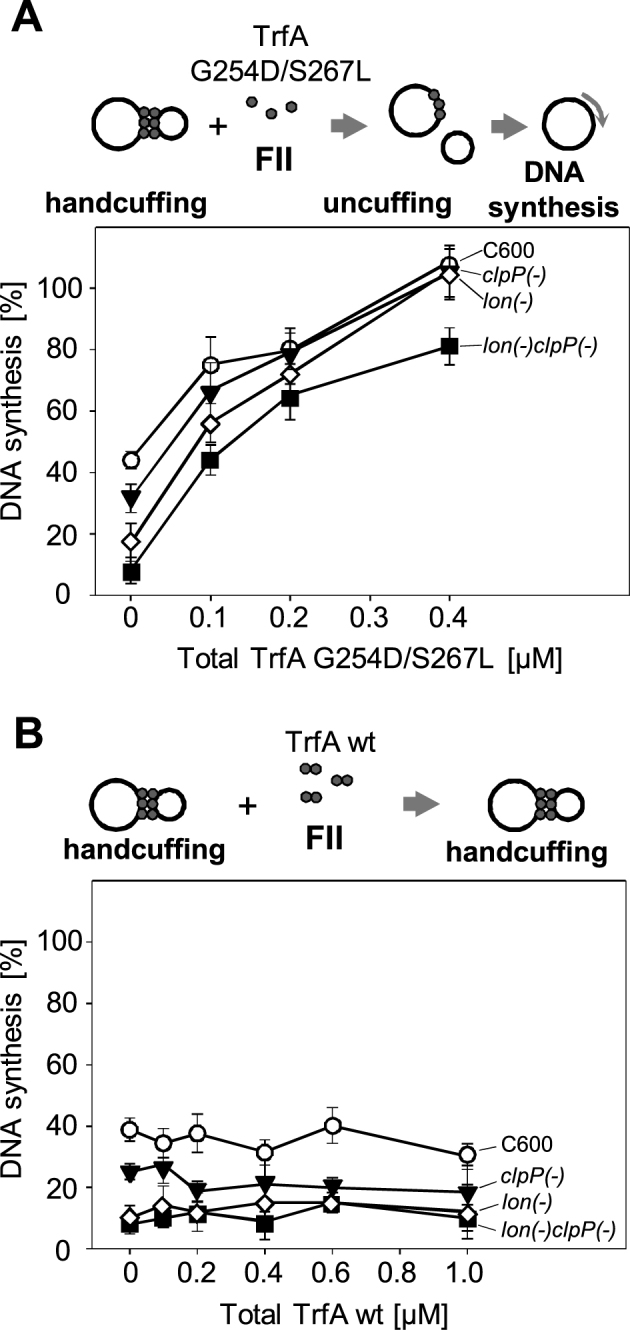
Monomeric form of TrfA protein re-initiates the RK2 plasmid replication. The influence of the monomeric (A) and wild-type form of TrfA protein (B), on handcuff structures, was tested in in vitro replication assay (FII). Reactions were performer using crude extracts obtained from E. coli C600 (○) or its protease-defective derivatives: clpP(−) (▾), lon(−) (⋄) and lon(−)clpP(−) (▪), as described under Materials and methods. All crude extracts were active for RK2 replication. Reactions contained 250 ng of supercoiled DNA template and 500 ng of wild-type TrfA protein. The starting points are the reactions in which, due to handcuffing, the maximum inhibition of DNA synthesis was observed. Formation of handcuff complexes was obtained through the addition of pBK20 iteron-containing plasmid in 2:1 molar ratio, in relation to pTJS42 RK2 mini-replicon. The influence of particular forms of TrfA protein was tested by titration of individual reactions by increasing concentration of monomeric TrfA (0.1, 0.2 and 0.4 μM) (A) or wild-type TrfA form (0.2, 0.4, 0.6 and 1.0 μM) (B). The error bars are derived from three repetitions of each reaction.
DISCUSSION
Application of ligation assay and NA assay with continuous buffer rinsing and utilization of TrfA protein conformational variants allowed us to investigate both Rep dimers and monomers contribution to the handcuff complex formation as well as the dynamics of the handcuff structure. During our experiments, the wild-type TrfA, which mainly exists as dimer in solution (6), or the constitutively dimeric variant TrfA S257F (16), but not the protein's monomeric variant TrfA G254D/S267L, contribute to forming the handcuff. Moreover, any addition of the monomeric TrfA to the reactions resulted in destabilization of the observed handcuff structure. This data is consistent with the data reported previously, demonstrating that TrfA G254D/S267L is defective in formation of the handcuff complexes in the ligase assay (19) and that the plasmid replication dependent on TrfA monomeric variants, TrfA G254D or TrfA G254D/S267L, is not inhibited by iterons added externally (19,35). Our data is also in line with the handcuff model proposed for the plasmids P1 (47) and R6K (11,12), where the Rep dimers are postulated to pair the plasmids origins. In contrast, our results do not support the concept that, apart from dimers, also monomers are involved in the handcuff formation, which was proposed on the basis that the constitutively dimeric TrfA S257F suppresses the run-away phenotype of TrfA G254D/S267L (16). This suppression was most likely due to handcuff formed by TrfA S257F as demonstrated during our experiments. Due to dimeric conformation of this TrfA variant it is able to interact with iteron containing dsDNA only to some extent (23) and this interaction does not result in the plasmid DNA replication initiation (16). Our experiment demonstrates clearly that TrfA S257F is able to handcuff two plasmid molecules. We cannot exclude that during the handcuff structure formation, TrfA dimers conformation is being altered. The dimerization interface of TrfA dimer could be different, when comparing to the interface of the TrfA protomers interacting within the RK2 handcuff complex. During plasmid pPS10 origin pairing the process is facilitated by zipping-up the DNA-bound RepA monomers (13). Crosslinking studies and Mass Spectrometry analysis revealed that the nature of RepA protomers interaction within a dimer and within the handcuff complexes is different. It might be a general feature also for other plasmid systems, including RK2. It is also possible that the Rep dimers can form even higher ordered quaternary structures when complexed in handcuff. We cannot exclude that TrfA molecules that handcuff RK2 plasmid molecules might form some kind of oligomer or filament assembled on plasmid DNA. By using immuno-electron microscopy analysis it has been recently demonstrated that RepA protein molecules engaged in pPS10 plasmid handcuff structure create amyloidogenic oligomers (56).
Results obtained with the use of the NA assay show high stability of the RK2 handcuff indicating that disruption of such complexes most likely does not occur through spontaneous dissociation caused by dilution of plasmid molecules during cell growth and division. The high stability of handcuff is reasonable, because handcuffed plasmid molecules are bridged together through several binding sites, iterons, and several Rep protein molecules. We assume that the handcuff needs to be actively triggered to enable the complex disruption and subsequent re-initiation of the plasmid replication. This raises questions about factors influencing the handcuff stability and the causes of the handcuff disruption before the re-initiation of the replication process.
Our investigations with the use of purified chaperone proteins revealed no measurable effects on the RK2 handcuff. The conformational activation by chaperones has been described for many Rep proteins including TrfA (4,24,57–61). Although both ClpX (22) and ClpA (this work) conformationally activate TrfA, the activity of those chaperones did not contribute to uncuffing of the coupled plasmid origins. Most likely the chaperones are not able to access and/or conformationally activate TrfA when the protein is involved in the handcuff complex. When we tested the effects of proteases on the RK2 handcuff, only the ClpAP and Lon proteases acted on the preformed handcuff complex and caused its disruption. No effects were observed when ClpXP was added to the handcuff complex. These results are consistent with the previous data (25) where we had shown that DNA has stimulatory effect on TrfA proteolysis by both ClpAP and Lon, while the presence of DNA in the reaction inhibits the ClpXP proteolytic activity. It is interesting that the ClpAP proteolytic complex recognizes and degrades TrfA forming the handcuff structure, but the ClpA alone is not able to conduct the uncuffing reaction. It could be conceived that the ClpA alone does not affect TrfA, involved in the handcuff, however, when coupled with proteolytic subunit ClpP, the degradation of TrfA is sufficient to disrupt the handcuff. Clearly, it's the protease activity, but not a chaperone activity itself, that is required for the decomposition of the handcuff nucleoprotein structure.
As mentioned above, once the handcuff complex is formed it cannot be disrupted by chaperones, however, as we demonstrated, chaperones can act on TrfA free dimers which are not engaged in the handcuff complex. The monomerized TrfA is then capable of disrupting the handcuff complex. Furthermore, addition of the monomeric protein TrfA G254D/S267L resulted in the plasmid uncuffing. The staged ligation and NA assays demonstrated that TrfA monomers displace the wild-type TrfA molecules which are involved in the handcuff, thus causing the release of the handcuffed DNA molecules. It could be considered whether handcuff disassembling by TrfA monomers requires some kind of dynamic change(s) within oligomeric/filamentous structure on plasmid DNA that involves specific protein-protein interactions. During our experiments the addition of the dimeric wild-type TrfA did not result in the displacement of the wild-type TrfA molecules forming the handcuff nucleoprotein structure. Those results altogether indicate a novel activity of the TrfA monomers as a positive DNA replication regulator. In addition to the interaction with the iterons (20,62) and ssDNA (33) required for the plasmid replication initiation, another important function of the monomeric TrfA protein is the RK2 handcuff disruption. This function is crucial for the process of replication re-initiation. Displacement of TrfA molecules forming the handcuff by the replicationally proficient TrfA monomers results in replication re-initiation observed in the in vitro replication experiment, where the addition of TrfA G254D/S267L to the assembled handcuff complex re-initiated the DNA synthesis. This proves that the TrfA monomers control both DNA replication initiation and the uncoupling of the handcuffed plasmids.
Our in vitro data showing proteases and TrfA monomers as factors disrupting the RK2 handcuff complex is consistent with the in vivo observations. It must be pointed out that the redundancy of proteases and TrfA monomers in the handcuff decomposition made the in vivo tests difficult. It has been previously shown that RK2 derivatives, which have substitutions in the trfA gene resulting in monomeric protein TrfA G254D/S267L, lead to uncontrolled run-away replication (16,19). Our transformation tests indicate that Lon is an important factor affecting plasmid DNA metabolism. Moreover, RK2 mini-replicons were less stable in E. coli lon(−) and clpP(−) mutants. Interestingly, we observed that protease deficiency resulted in a decreased number of plasmid foci with more plasmid molecules clustered together, thus, most likely, causing an unequal plasmid distribution to daughter cells and, consequently, plasmid loss. It also indicates functional connection between the handcuff and plasmid foci formation. The nature and structural basis of the latter one is still ambiguous.
An important question arises: why both proteases and Rep monomers are involved in the uncuffing of the plasmid molecules? We assume that the Rep monomers are the key factors in the uncuffing reaction and that the interplay between Rep dimers and monomers is most critical for hampering DNA replication. The monomer-dimer paradox in control of the plasmid DNA replication by iterons has been discussed in an excellent micro-review by Chattoraj (3). A spontaneous dissociation of the handcuff complexes, increasing Rep concentration to saturate the iterons and active dissociation of the handcuff were considered as mechanisms for the handcuff reversal. We provide evidence that the replicationally-potent Rep monomers actively displace Rep particles involved in the handcuff, which releases plasmids and allows re-initiation of DNA replication. Formation of handcuff is facilitated by Rep dimers when plasmid copy number is elevated and no Rep monomers are present in cytosol. The ratio between Rep dimers, Rep monomers and plasmid molecules would be a key for controlling handcuffing reversal and plasmid DNA synthesis. The important factor is regulation of expression of rep that affects Rep concentration. In the RK2 system we cannot forget about kor regulation, the influence of which is not fully understood, but certainly has an effect on TrfA concentration. The RK2 replication is primarily controlled by maintaining the TrfA in rate-limiting amounts. This is achieved at least in part through transcriptional repression by korA and korB (63,64). Removal of korB from mini-replicons results in an increase in plasmid copy number (49,65).
What is the relevance of proteases for the process of handcuff disruption? Under vegetative conditions proteases would disrupt the handcuff constitutively in a very limited manner sustaining some kind of equilibrium. However, under stress conditions, when chaperones and proteases are induced, it most likely results in increased Rep monomerization, proteolysis and handcuff disruption. Reversal of the handcuffing should be beneficial for plasmid stability in cells under stress condition. It would allow re-initiation of plasmid replication, increase of the plasmid copy and foci numbers, and increase the chance of equal partitioning of plasmids into newborn cells. Investigation of plasmids under stress conditions would verify these possibilities. Although our study concerned plasmid handcuff, it is of particular interest to test if proteolysis and displacement of regulatory proteins by their specific conformational variants could be involved in the regulation of other replicons or in any other processes in which DNA pairing was observed.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Professors Bernd Bukau and David Sherratt for providing strains and plasmids. We also thank dr. Magdalena Rajewska and Marta Gross for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Polish National Science Centre [2012/04/A/NZ1/00048]. Funding for open access charge: Polish National Science Centre [2012/04/A/NZ1/00048].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ingmer H., Fong E.L., Cohen S.N.. Monomer-dimer equilibrium of the pSC101 RepA protein. J. Mol. Biol. 1995; 250:309–314. [DOI] [PubMed] [Google Scholar]
- 2. Kolatka K., Kubik S., Rajewska M., Konieczny I.. Replication and partitioning of the broad-host-range plasmid RK2. Plasmid. 2010; 64:119–134. [DOI] [PubMed] [Google Scholar]
- 3. Chattoraj D.K. Control of plasmid DNA replication by iterons: no longer paradoxical. Mol. Microbiol. 2000; 37:467–476. [DOI] [PubMed] [Google Scholar]
- 4. Wickner S., Hoskins J., McKenney K.. Monomerization of RepA dimers by heat shock proteins activates binding to DNA replication origin. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:7903–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishiai M., Wada C., Kawasaki Y., Yura T.. Replication initiator protein RepE of mini-F plasmid: functional differentiation between monomers (initiator) and dimers (autogenous repressor). Proc. Natl. Acad. Sci. U.S.A. 1994; 91:3839–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Toukdarian A.E., Helinski D.R., Perri S.. The plasmid RK2 initiation protein binds to the origin of replication as a monomer. J. Biol. Chem. 1996; 271:7072–7078. [DOI] [PubMed] [Google Scholar]
- 7. Kruger R., Konieczny I., Filutowicz M.. Monomer/dimer ratios of replication protein modulate the DNA strand-opening in a replication origin. J. Mol. Biol. 2001; 306:945–955. [DOI] [PubMed] [Google Scholar]
- 8. Garcia de Viedma D., Serrano-Lopez A., Diaz-Orejas R.. Specific binding of the replication protein of plasmid pPS10 to direct and inverted repeats is mediated by an HTH motif. Nucleic Acids Res. 1995; 23:5048–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ingmer H., Cohen S.N.. The pSC101 par locus alters protein-DNA interactions in vivo at the plasmid replication origin. J. Bacteriol. 1993; 175:6046–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. York D., Filutowicz M.. Autoregulation-deficient mutant of the plasmid R6K-encoded pi protein distinguishes between palindromic and nonpalindromic binding sites. J. Biol. Chem. 1993; 268:21854–21861. [PubMed] [Google Scholar]
- 11. Urh M., Wu J., Wu J., Forest K., Inman R.B., Filutowicz M.. Assemblies of replication initiator protein on symmetric and asymmetric DNA sequences depend on multiple protein oligomerization surfaces. J. Mol. Biol. 1998; 283:619–631. [DOI] [PubMed] [Google Scholar]
- 12. Kunnimalaiyaan S., Inman R.B., Rakowski S.A., Filutowicz M.. Role of pi dimers in coupling (‘handcuffing’) of plasmid R6K's gamma ori iterons. J. Bacteriol. 2005; 187:3779–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gasset-Rosa F., Diaz-Lopez T., Lurz R., Prieto A., Fernandez-Tresguerres M.E., Giraldo R.. Negative regulation of pPS10 plasmid replication: origin pairing by zipping-up DNA-bound RepA monomers. Mol. Microbiol. 2008; 68:560–572. [DOI] [PubMed] [Google Scholar]
- 14. Giraldo R., Andreu J.M., Diaz-Orejas R.. Protein domains and conformational changes in the activation of RepA, a DNA replication initiator. EMBO J. 1998; 17:4511–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zzaman S., Bastia D.. Oligomeric initiator protein-mediated DNA looping negatively regulates plasmid replication in vitro by preventing origin melting. Mol. Cell. 2005; 20:833–843. [DOI] [PubMed] [Google Scholar]
- 16. Toukdarian A.E., Helinski D.R.. TrfA dimers play a role in copy-number control of RK2 replication. Gene. 1998; 223:205–211. [DOI] [PubMed] [Google Scholar]
- 17. Kues U., Stahl U.. Replication of plasmids in gram-negative bacteria. Microbiol. Rev. 1989; 53:491–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas C.a.H.D. Thomas C.M. Promiscuous Plasmids of Gram-negative Bacteria. 1989; London: Academic Press Inc. Ltd; 1–25. [Google Scholar]
- 19. Blasina A., Kittell B.L., Toukdarian A.E., Helinski D.R.. Copy-up mutants of the plasmid RK2 replication initiation protein are defective in coupling RK2 replication origins. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:3559–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perri S., Helinski D.R., Toukdarian A.. Interactions of plasmid-encoded replication initiation proteins with the origin of DNA replication in the broad host range plasmid RK2. J. Biol. Chem. 1991; 266:12536–12543. [PubMed] [Google Scholar]
- 21. Lin J., Helinski D.R.. Analysis of mutations in trfA, the replication initiation gene of the broad-host-range plasmid RK2. J. Bacteriol. 1992; 174:4110–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konieczny I., Helinski D.R.. The replication initiation protein of the broad-host-range plasmid RK2 is activated by the ClpX chaperone. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:14378–14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pierechod M., Nowak A., Saari A., Purta E., Bujnicki J.M., Konieczny I.. Conformation of a plasmid replication initiator protein affects its proteolysis by ClpXP system. Protein Sci. 2009; 18:637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Konieczny I., Liberek K.. Cooperative action of Escherichia coli ClpB protein and DnaK chaperone in the activation of a replication initiation protein. J. Biol. Chem. 2002; 277:18483–18488. [DOI] [PubMed] [Google Scholar]
- 25. Kubik S., Wegrzyn K., Pierechod M., Konieczny I.. Opposing effects of DNA on proteolysis of a replication initiator. Nucleic Acids Res. 2012; 40:1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Komori H., Matsunaga F., Higuchi Y., Ishiai M., Wada C., Miki K.. Crystal structure of a prokaryotic replication initiator protein bound to DNA at 2.6 A resolution. EMBO J. 1999; 18:4597–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giraldo R. Common domains in the initiators of DNA replication in Bacteria, Archaea and Eukarya: combined structural, functional and phylogenetic perspectives. FEMS Microbiol. Rev. 2003; 26:533–554. [DOI] [PubMed] [Google Scholar]
- 28. Giraldo R., Fernandez-Tornero C., Evans P.R., Diaz-Orejas R., Romero A.. A conformational switch between transcriptional repression and replication initiation in the RepA dimerization domain. Nat. Struct. Biol. 2003; 10:565–571. [DOI] [PubMed] [Google Scholar]
- 29. Giraldo R., Fernandez-Tresguerres M.E.. Twenty years of the pPS10 replicon: insights on the molecular mechanism for the activation of DNA replication in iteron-containing bacterial plasmids. Plasmid. 2004; 52:69–83. [DOI] [PubMed] [Google Scholar]
- 30. Sharma S., Sathyanarayana B.K., Bird J.G., Hoskins J.R., Lee B., Wickner S.. Plasmid P1 RepA is homologous to the F plasmid RepE class of initiators. J. Biol. Chem. 2004; 279:6027–6034. [DOI] [PubMed] [Google Scholar]
- 31. Swan M.K., Bastia D., Davies C.. Crystal structure of pi initiator protein-iteron complex of plasmid R6K: implications for initiation of plasmid DNA replication. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18481–18486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Durland R.H., Toukdarian A., Fang F., Helinski D.R.. Mutations in the trfA replication gene of the broad-host-range plasmid RK2 result in elevated plasmid copy numbers. J. Bacteriol. 1990; 172:3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wegrzyn K., Fuentes-Perez M.E., Bury K., Rajewska M., Moreno-Herrero F., Konieczny I.. Sequence-specific interactions of Rep proteins with ssDNA in the AT-rich region of the plasmid replication origin. Nucleic Acids Res. 2014; 42:7807–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wawrzycka A., Gross M., Wasaznik A., Konieczny I.. Plasmid replication initiator interactions with origin 13-mers and polymerase subunits contribute to strand-specific replisome assembly. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E4188–E4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kittell B.L., Helinski D.R.. Iteron inhibition of plasmid RK2 replication in vitro: evidence for intermolecular coupling of replication origins as a mechanism for RK2 replication control. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sambrook J., Fritsch E.F., Maniatis T.. Molecular Cloning: a Laboratory Manual. 1989; 2nd edn, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 37. Thompson M.W., Maurizi M.R.. Activity and specificity of Escherichia coli ClpAP protease in cleaving model peptide substrates. J. Biol. Chem. 1994; 269:18201–18208. [PubMed] [Google Scholar]
- 38. Maurizi M.R., Thompson M.W., Singh S.K., Kim S.H.. Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli. Methods Enzymol. 1994; 244:314–331. [DOI] [PubMed] [Google Scholar]
- 39. Goldberg A.L., Moerschell R.P., Chung C.H., Maurizi M.R.. ATP-dependent protease La (lon) from Escherichia coli. Methods Enzymol. 1994; 244:350–375. [DOI] [PubMed] [Google Scholar]
- 40. McEachern M.J., Bott M.A., Tooker P.A., Helinski D.R.. Negative control of plasmid R6K replication: possible role of intermolecular coupling of replication origins. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:7942–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gilchrist A., Smit J.. Transformation of freshwater and marine caulobacters by electroporation. J. Bacteriol. 1991; 173:921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kowalczyk L., Rajewska M., Konieczny I.. Positioning and the specific sequence of each 13-mer motif are critical for activity of the plasmid RK2 replication origin. Mol. Microbiol. 2005; 57:1439–1449. [DOI] [PubMed] [Google Scholar]
- 43. Easter C.L., Sobecky P.A., Helinski D.R.. Contribution of different segments of the par region to stable maintenance of the broad-host-range plasmid RK2. J. Bacteriol. 1997; 179:6472–6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pogliano J., Ho T.Q., Zhong Z., Helinski D.R.. Multicopy plasmids are clustered and localized in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:4486–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ruiz-Barba J.L., Maldonado A., Jimenez-Diaz R.. Small-scale total DNA extraction from bacteria and yeast for PCR applications. Anal Biochem. 2005; 347:333–335. [DOI] [PubMed] [Google Scholar]
- 46. Urh M., Wu J., Forest K., Inman R.B., Filutowicz M.. Assemblies of replication initiator protein on symmetric and asymmetric DNA sequences depend on multiple protein oligomerization surfaces. J. Mol. Biol. 1998; 283:619–631. [DOI] [PubMed] [Google Scholar]
- 47. Das N., Chattoraj D.K.. Origin pairing ('handcuffing') and unpairing in the control of P1 plasmid replication. Mol. Microbiol. 2004; 54:836–849. [DOI] [PubMed] [Google Scholar]
- 48. Kubik S., Wegrzyn K., Pierechod M., Konieczny I.. Opposing effects of DNA on proteolysis of a replication initiator. Nucleic Acids Res. 40:1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Durland R.H., Helinski D.R.. Replication of the broad-host-range plasmid RK2: direct measurement of intracellular concentrations of the essential TrfA replication proteins and their effect on plasmid copy number. J. Bacteriol. 1990; 172:3849–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fang F.C., Helinski D.R.. Broad-host-range properties of plasmid RK2: importance of overlapping genes encoding the plasmid replication initiation protein TrfA. J. Bacteriol. 1991; 173:5861–5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kolatka K., Witosinska M., Pierechod M., Konieczny I.. Bacterial partitioning proteins affect the subcellular location of broad-host-range plasmid RK2. Microbiology. 2008; 154:2847–2856. [DOI] [PubMed] [Google Scholar]
- 52. Gottesman S., Halpern E., Trisler P.. Role of sulA and sulB in filamentation by lon mutants of Escherichia coli K-12. J. Bacteriol. 1981; 148:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mizusawa S., Gottesman S.. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pogliano J., Pogliano K., Weiss D.S., Losick R., Beckwith J.. Inactivation of FtsI inhibits constriction of the FtsZ cytokinetic ring and delays the assembly of FtsZ rings at potential division sites. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ho T.Q., Zhong Z., Aung S., Pogliano J.. Compatible bacterial plasmids are targeted to independent cellular locations in Escherichia coli. EMBO J. 2002; 21:1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Molina-Garcia L., Gasset-Rosa F., Moreno-Del Alamo M., Fernandez-Tresguerres M.E., Moreno-Diaz de la Espina S., Lurz R., Giraldo R.. Functional amyloids as inhibitors of plasmid DNA replication. Sci. Rep. 2016; 6:25425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kawasaki Y., Wada C., Yura T.. Roles of Escherichia coli heat shock proteins DnaK, DnaJ and GrpE in mini-F plasmid replication. Mol. Gen. Genet. 1990; 220:277–282. [DOI] [PubMed] [Google Scholar]
- 58. Wickner S., Skowyra D., Hoskins J., McKenney K.. DnaJ, DnaK, and GrpE heat shock proteins are required in oriP1 DNA replication solely at the RepA monomerization step. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:10345–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wickner S., Gottesman S., Skowyra D., Hoskins J., McKenney K., Maurizi M.R.. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:12218–12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. DasGupta S., Mukhopadhyay G., Papp P.P., Lewis M.S., Chattoraj D.K.. Activation of DNA binding by the monomeric form of the P1 replication initiator RepA by heat shock proteins DnaJ and DnaK. J. Mol. Biol. 1993; 232:23–34. [DOI] [PubMed] [Google Scholar]
- 61. Sozhamannan S., Chattoraj D.K.. Heat shock proteins DnaJ, DnaK, and GrpE stimulate P1 plasmid replication by promoting initiator binding to the origin. J. Bacteriol. 1993; 175:3546–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Perri S., Helinski D.R.. DNA sequence requirements for interaction of the RK2 replication initiation protein with plasmid origin repeats. J. Biol. Chem. 1993; 268:3662–3669. [PubMed] [Google Scholar]
- 63. Schreiner H.C., Bechhofer D.H., Pohlman R.F., Young C., Borden P.A., Figurski D.H.. Replication control in promiscuous plasmid RK2: kil and kor functions affect expression of the essential replication gene trfA. J. Bacteriol. 1985; 163:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thomas C.M., Cross M.A., Hussain A.A., Smith C.A.. Analysis of copy number control elements in the region of the vegetative replication origin of the broad host range plasmid RK2. EMBO J. 1984; 3:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thomas C.M., Hussain A.A.. The korB gene of broad host range plasmid RK2 is a major copy number control element which may act together with trfB by limiting trfA expression. EMBO J. 1984; 3:1513–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.