

Abstract
The central photochemical reaction in photosystem II of green algae and plants and the reaction center of some photosynthetic bacteria involves a one-electron transfer from a light-activated chlorin complex to a bound quinone molecule. Through protein engineering, we have been able to modify a protein to mimic this reaction. A unique quinone-binding site was engineered into the Escherichia coli cytochrome b562 by introducing a cysteine within the hydrophobic interior of the protein. Various quinones, such as p-benzoquinone and 2,3-dimethoxy-5-methyl-1,4-benzoquinone, were then covalently attached to the protein through a cysteine sulfur addition reaction to the quinone ring. The cysteine placement was designed to bind the quinone ≈10 Å from the edge of the bound porphyrin. Fluorescence measurements confirmed that the bound hydroquinone is incorporated toward the protein's hydrophobic interior and is partially solvent-shielded. The bound quinones remain redox-active and can be oxidized and rereduced in a two-electron process at neutral pH. The semiquinone can be generated at high pH by a one-electron reduction, and the midpoint potential of this can be adjusted by ≈500 mV by binding different quinones to the protein. The heme-binding site of the modified cytochrome was then reconstituted with the chlorophyll analogue zinc chlorin e6. By using EPR and fast optical techniques, we show that, in the various chlorin–protein–quinone complexes, light-induced electron transfer can occur from the chlorin to the bound oxidized quinone but not the hydroquinone, with electron transfer rates in the order of 108 s–1.
Keywords: photosynthetic reaction center, artificial photosynthesis, chlorophyll analog, zinc chlorin, cysteine
All of the energy needs of plants and photoautotrophic bacteria are met by the light reactions of photosynthesis. The primary photochemistry involves the absorption of visible photons and the subsequent conversion of the absorbed energy into chemical potential energy by the formation of a charge-separated state. This photochemistry takes place in membrane-bound protein complexes called reaction centers (RCs). The best understood of these systems are the bacterial RCs from purple and green filamentous bacteria. The primary photochemistry that occurs in the bacterial RC is also common to the RC in photosystem II of cyanobacteria and higher plants (reviewed in refs. 1–3). The photochemical reactions in the RCs occur through the transfer of an electron from a light-excited (bacterio)chlorophyll [(B)Chl] through the protein, by means of a (bacterio)pheophytin, to a bound quinone molecule. The cofactors are ≈10 Å apart so that the electron transfer (ET) reaction is nonadiabatic, occurring through a quantum tunneling mechanism (4–6). This ET reaction has been mimicked by using organic “diad” molecules consisting of a covalently linked, light-activated electron donor, such as a porphyrin, and an electron acceptor, such as a quinone, for several decades with considerable success (7–10).
The protein environment plays a major role in modulating the redox properties of the cofactors (11, 12) and in the ET reaction (6, 13). It would then seem to be useful to use a protein-based system to mimic or study this photochemistry. However, although quinones are common biological redox-active cofactors, we are not aware of any previous work using them as bound electron donor/acceptors in designed or engineered proteins. As yet, there is no general structural motif for a quinone-binding site (14). The best characterized binding site is the QA site of the bacterial RC (15), which has recently been shown to be structurally similar to the QA site of photosystem II (16). Quinone binding is stabilized by hydrogen-bonding to the quinone carbonyl oxygen, van der Waal's contact between the protein and quinone head group and especially by hydrophobic binding of the large quinone phytol tail (17). The de novo design of a binding site that mimics all of the features of the natural system is far too complex. To simplify this, we took a different approach in which the quinone is bound covalently.
It has been long known that unsubstituted quinones such as para-benzoquinone (pBQ) form adducts with thiol-containing compounds (18) including cysteine (19). The reaction of cysteine and 2,3-dimethoxy-5-methyl-1,4-benzoquinone (CoQ0, for coenzyme Q0) (ubiquinone lacking the phytol tail) is thought to occur according to the following reaction scheme. Although the quinones associated with photosynthetic RCs are not covalently attached to the protein, this is not expected to have a major influence on the redox properties of the quinones (20). Most model protein systems previously used for ET have relied on the covalent attachment of one or both of the donor/acceptor molecules to the surface of the protein (21). As a result, the donor/acceptor molecules are not shielded from the high dielectric solvent. The solvent exposure is expected to influence the ET event as solvent reorganization energy will play a major role. By attaching the quinone to a cysteine within the interior of the protein, we are able to bury the donor and acceptor within the interior of the protein, and, thus, the route of ET is expected to be free of the solvent.
Figure 1.

The photosynthetic RCs all have a dimer or oligomer of excitonically coupled (B)Chl which, upon light excitation, donates an electron to a nearby acceptor. A principal reason for the use of multiple (B)Chl molecules is that their associated lower energy allows the effective trapping of light energy that is funneled in to the reaction center from higher-energy (B)Chls in the light-harvesting antennae systems. Although there is good reason for the use of multiple (B)Chl molecules in the natural systems, the added complexity of a model system that utilizes a (B)Chl dimer is counterproductive. We have previously demonstrated the possibility of using de novo-designed, heme-binding peptides to bind chlorophyll analogues, such as zinc chlorin e6 (Zn-Ce6), and showed light-induced ET from the bound Zn-Ce6 to various quinones in solution (22). These de novo-designed peptides are based on the four-helix bundle motif that is perhaps best described by the Escherichia coli cytochrome b562, and it is this protein that we have chosen to use in this work.
Cytochrome b562 is a small, soluble, four-helix-bundle protein with a single b-type heme cofactor originally described by Itagaki and Hagar (23). The protein has been well characterized both structurally (24, 25) and biophysically (see, for example, refs. 26 and 27). In b-type cytochromes, the heme is not covalently attached to the protein, which is the case in the c-type cytochromes. This noncovalent attachment means that the heme is easily removed and apocytochrome b562 can be reconstituted with a range of nonheme porphyrins (28). In this work we have been able to bind Zn-Ce6 to apocytochrome b562 and demonstrate ET from this bound chlorin to a quinone molecule bound in the engineered quinone binding site, thus mimicking the primary photochemistry of pheophytin/quinone-type RCs.
Materials and Methods
Sample Preparation. The construction of the cytochrome b562 H63N mutant has been described in ref. 29. The H63N-pET 30 Xa/LIC plasmid was point-mutated with the following primers: CAATTTAAAA GTGTGCGAAA AAGCGG and CCGCTTTTTC GCACACTTTT AAATTGTCGT TGAGGG. The I17C protein was expressed and purified as for other cytochrome b562 mutants described in ref. 29. About 30% purified protein had bound heme, which was removed by using acidification and methyl ethyl ketone extraction (30). Subsequently, heme absorbance was not detected in the apoprotein, which had Abs277 nm/Abs418 nm > 15. The protein was treated with DTT at pH 9.0 and then dialyzed against 100 mM KCl/1 mM sodium acetate, pH 5.0. The protein was generally considered pure and not cross-linked at this stage as judged by nonreducing SDS/PAGE. Except for binding titrations or hydroquinone fluorescence measurements, the quinones were bound to apo-I17C by adding a 5-fold molar excess of quinone to the protein at pH 7.0, which was allowed to react for ≥30 min with gentle mixing. The unbound quinone was then removed by dialysis. After dialysis there was no detectable unbound quinone as measured by EPR. All bound quinone was in the oxidized state as measured by absorbance and fluorescence. The apoprotein was reconstituted with freshly prepared hemin chloride, Zn-Ce6, or other porphyrins by standard literature methods (31, 32) essentially as described in ref. 29. Guanidine denaturation was also performed as described in ref. 29. All reagents were reagent grade. Zn-Ce6 was prepared as described in ref. 22, and the quinones were purchased from Sigma and used without further purification.
Redox Measurements. Chemical redox titrations were performed essentially as described by Dutton (33) and were made anaerobic by continuously flushing the sample with nitrogen. These experiments were performed with ≈100 μM CoQ0 or I17C–CoQ0 and 5 μM (each) phenazine methosulphate [midpoint potential (Em), +80 mV], diaminodurol (Em, +260 mV), pBQ (Em, +295 mV), and ferricyanide (Em, +430 mV) as mediators in 100 mM KCl/50 mM potassium phosphate, pH 7.0. Measurements were made against a KCl-saturated calomel electrode and a Pt counter electrode which was calibrated against 1:1 ferri/ferrocyanide. Data were fit to a Nernst curve,
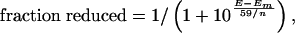 |
where E is the solution potential in mV versus the standard hydrogen electrode at room temperature and n is the number of electrons transferred in the reaction.
Fluorescence. Steady-state fluorescence spectra were acquired on a Spex FluoroMax-3 (Jobin Yvon Horiba, Edison, NJ). Fluorescence decays kinetics were obtained by the time correlated single photon counting technique as outlined in refs 34 and 35, with a Rhodamine 6G dye laser pumped by a SESAM (semiconductor saturable absorber mirror) mode-locked, diode-pumped, solid-state laser (Cheetah-X, Time-Bandwidth Products, Zurich) at ≈600 nm and with emission monitored at 650 nm. The observed decay curves were analyzed by nonlinear least squares iterative reconvolution into multicomponent exponential functions by using a repeated reconvolution program (34).
EPR and Molecular Modeling. X-band EPR spectra were acquired at room temperature on a ESP300E spectrometer equipped with a TM011 cavity (Bruker, Billerica, MA). Generally, the spectra were acquired at 1–4 mW microwave power with 1.0 G modulation amplitude and 100 KHz modulation frequency. Typically 5–50 scans were averaged. Samples contained 200–500 μM protein and were made anaerobic by the addition of glucose oxidase, glucose, and catalase. The light-minus-dark measurements were made essentially as described in ref. 22. A fiberoptic cable was used to illuminate the sample directly in the EPR cavity with saturating 10-μs flashes from an EG & G ElectroOptics (Salem, MA) flash lamp. The H63N and I17C mutations and pBQ-I17C complexes were modeled by energy minimization in a periodic box with the amber94 forcefield in hyperchem (Hypercube, Gainesville, FL).
Results and Discussion
Wild-type cytochrome b562 has a solvent-exposed histidine residue at position 67, which we previously replaced with an asparagine to create the H63N mutant (29). The rationale for this mutant was to prevent any adventitious ligation of porphyrins, such as Zn-Ce6, by this residue upon reconstitution of the apoprotein. H63N was then mutated at amino acid position 17, replacing isoleucine with a cysteine, which created the I17C mutant. This residue was chosen because it is not solvent-exposed in the holoprotein but reasonably accessible in the apoprotein when the porphyrin is removed. The equivalent residue in a structural homologue of cytochrome b562, cytochrome c prime, is tryptophan. A cysteine–benzoquinone conjugate is of similar size to tryptophan, so it was estimated that cytochrome b562 could accommodate the quinone in this position. Additionally, molecular modeling of the protein (Fig. 1) suggested that the edge-to-edge distance between the bound porphyrin and quinone would be ≈10 Å, which is roughly the average distance between cofactors in the photosynthetic RCs and a feasible distance over which electron tunneling can occur (13). The absorbance, redox, and EPR properties of the heme-bound holo-I17C protein are indistinguishable from those of the H63N or wild-type protein, suggesting that the I17C mutation has not disrupted the heme binding site. Approximately 30% of the purified protein contained bound heme, which was successfully removed by the method described in ref. 30, and no heme was detected in the apoprotein after this treatment. The apo-I17C protein had a tendency to cross-link during purification, which was reversed by treatment with DTT, indicating that the cysteine residue was at least partially solvent-exposed in the apoprotein as expected.
Fig. 1.

Molecular model of quinone-bound cytochrome b562 I17C showing the porphyrin-binding site and cysteine-bound benzoquinone.
Cysteine-reacted quinone has a characteristic absorbance spectra, with a peak at ≈303 nm (19, 20) which was used to monitor the titration of CoQ0 into apo-I17C (shown in Fig. 2). Quinone binding occurred with a concomitant loss of free thiol (cysteine) as measured by Ellman's reagent (data not shown), and no binding was observed when the apoprotein was cross-linked before the addition of the quinone. There was no cross-linking of the protein after reaction with CoQ0, and the loss of reduced cysteine was attributed to the covalent attachment of the quinone. The absorbance at ≈303 nm was thought to be caused by the doubly reduced (hydroquinone) cysteine–quinone conjugate (19) and was consistent with the reaction scheme outlined above. Although the sulfur addition to the quinone caused the reduction of the quinone to hydroquinone the bound quinone can be reoxidized. The 2-electron equilibrium oxidation/reduction of I17C-bound CoQ0 occured with an Em of +205 mV at pH 7 (Fig. 3). The Em of CoQ0 in solution was similarly measured to be +180 mV (Fig. 3), which is in reasonable agreement with reported values [+162 mV (36)]. Sulfur addition to benzoquinones has been shown to have little effect on their redox properties (reviewed in ref. 20); thus, the effect of the protein environment on the redox properties of CoQ0 appeared to be minimal (about +25 mV). No semiquinone was observed during either redox titration and the stability constant for the bound semiquinone is thought to be «1, as is the case for ubiquinone in solution (37). Because the Em of CoQ0 does not change substantially upon binding I17C, we assume that the one-electron reduction potential (E1) does not change significantly either. The E1 of ubiquinone-10 in water has been estimated to be –100 to –120 mV (38). The effect of a prenyl group (phytol tail) on E1 is similar to the effect of a methyl group (39), which lowers the E1 of pBQ by ≈80 mV (39, 40). Thus, we can estimate an E1 of –30 mV for CoQ0 in solution and a similar value for I17C-bound CoQ0. It may be possible to directly measure this value at high pH by using EPR.
Fig. 2.

Absorbance spectra of CoQ0 (dotted line) and the hydroquinone form of I17C-bound CoQ0 (solid line). (Inset) A titration of CoQ0 into 15 μM apo-I17C as measured by absorbance at 300 nm. All samples were in 50 mM potassium phosphate/100 mM KCl, pH 7.0.
Fig. 3.

Redox titration curves of I17C-bound CoQ0 (•) and CoQ0 in solution (□) as measured optically in 50 mM potassium phosphate/100 mM KCl, pH 7.0. The data were fit by a Nernst curve with Em of +205 mV (n = 1.9 ± 0.1) for I17C-bound CoQ0 and Em of +180 mV (n = 2.1 ± 0.1) for CoQ0 in solution. (Inset) Oxidized-minus-reduced absorbance difference spectrum of the I17C–CoQ0 titration.
Hydroquinone fluoresces in aqueous solution with an excitation peak at 290 nm and an emission peak at 330 nm (41). Upon the stoichiometric addition of pBQ to I17C, a fluorescent species was observed with these spectral attributes, confirming the formation of bound hydroquinone (see Fig. 7, which is published as supporting information on the PNAS web site). The binding of ferric heme to pBQ-bound I17C caused a ≈30% quenching of the hydroquinone fluorescence. Quenching of fluorescent protein side-chains, such as tryptophan, by heme through resonance energy transfer is well characterized (see ref. 42 and references within), and a similar mechanism is expected to occur for the hydroquinone. The fluorescence of bound hydroquinone was used to determine the extent of solvent shielding conferred by the protein by using sodium iodide as a quencher. Charged iodide will only quench solvent-exposed chromophores, and, thus, if the bound hydroquinone is buried within the protein, then quenching will not occur. Hydroquinone-bound glutathione (GS-pBQ), which also has a cysteine–quinone bond, was prepared as a minimal peptide–quinone model, in which the quinone is solvent-exposed. GS-pBQ has a Stern–Volmer quenching constant (KSV) of 1.9 ± 0.1 M–1, whereas native apo-I17C- pBQ (no heme bound) has a KSV of 0.69 ± 0.05 M–1(see Fig. 7). Denatured apo-I17C- pBQ (in 4 M guanidine) has a KSV of 2.0 ± 0.1 M–1 similar to GS-pBQ. These findings suggest that the native protein confers some solvent shielding to the quinone, which is lost upon denaturation of the protein in guanidine. The binding of pBQ to I17C does not significantly effect the stability of the ferric heme-bound protein toward denaturation by guanidine at pH 7.0 [ΔGH2O of 34 ± 3 and 31.0 ± 1kJ·mol–1 for heme-bound I17C and heme- and quinone-bound I17C, respectively (see Fig. 8, which is published as supporting information on the PNAS web site)]. The equivalent stability of H63N is 29 ± 3 kJ·mol–1 (29), which indicates that the I17C mutation is not significantly destabilizing to the cytochrome and quinone-binding is, likewise, minimally destabilizing.
The apo-I17C and CoQ0-bound protein can be reconstituted with the light-active chlorophyll analogue Zn-Ce6. We have previously shown that Zn-Ce6 can bind to de novo-designed peptides and is ligated by a single histidine, which causes a characteristic red-shifting of the absorbance bands (22). Because of the H63N mutation, the only remaining histidine in I17C is at position 102 and is one of the heme ligands within the binding pocket of the cytochrome. The absorbance spectra of Zn-Ce6 in solution and bound to CoQ0-bound I17C are shown in Fig. 4. The binding of Zn-Ce6 to apo-I17C is tight, with Kd ≈ 25 nM and a stoichiometry of 1:1. The absorbance spectra of Zn-Ce6 bound to I17C and to CoQ0-bound I17C (denoted Zn-Ce6–I17C–CoQ0) are indistinguishable, suggesting that there is no ground-state coupling between the chlorin and quinone. The fluorescence intensity of Zn-Ce6 bound to CoQ0-bound I17C increases by ≈20% when the quinone is reduced with dithionite (Fig. 4 Inset). Thus, in Zn-Ce6–I17C–CoQ0, there is a ≈20% quenching of the chlorin fluorescence by the quinone if it is oxidized. The quenching could be either due to Förster energy transfer (the absence of ground-state coupling rules out a Dexter mechanism) or ET. Energy transfer is not energetically feasible, and because the quenching is only observed in the presence of the oxidized quinone, which is a classic electron acceptor, we favored the later explanation of ET.
Fig. 4.

Absorbance spectra of Zn-Ce6 (dotted line) and Zn-Ce6–I17C–CoQ0 (solid line). (Inset) Fluorescence of Zn-Ce6–I17C–CoQ0 in the absence (solid line) and presence (dotted line) of sodium dithionite.
If there is light-induced ET from Zn-Ce6 to the quinone in Zn-Ce6–I17C–CoQ0, then the chlorin cation and semiquinone radicals will be formed upon irradiation of Zn-Ce6–I17C–CoQ0. These radicals can be detected by EPR. The room temperature X-band EPR spectra of CoQ0 in solution and bound to I17C at pH 9.0 are shown in Fig. 5. At pH 7.0, there is no detectable semiquinone consistent with the lack of any observed semiquinone during the potentiometry experiments (Fig. 3). The five-line spectrum of CoQ0 in solution (Fig. 5, spectrum D) is consistent with the semiquinone anion, whereas the four-line spectrum of I17C-bound CoQ0 (Fig. 5, spectrum C) is similar to that reported for the glutathione-bound semi-CoQ0 anion (43). The power saturation behavior of semi-CoQ0 does not change upon binding to apo-I17C (power of half saturation (P1/2) of semi-CoQ0, 27 ± 1 mW; P1/2 of semi-CoQ0-I17C, 25 ± 2 mW) (see Fig. 9, which is published as supporting information on the PNAS web site, and ref. 44). Under illumination, in the presence of an electron acceptor, Zn-Ce6 forms a porphyrin π cation (22), which is shown in Fig. 5 (spectrum A) for I17C-bound Zn-Ce6. The EPR spectrum of Zn-Ce6–I17C–CoQ0 bound to I17C under illumination at pH 7 is also shown in Fig. 5, spectrum B. In the dark, there is no detectable EPR signal from this sample. The spectrum in Fig. 5 (spectrum B) is a composite of the Zn-Ce6 cation and semi-CoQ0 anion and confirms that light-induced ET occurs from Zn-Ce6 to CoQ0. The power saturation behavior of the Zn-Ce6 cation changes in the presence of the bound semiquinone, with the P1/2 increasing from 86 ± 4 to 198 ± 12 mW (see Fig. 9). This relaxation is likely due to the close proximity of the semiquinone radical, which is acting as a relaxer. The distance, r, between Zn-Ce6+ and the relaxer is related to the power saturation relaxation by ΔP1/2 ∝ S2/r6 (45), where S is the spin of Zn-Ce6+ (S = 1/2). To achieve a ΔP1/2 of >100 mW, r must be quite small, which is consistent with the model shown in Fig. 1, where the quinone is ≈10 Å from the bound chlorin. The ET kinetics in Zn-Ce6–I17C–CoQ0 were investigated by using time-resolved EPR of the light-induced Zn-Ce6 cation, but the forward and back reactions were not resolved because of instrument limitations (≈20-μs resolution).
Fig. 5.

Room temperature X-band EPR spectra. Spectrum A is a light-minus-dark spectrum of the I17C-bound Zn-Ce6 cation; spectrum B is a light-minus-dark spectrum of Zn-Ce6–I17C–CoQ0 at pH 7; spectrum C shows I17C-bound semi-CoQ0 (no chlorin) at pH 9; and spectrum D shows semi-CoQ0 in solution at pH 9.0. Note that the dotted lines allow the visual alignment of the semiquinone contribution to spectrum B.
To resolve the rate of the forward ET reaction, the Zn-Ce6 fluorescence lifetime was measured. The fluorescence lifetime of I17C-bound Zn-Ce6 decreased in the presence of bound CoQ0 (Fig. 6 and Table 1), which allowed the calculation of the rate of ET (kET)of8.1 × 107 s–1. If the quinone in Zn-Ce6–I17C–CoQ0 is reduced with dithionite, the lifetime of bound Zn-Ce6 increases to a similar value to that of quinoneless I17C-bound Zn-Ce6. This result is consistent with an absence of ET to the bound hydroquinone, which is not typically an electron acceptor. The efficiency of the ET reaction is ≈20% and in good agreement with steady-state fluorescence quenching (Fig. 4). The effect of the driving force (ΔG0) on this ET reaction was investigated by measuring the fluorescence decay kinetics of this system when CoQ0 was substituted for several other quinones. The E1 values of the parent quinones chosen spanned >500 mV in water; yet only a ≈2.5-fold change in kET was observed (Table 1). In Marcus theory, kET ∝ exp[–(ΔG0 + λ)2/λ], where λ is the reorganization energy (reviewed in ref. 5). In the Zn-Ce6–I17C–quinone system, the electron is transferred from the excited state of Zn-Ce6, which has an energy (E0,0) of 1.91 eV (1 eV = 1.602 × 10–19 J), the average energy of the QY absorbance band and fluorescence emission band (5). The driving force can be approximated as ΔG0 = ΔE – E0,0, where ΔE is the difference between the oxidation potential of Zn-Ce6 and the E1 of the quinone. Attempts to measure the oxidation potential of I17C-bound Zn-Ce6 were hampered by its apparently high value. The oxidation potential of Zn-Ce6 should be similar to that of Zn-Chl a, which is about +1.1 V (46). By using the E1 values in Table 1 and an oxidation potential of I17C-bound Zn-Ce6 of + 1.1 V, we calculated a range of ΔG0 values from about –0.7 to –1.3 eV (Table 1). The driving force is thus large, and the small change in kET suggests that the ET reactions occurred toward the top of a broad (large λ) Marcus curve (where –ΔG0 ≈ λ and kET is maximal). The data can be fit to Dutton's ruler (47), an empirical Marcus relationship relating kET to ΔG0, λ, and the edge-to-edge cofactor distance in a protein. The fit is poor but yields λ ≈ 1eV and the distance between Zn-Ce6 and CoQ0 ≈ 11 Å (see Fig. 10, which is published as supporting information on the PNAS web site) in good agreement with the molecular model shown in Fig. 1.
Fig. 6.

Zn-Ce6 fluorescence decay curves for I17C-bound Zn-Ce6 (curve A), Zn-Ce6–I17C–CoQ0 (curve B), and dithionite-reduced Zn-Ce6–I17C–CoQ0 (curve C). The instrument response (curve D) is also shown. The Zn-Ce6 lifetimes are given in Table 1.
Table 1. Zn-Ce6 fluorescence lifetimes, τ, kET, and estimated E1 and ΔG0 values for some Zn-Ce6–I17C–quinone complexes.
| Quinone | τ, ns | kET, s-1 | E1, mV | ΔG0, eV |
|---|---|---|---|---|
| None | 3.01 | — | — | — |
| CoQ0 | 2.42 | 8.1 × 107 | -30 | -0.78 |
| CoQ0 reduced | 3.07 | 0 | — | — |
| DCBQ | 2.50 | 6.8 × 107 | +470 | -1.28 |
| pBQ | 2.47 | 7.3 × 107 | +99 | -0.91 |
| 2,5-DMBQ | 1.97 | 1.8 × 108 | -66 | -0.74 |
| 2,6-DMBQ | 2.18 | 1.3 × 108 | -88 | -0.73 |
kET values were calculated from the fluorescence lifetime by kET = τ-1 (Zn-Ce6-I17C-quinone) - τ-1 (Zn-Ce6-I17C). E1 values are for parent quinones in water at pH 7 and were taken from ref. 40 or (for CoQ0) described in the text. DCBQ, 2,6-dichloro-1,4-benzoquinine; DMBQ, dimethyl-1,4-benzoquinone.
Conclusions
The design reported here not only offers another option for the study of protein ET reactions but could also be utilized to mimic quinoproteins and quinohemoproteins, which are emerging as an important class of industrially useful enzymes (48). It is clear that the protein environment around the bound quinone in I17C is significantly different from that of the QA-binding site in the bacterial RC and photosystem II. The modification of I17C by introducing neighboring residues capable of forming hydrogen bonds with carbonyl oxygens of the bound quinone or π-stacking with the quinone head group should create a more native-like system. Detailed structural information is also required to better understand the effect of quinone binding on the structure of the protein. The crystallization of a heme–protein–quinone complex for x-ray diffraction studies is one method of obtaining such information. The efficiency of ET in the various Zn-Ce6–I17C–quinone complexes is 15–35%, but the ability to harness this energy depends on how well the charge-separated state can be stabilized. The consequence of having such a large ΔG0 is the back reaction is expected to occur near the top of the Marcus curve, which causes a short-lived charge-separated state. This situation is consistent with τ < 20 μs, as measured by EPR. Despite our attempts to bury the quinone within the interior of the protein, it remains partially solvent-exposed. As a result, the reorganization energy associated with the ET reaction is large. We evaluated the reorganization energy by using continuum electrostatics calculations [as per the method of Sharp (49)] and the solvent reorganization energy has been estimated as ≈1.2 eV. The reorganization energy associated with the reduction of the quinone, estimated by using standard quantum chemical techniques, was ≈0.3 eV, and, because changes in bond lengths in the chlorin are assumed to be negligible during chlorin oxidation, the total reorganization energy is ≈1.5 eV. It may be possible to reduce λ by further shielding the bound quinone from the solvent in site-directed mutants, but it is possible that cytochrome b562 is simply too small to bury the cofactors far enough away from the solvent to prevent wide-spread solvent reorganization during the ET reaction. Despite its limitations, we have a working model of a reaction center. By using a protein scaffold, it is possible to investigate the role of the protein in a type of ET reaction previously only studied in organic molecules or large natural RCs.
Supplementary Material
Acknowledgments
We thank Karin Ahrling and Reza Razeghifard for useful discussions. This work was supported in part by Australian Research Council Discovery Project Grant DP045021. S.H. was supported by an Australian National University Graduate School Scholarship. B.B.W. was supported by an Australian Postgraduate Award.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: RC, reaction center; CoQ0, 2,3-dimethoxy-5-methyl-1,4-benzoquinone; E1, quinone one-electron reduction potential; ET, electron transfer; pBQ, para-benzoquinone; Zn-Ce6, zinc-chlorin e6; Em; midpoint potential; (B)Chl, (bacterio)chlorophyll; KSV, Stern–Volmer quenching constant.
References
- 1.Allen, J. P. & Williams, J. C. (1998) FEBS Lett. 438, 5–9. [DOI] [PubMed] [Google Scholar]
- 2.Diner, B. A. & Rappaport, F. (2002) Annu. Rev. Plant Biol. 53, 551–580. [DOI] [PubMed] [Google Scholar]
- 3.Okamura, M. Y., Paddick, M. L, Graige, M. S. & Feher, G. (2000) Biochim. Biophys. Acta 1458, 148–163. [DOI] [PubMed] [Google Scholar]
- 4.DeVault, D., Parkes, J. H. & Chance, B. (1967) Nature 215, 642–624. [DOI] [PubMed] [Google Scholar]
- 5.Marcus, R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811, 265–322. [Google Scholar]
- 6.Winkler, J. R., Di Bilio, A. J., Farrow, N. A., Richards, J. H. & Gray, H. B. (1991) Pure Appl. Chem. 71, 1753–1764. [Google Scholar]
- 7.Ho, T.-F., McIntosh, A. R. & Bolton, J. R. (1980) Nature 286, 254–256. [Google Scholar]
- 8.Kurreck, H. & Huber, M. (1995) Angew. Chem. Int. Ed. Engl. 34, 849–866. [Google Scholar]
- 9.Gust, D., Moore, T. A. & Moore, A. L. (1993) Acc. Chem. Res. 26, 198–205. [Google Scholar]
- 10.Gust, D., Moore, T. A. & Moore, A. L. (2001) Acc. Chem. Res. 34, 40–48. [DOI] [PubMed] [Google Scholar]
- 11.Ivancich, A., Artz, K., Williams, J. C., Allen, J. P. & Mattioli, T. A. (1998) Biochemistry 37, 11812–11820. [DOI] [PubMed] [Google Scholar]
- 12.Rinyu, L., Martin, E. W., Takahashi, E., Maroti, P. & Wraight, C. A. (2004) Biochim. Biophys. Acta 1655, 93–101. [DOI] [PubMed] [Google Scholar]
- 13.Page, C. C., Moser, C. C., Chen, X. & Dutton, P. L. (1999) Nature 402, 47–52. [DOI] [PubMed] [Google Scholar]
- 14.Fisher, N. & Rich, P. R. (2000) J. Mol. Biol. 296, 1153–1162. [DOI] [PubMed] [Google Scholar]
- 15.Allen, J. P., Feher, G., Yeates, T. O., Komiya, H. & Rees, D. C. (1988) Proc. Natl. Acad. Sci. USA 85, 8487–8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreira, K. N., Iverson, T. M., Maghlaoui, K., Barber, J. & Iwata, S. (2004) Science 303, 1831–1838. [DOI] [PubMed] [Google Scholar]
- 17.Warncke, K., Gunner, M. R., Braun, B. S., Gu, L., Yu, C.-A., Bruce, J. M. & Dutton, P.L. (1994) Biochemistry 33, 7830–7841. [DOI] [PubMed] [Google Scholar]
- 18.Snell, J. M. & Weissberger, A. (1939) J. Am. Chem. Soc. 61, 450–453. [Google Scholar]
- 19.Redfearn, E. R. (1965), in Biochemisty of Quinones, ed. Morton, R. A. (Academic, New York), pp. 149–181.
- 20.Brunmark, A. & Cadenas, E. (1989) Free Radic. Biol. Med. 7, 435–477. [DOI] [PubMed] [Google Scholar]
- 21.Bjerrum, M. J., Casimiro, D. R., Chang, I. J., Di Bilio, A. J., Gray, H. B., Hill, M. G., Langen, R., Mines, G. A., Skov, L. K., Winkler, J. R. & Wuttke, D. S. (1995) J. Bioenerg. Biomembr. 27, 295–2930. [DOI] [PubMed] [Google Scholar]
- 22.Razeghifard, M. R. & Wydrzynski, T. (2003) Biochemistry 42, 1024–1034. [DOI] [PubMed] [Google Scholar]
- 23.Itagaki, E. & Hagar, L. P. (1966) J. Biol. Chem. 241, 3687–3695. [PubMed] [Google Scholar]
- 24.Hamada, K., Bethge, P. H. & Mathews, F.S. (1995) J. Mol. Biol. 247, 947–962. [DOI] [PubMed] [Google Scholar]
- 25.Arnesano, F., Banci, L., Bertini, I., Faraone-Mennella, J., Rosato, A., Barker, P. D. & Fersht, A. R. (1999) Biochemistry 38, 8657–8670. [DOI] [PubMed] [Google Scholar]
- 26.Moore, G. R., Williams, R. J. P., Peterson, J., Thomson, A. J. & Mathews, F. S. (1985) Biochim. Biophys. Acta 829, 83–96. [DOI] [PubMed] [Google Scholar]
- 27.Robinson, C. R., Liu, Y., Thomson, A. J., Sturtevant, J. M. & Sligar, S. G. (1997) Biochemistry 36, 16141–16146. [DOI] [PubMed] [Google Scholar]
- 28.Ishida, Y., Konishi, K., Aida, T. & Nagamune, T. (1998) Chem. Eur. J. 4, 1148–1152. [Google Scholar]
- 29.Hay, S. & Wydrzynski, T. (2004) Biochemistry, in press. [DOI] [PubMed]
- 30.Teale, F. W. J. (1959) J. Biochem. Biophys. Acta 35, 543. [DOI] [PubMed] [Google Scholar]
- 31.Gibney, B. R. & Dutton, P. L. (2001) in Advances in Inorganic Chemistry, eds. Mauk, A. & Sykes, A. (Academic, New York), Vol 51, pp. 409–455. [Google Scholar]
- 32.Lombardi, A., Nastri., F. & Pavone, V. (2001) Chem. Rev. 101, 3165–3189. [DOI] [PubMed] [Google Scholar]
- 33.Dutton, P. L. (1978) Methods Enzymol. 54, 411–435. [DOI] [PubMed] [Google Scholar]
- 34.Ghiggino, K. P. & Smith, T. A. (1993) Prog. React. Kinet. Mech. 18, 375–436. [Google Scholar]
- 35.Sisido, M., Hoshino, S., Kusano, H., Kuragaki, M., Makino, M., Sasaki, H., Smith, T. A. & Ghiggino, K. P. (2001) J. Phys. Chem. B 105, 10407–10415. [Google Scholar]
- 36.Lemma, E., Unden, G. & Kroger, A. (1990) Arch. Microbiol. 155, 62–67. [DOI] [PubMed] [Google Scholar]
- 37.Hastings, S. F., Kaysser, T. M., Jiang, F., Salerno, J. C., Gennis, R. B. & Ingledew, W. J. (1998) Eur. J. Biochem. 255, 317–323. [DOI] [PubMed] [Google Scholar]
- 38.Wraight, C. A. (1998) in Photosynthesis: Mechanisms and Effects, ed. Garab, G. (Kluwer, Dordrecht, The Netherlands), Vol 2, pp. 693–689. [Google Scholar]
- 39.Prince, R. C., Dutton, P. L. & Bruce, J. M. (1983) FEBS Lett. 160, 273–276. [Google Scholar]
- 40.Depew, M. C. & Wan, J. K. S. (1988) in The Chemistry of the Quinonoid Compounds, eds. Patai, S. & Rappoport, Z. (Wiley, New York), Vol. 2, pp. 899–962. [Google Scholar]
- 41.Tchaikovskaya, O. N., Sokolova, I. V., Kuznetsova, R. T., Swetlitchnyi, V. A., Kopylova, T. N. & Mayer, G. V. (2000) J. Fluoresc. 10, 403–408. [Google Scholar]
- 42.Willis, K. J., Szabo, A. G., Zuker, M., Ridgeway, J. M. & Alpert, B. (1990) Biochemistry 29, 5270–5275. [DOI] [PubMed] [Google Scholar]
- 43.Guo, Q., Corbett, J. T., Yue, G., Fann, Y. C., Qian, S. Y., Tomer, K. B. & Mason, R. P. (2002) J. Biol. Chem. 277, 6104–6110. [DOI] [PubMed] [Google Scholar]
- 44.Rupp, H., Rao, K. K., Hall, D. O. & Cammack, R. (1978) Biochim. Biophys. Acta 537, 255–269. [DOI] [PubMed] [Google Scholar]
- 45.Abragam, A. & Bleaney, B. (1986) Electron Paramagnetic Resonance of Transition Ions (Dover, Mineola, NY).
- 46.Watanabe, T. & Kobayashi, M. (1991) in Chlorophylls, ed. Scheer, H. (CRC, Boca Raton, FL), pp. 288–315.
- 47.Moser, C. C. & Dutton, P. L. (1992) Biochim. Biophys. Acta 1101, 171–176. [PubMed] [Google Scholar]
- 48.Anthony, C. (1996) Biochem. J. 320, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharp, K. A. (1998) Biophys. J. 74, 1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}