

Summary
In mammalian cells, the second messenger (2′-5′, 3′-5′) cyclic GMP-AMP (2′, 3′-cGAMP), is produced by the cytosolic DNA sensor cGAMP synthase (cGAS), and subsequently bound by stimulator of interferon genes (STING) to trigger interferon response. Thus, the cGAS-cGAMP-STING pathway plays a critical role in pathogen detection, as well as pathophysiological conditions including cancer and autoimmune disorders. However, studying and targeting this immune signaling pathway has been challenging due to the absence of tools for high-throughput analysis. We have engineered an RNA-based fluorescent biosensor that responds to 2′, 3′-cGAMP. The resulting “mix-and-go” cGAS activity assay shows excellent statistical reliability as a high-throughput screening (HTS) assay and distinguishes between direct and indirect cGAS inhibitors. Furthermore, the biosensor enables quantitation of 2′, 3′-cGAMP in mammalian cell lysates. We envision this biosensor-based assay as a resource to study the cGAS-cGAMP-STING pathway in the context of infectious diseases, cancer immunotherapy, and autoimmune diseases.
In Brief (eTOC Blurb)
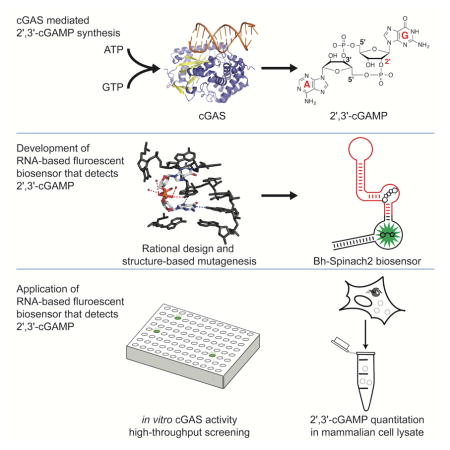
Bose et. al. developed a fluorescent biosensor for the mammalian immune signal 2′, 3′-cGAMP that enables high-throughput screening assays and quantitation of cGAS enzyme activity. Biosensor-based assays revealed that DNA intercalators indirectly modulate cGAS activity and quantitated the amount of 2′, 3′-cGAMP produced in DNA-stimulated L929 cells.
Introduction
The mammalian innate immune system uses pattern recognition receptors (PRRs) to sense extracellular or intracellular pathogens by recognizing pathogen associated molecular patterns (PAMPs) to trigger the proper immune response. Nucleic acid sensors are one important class of PRRs that recognize foreign DNA or RNA upon microbial infection and other patho-physiological conditions. The cGAS/STING pathway was recently discovered as an important cytosolic immune surveillance pathway (Ishikawa and Barber, 2008; Sun et al., 2013). cGAS is a universal DNA sensor that is activated upon binding to cytosolic DNA to produce the signaling molecule (2′-5′, 3′-5′) cyclic GMP-AMP (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013b). Acting as a second messenger during microbial infection, 2′, 3′-cGAMP binds and activates STING, leading to production of type I interferon (IFN) and other co-stimulatory molecules that trigger the immune response (Ahn et al., 2014; Gao et al., 2013a; Hansen et al., 2014; Watson et al., 2015). STING also has been shown to recognize bacterial-derived cyclic dinucleotides and elicit the type I IFN response (Barker et al., 2013; Burdette et al., 2011). Natural variants in human STING have differential specificity in recognizing cyclic dinucleotides. While human R232 STING protein (and the mouse R231 counterpart) can be stimulated by 2′, 3′-cGAMP, bacterial 3′, 3′-cGAMP and bacterial c-di-GMP (Gao et al., 2013a), human H232 STING is only responsive to 2′, 3′-cGAMP (Diner et al., 2013; Gao et al., 2013a; Zhang et al., 2013).
Besides its role in infectious disease, the cGAS/STING pathway has emerged as a promising new target for cancer immunotherapy and autoimmune diseases (Ahn et al., 2015; Baird et al., 2016; Corrales and Gajewski, 2015; Corrales et al., 2015; Curran et al., 2015; Demaria et al., 2015; Deng et al., 2014; Woo et al., 2015b; Woo et al., 2014). DNA fragments present in the tumor microenvironment are proposed to activate cGAS in dendritic cells (DC), followed by IFN-induced DC maturation and activation of a potent and beneficial immune response against cancer cells (Woo et al., 2015a). In a separate context, dysregulation of the cGAS/STING pathway has been implicated in self DNA triggered inflammatory and autoimmune disorders, such as systemic lupus erythematosus (SLE) and Aicardi-Goutieres syndrome (Ahn et al., 2012; Ahn et al., 2014; Gao et al., 2015).
With increasing evidence that the cGAS/STING pathway is involved in immune responses to microbial pathogens and cancer cells, and may play a role in autoimmune disorders, it is critical to explore its regulation and activation status. However, deconvoluting the role of the cGAS/STING pathway in immune responses is made difficult by the presence of multiple surveillance pathways that trigger the same IFN signal downstream. For example, one of the most commonly used and sensitive methods to detect activation of the cGAS/STING pathway is a luciferase reporter fused to an IFN-β promoter (Diner et al., 2013). However, other DNA sensors (e.g. TLR9), RNA sensors (e.g. RIG, MDA5) and immune modulators also activate expression of this reporter as all of them activate downstream IFN production (Shrivastav and Niewold, 2013). Additionally, the assay requires transfection of the reporter DNA, and this foreign DNA can activate cGAS and other DNA sensing pathways, which can mask the underlying physiology. Thus, it would be advantageous to have a direct method of detecting and quantifying 2′, 3′-cGAMP rather than downstream signals, for diagnostic purposes and for clearly distinguishing cGAS/STING from other nucleic acid sensing pathways, in order to determine its contribution to the overall immune response and expand our understanding of its regulation.
Furthermore, a detection method for 2′, 3′-cGAMP that is adaptable to a high-throughput screening format is desirable for biomedical applications, as efforts are underway to develop small molecule modulators of the cGAS/STING pathway for cancer immunotherapy and vaccine development. While current efforts have focused on targeting STING with natural and unnatural cyclic dinucleotides and the small molecule DMXAA (Baird et al., 2016; Chandra et al., 2014; Corrales et al., 2015; Demaria et al., 2015; Deng et al., 2014; Downey et al., 2014; Kobayashi et al., 2015; Nakamura et al., 2015; Zhang et al., 2015), cGAS may be an advantageous drug target because activating the enzyme can produce an amplified signal relative to the STING receptor-small molecule interaction. cGAS also may be an attractive target for small molecule inhibition in the case of autoimmune diseases.
Unfortunately, current in vitro methods to detect 2′, 3′-cGAMP have specific limitations to their utility. Thin layer chromatography (TLC) assays utilizing radioactive nucleotide substrates to detect 2′, 3′-cGAMP produced by cGAS in vitro cannot be used to quantify endogenous 2′, 3′-cGAMP levels inside the cell or in cell lysates. Liquid chromatography-mass spectrometry (LC-MS) has been used to detect cyclic dinucleotides in bacterial cell extracts (Burhenne and Kaever, 2013; Waters, 2010) and mammalian cell extracts (Wu et al., 2013). Importantly, however, neither of these methods are readily adapted to high-throughput screening formats, nor offer the potential for live cell imaging.
Both fundamental and applied studies of the cGAS/STING immune signaling pathway would greatly benefit from a direct and high-throughput method to assay cGAS activity. However, the natural protein receptor for 2′, 3′-cGAMP, STING, is poorly suited for engineering a fluorescent biosensor, because it functions as a homodimer and its structure precludes facile connection of the protein chains or circular permutation. Overexpression of STING-based constructs in vivo also may activate downstream responses, which would be an undesired physiological effect. To our knowledge, no other receptors for 2′, 3′-cGAMP have been reported.
Here we describe the development of fluorescent biosensors that exhibit turn-on response to 2′, 3′-cGAMP. We designed the biosensors by making rational mutations to natural riboswitch aptamers of the GEMM-II class that recognize the related molecule 3′, 3′-cyclic di-GMP (c-di-GMP) (Lee et al., 2010). Using one of these 2′, 3′-cGAMP biosensors, we showed direct detection of cGAS enzymatic activity by fluorescence readout in a 384-well format, fluorescence-based detection of overexpressed cGAS activity in bacterial lysates by plate-reader and in live bacteria by flow cytometry, and fluorescence-based quantitation of endogenous cGAS activity in DNA-stimulated lysates from the L929 mammalian cell line. Our in vitro enzymatic assays showed that nucleic acid intercalators can indirectly inhibit cGAS activity. Finally, analysis of mammalian cell lysates revealed that 60 attomoles, or 6 million molecules of 2′, 3′-cGAMP, are produced on average per cell upon DNA stimulation in the L929 cell line.
Results
Engineering a fluorescent biosensor to sense 2′, 3′-cGAMP
Previously, our lab developed RNA-based fluorescent biosensors selective for c-di-GMP or 3′, 3′-cGAMP based on natural GEMM-I riboswitches (Kellenberger et al., 2015; Kellenberger et al., 2013). However, these biosensors did not respond at all to 2′, 3′-cGAMP, even though some of the parent riboswitch aptamers exhibited very high specificity and affinity to 3′, 3′-cGAMP (Kellenberger et al., 2015; Ren et al., 2015). X-ray crystal structures of ligand-bound GEMM-I riboswitches showed extensive interactions with the 2′-hydroxyls and phosphodiester oxygens of c-di-GMP or 3′, 3′-cGAMP (Fig. S1A, B) (Ren et al., 2015; Shanahan et al., 2011; Smith et al., 2011). These interactions would be disrupted by the 2′-5′ linkage present in 2′, 3′-cGAMP.
We instead reasoned that the GEMM-II riboswitch scaffold may be more amenable for engineering a 2′, 3′-cGAMP biosensor (Fig. 1A). The canonical ligand for this riboswitch class is c-di-GMP, however x-ray crystal structures showed that there are few canonical Watson-Crick or Hoogsteen pairing interactions between ligand nucleobases and the riboswitch, and few hydrogen bonds between the ligand backbone and the riboswitch (Fig. S1A, B). This suggests that GEMM-II may have some flexibility in base recognition. Furthermore, the GEMM-II riboswitch aptamer from Clostridium acetobutylicum has been reported to accept c-di-GMP analogs with modifications to the ribose and phosphates (Shanahan et al., 2011; Smith et al., 2011), which was promising for tolerating changes to the backbone linkage.
Figure 1. Development of RNA-based fluorescent biosensors for 2′, 3′-cGAMP.

(A) Chemical structures of 3′, 3′-cGAMP, 2′, 3′-cGAMP, and DFHBI. Secondary structure model of GEMM-II riboswitch-based biosensors that detect 2′, 3′-cGAMP. Key mutations that led to first-generation biosensors are boxed and labeled. (B) In vitro fluorescence activation and binding affinity measurements for RNA-based biosensors with 2′, 3′-cGAMP. (C) Analysis of the effect of binding pocket mutations on biosensor response to different cyclic dinucleotides. (D) Hydrogen bonding interactions between the GEMM-II riboswitch and Gα and Gβ of c-di-GMP (adapted from (Shanahan et al., 2011)) and proposed effect of G103A mutation on recognition of the adenine in 2′, 3′-cGAMP. Error bars indicate standard deviations for three independent replicates.
Four characterized GEMM-II riboswitches (Lee et al., 2010; Smith et al., 2011) were picked to design 16 biosensor candidates with varied length transducer stems derived from the natural riboswitch P1 stems (Table S1). These biosensors were initially screened for fluorescence turn-on and binding affinity to c-di-GMP, the canonical ligand (Fig. S1C), then the four most promising biosensors were further profiled for response to c-di-GMP, 3′, 3′-cGAMP, and 2′, 3′-cGAMP (Fig. S1D). One of the candidates that incorporated a GEMM-II riboswitch from Bacillus halodurans C-125, Bh P1-5 delC, exhibited fluorescence response to micromolar concentrations of 2′, 3′-cGAMP. The name indicates that the riboswitch portion of the biosensor is fused to the Spinach2 aptamer via a 5 base-paired P1 stem with deletion of a single C present in the natural stem bulge (Fig. 1A).
While Bh P1-5 delC showed good affinity for 2′, 3′-cGAMP (apparent dissociation constant or KD = 13.4 ± 0.9 μM) at 37 °C, 3 mM Mg2+ (Fig. 1B), the related Bh P1-6 showed lower background fluorescence and higher activation (5-fold) with c-di-GMP (Fig. S1C, D), which allowed us to analyze the effect of mutations on ligand selectivity. Two positions in the riboswitch binding pocket that make direct contacts with the ligand nucleobases were mutagenized and the fluorescence response to 2′, 3′-cGAMP was examined (Fig. 1C, D). Whereas mutations to A99, the position that recognizes Gα of c-di-GMP, did not improve response to 2′, 3′-cGAMP, we observed fluorescence activation by 2′, 3′-cGAMP with G103A, which mutates the position that recognizes Gβ of c-di-GMP.
The effect of the G103A mutation can be rationalized as establishing a hydrogen bond with the A base of 3′, 3′-cGAMP or 2′, 3′-cGAMP, and disfavoring interaction with the G base of c-di-GMP. Accordingly, we found that the cyclic dinucleotide selectivity profile for Bh P1-6 G103A biosensor was switched relative to wild-type Bh P1-6 (Table S2). Binding affinity to 3′, 3′-cGAMP was increased by more than 10-fold (KD from >11 μM to 0.8 μM), whereas binding affinity to c-di-GMP was reduced by more than 125-fold (KD from 56 nM to >7 μM). Furthermore, improved binding extended to 2′, 3′-cGAMP, such that 200 nM of Bh P1-6 G103A showed 5-fold fluorescence activation in response to 20 μM ligand, whereas the wild-type biosensor had shown no response (Fig. 1B, S1).
We had expected that the same G103A mutation could be applied to the Bh P1-5 delC biosensor, but disappointingly, it resulted in complete loss of biosensor function (Fig. S1E). High fluorescence was observed even in the absence of ligand, which implies that this single nucleotide substitution, in conjunction with deleting one nucleotide in the stem bulge, favors formation of a stable transducer stem in the absence of ligand. Nevertheless, by combining rational design and structure-based mutagenesis, we have developed two fluorescent biosensors capable of detecting 2′, 3′-cGAMP at micromolar concentrations. A detailed comparison of their binding affinities and fluorescent turn-on properties is given in Table S2. Similar to STING, the native protein receptor for 2′, 3′-cGAMP, the biosensors also respond to the bacterial second messengers c-di-GMP and 3′, 3′-cGAMP, but otherwise exhibit high selectivity against other metabolites such as ATP and GTP (Fig. 2A). Importantly, whereas STING is a homodimeric protein that is not readily adaptable as a fluorescent biosensor, we have generated a first-generation fluorescent biosensor for 2′, 3′-cGAMP based on the GEMM-II riboswitch scaffold.
Figure 2. Application of fluorescent biosensor to cGAS enzyme activity assays.

(A) Bh P1-6 G103A biosensor detects 2′, 3′-cGAMP produced in the cGAS enzyme reaction (black). Enzyme reactions that lack one component served as negative controls (white). (B) In situ detection of cGAS activity by the RNA-based fluorescent biosensor. (C) Determination of the Z′ factors for cGAS activity assay using RNA-based biosensor in 384-well format. Error bars indicate standard deviations for three (A) or 16 (C) independent replicates.
Assaying cGAS activity in vitro using a fluorescent biosensor
Currently, the established method to assay cGAS activity in vitro is via thin layer chromatography analysis of radiolabeled nucleotides (Fig. S2A) (Diner et al., 2013). Although it is accurate and highly sensitive, this method uses radioactive materials, which poses increased safety concerns and hampers adaptation as a high-throughput screening assay. These issues could be overcome by using fluorescent biosensors to assay cGAS enzyme activity.
We analyzed enzymatic activity of the DNA sensor cGAS using the Bh P1-6 G103A biosensor, which has lower background fluorescence and higher turn-on. Upon binding to DNA, cGAS is activated to catalyze the 2′-5′ phosphodiester bond formation between GTP and ATP, followed by cyclization to form the 3′-5′ bond (Ablasser et al., 2013). As expected, the biosensor gives strong fluorescence signal only when all components of the reaction are present to enable robust production of 2′, 3′-cGAMP (Fig. 2A). Besides carrying out assays in a standard mix-and-go format, in which the enzyme reactions are performed separately from the biosensor detection reactions, we also showed that the biosensor can be employed for direct in situ detection, e.g. the biosensor functions in the enzymatic reaction buffer (Fig. 2B).
Minor fluorescence increases were also detected in control reactions without DNA or without ATP. The former was due to incomplete removal of DNA during purification of cGAS enzyme, resulting in the observed basal activation. The latter could be caused by the formation of either pppGp(2′-5′)G or 2′, 3′-c-di-GMP, which have been previously reported as minor products in the absence of ATP (Ablasser et al., 2013). We did not test biosensor response to these side products because of the difficulty in obtaining sufficient amounts for binding measurements, however the signal-to-background ratio is still quite good even when comparing the enzyme reactions with and without ATP (4.2x).
Due to the involvement of the cGAS/STING pathway in multiple pathophysiological conditions, a high-throughput screen for its modulators is highly desirable. Since the biosensor-based fluorescent assay can be performed in a 384-well plate and analyzed in a fluorescent plate reader, it is readily adapted to high-throughput screening for activators or inhibitors of cGAS. With 1.5 μM concentration of cGAS, we determined a Z′ factor of 0.83 and signal-to-background ratio of 8.2x for the assay in this format, where a Z′ factor >0.5 is considered an assay with excellent statistical reliability for HTS (Fig. 2C) (Zhang et al., 1999). In order to screen for high affinity inhibitor or activator compounds, the assay would need to be performed with nanomolar concentrations of enzyme. We obtained a Z′ factor of 0.51 and signal-to-background ratio of 4.5x using the RNA biosensor, 300 nM enzyme, and manual pipetting of all reagents in 384-well format (Fig. 2C). Thus, we expect that the current fluorescent biosensor enables HTS assays for moderate affinity (e.g. 300–500 nM dissociation constant) compounds that affect cGAS activity.
Revealing a limitation of the biosensor assay and an alternative mechanism of small molecule inhibition of cGAS
Quinacrine, a well-characterized anti-malarial drug, has been reported to inhibit cGAS activity, with an IC50 value of 13 μM measured by the radiolabeled TLC assay (An et al., 2015). We attempted to demonstrate inhibitor characterization using our fluorescent biosensor assay (Fig. S2B), but a control experiment revealed that the decrease in fluorescence signal was independent of enzyme activity (Fig. 3A). Quinacrine is a known DNA intercalator (Boer et al., 2009) and we reasoned that it may disrupt biosensor function by intercalating into the RNA helices (Sinha et al., 2007). This appeared to be the case, as we showed that quinacrine also decreased fluorescence of the dye-binding Spinach aptamer (Fig. 3A). This observation reveals an inherent limitation of RNA-based biosensors, which is that intercalating compounds may yield false-positive fluorescent changes. However, RNA-based biosensors have been successfully used to characterize inhibitors that specifically target proteins, including methyltransferases (Su et al., 2016) and RNA demethylases (Svensen and Jaffrey, 2016).
Figure 3. The effect of nucleic acid intercalators on the fluorescence of RNA-based biosensors and cGAS activity.

(A) Observation of dose-dependent loss of fluorescence in presence of QC for both the 2′, 3′-cGAMP biosensor and Spinach2 aptamer. (B) TLC-based activity assay to assess cGAS inhibition by quinacrine (QC), ethidium bromide (EtBr), actinomycin D (AD), and methylene blue (MB).
By the same logic, we realized that quinacrine likely inhibits cGAS activity indirectly, by interfering with DNA binding. Previous studies inferred that quinacrine intercalation would lengthen the B-form DNA helix based on circular dichroism measurements, but otherwise would not grossly alter conformation (Hossain et al., 2008). To support our hypothesis about mechanism of action, we tested other DNA intercalators for effects on cGAS activity. The radiolabeled TLC assay was employed in order to avoid possible complications in data interpretation from compounds interacting with the RNA biosensor. Quinacrine and ethidium bromide have similar DNA binding modes (LePecq and Paoletti, 1967), and both fully inhibited cGAS activity at 100 μM concentration. Actinomycin D, which binds to single-stranded and double-stranded DNA, showed partial inhibition at that concentration, whereas methylene blue, which intercalates in a completely different orientation than quinacrine (Hossain et al., 2008), had no effect (Fig. 3B). These results provide strong evidence that nucleic acid intercalators are a class of small molecule compounds capable of inhibiting cGAS. Our results and those from the original study with quinacrine (An et al., 2015) do not distinguish whether inhibition is via a direct or indirect mechanism, but we favor the latter, e.g. these compounds intercalate and shift the DNA helix conformation sufficiently to reduce binding to cGAS enzyme. We can easily analyze the enzyme-independent effect of compounds on fluorescence of the RNA-based biosensors in control reactions, which allows us to identify compounds in the screen that likely interact with the DNA instead of the enzyme directly, a potential source for false positive hits. Overall, our analysis of the limitation of our RNA-based biosensor’s capacity to characterize cGAS inhibition by intercalating compounds led to an important finding on alternate mechanisms of cGAS inhibition by small molecules, and revealed the advantage of using the RNA biosensor to distinguish nucleic acid intercalators from direct cGAS inhibitors.
Quantitating 2′, 3′-cGAMP in lysates of DNA-stimulated mammalian cells
A critical aim is to detect cGAS activation in mammalian cells. Based on our positive results in detecting cGAS overexpression in bacteria (Fig S3), we considered applying the fluorescent biosensor to detect cellular cGAMP levels in mammalian cell lysates. 2′, 3′-cGAMP is produced by cGAS+ mammalian cells as part of a cytosolic immune surveillance pathway for foreign DNA, which could be caused by viral or bacterial infection, and alternatively, by leakage of damaged nuclear or mitochondrial DNA (Gao et al., 2013a; Hansen et al., 2014; Li et al., 2016; Ma and Damania, 2016; Watson et al., 2015; White et al., 2014; Woo et al., 2015a). A standard method to simulate these conditions is to transfect cells with double-stranded DNA, however it has not been straightforward to directly measure the levels of 2′, 3′-cGAMP produced. Besides mass spectrometry, which is low throughput, the main method to study this pathway involves an IFN-β reporter assay, which is highly sensitive, but has drawbacks in that it requires prior transfection of the reporter DNA and is not specific to the cGAS-STING pathway, as other immune signaling also stimulate interferon production and activate the promoter controlling reporter expression.
We evaluated the ability of the fluorescent biosensor to detect 2′, 3′-cGAMP produced upon DNA stimulation of cGAS+ L929 cells (Sun et al., 2013) In comparison, we also analyzed mock-transfected L929 cells, which express functional cGAS, and HEK293T cells, which are cGAS null, under identical conditions. In vitro measurements were performed by adding aliquots of cell lysate to the fluorescent biosensor reaction in a 96-well plate reader (Fig. 4A). Fluorescence activation was observed for DNA-stimulated over mock-stimulated L929 cells, with increasing turn-on signal upon addition of 1 to 5 μL of concentrated cell lysate (3x105 cells per μL of concentrated cell extract) (Fig. 4B). In contrast, no significant fluorescence change between DNA-stimulated and mock-stimulated was observed for HEK293T cells (Fig. 4C). Thus, we showed that our first-generation biosensor has sufficient sensitivity to detect endogenous 2′, 3′-cGAMP produced in response to DNA activation of cGAS enzyme in human cell lysates.
Figure 4. Application of fluorescent biosensor to detect 2′, 3′-cGAMP in mammalian cell extracts.

(A) Schematic representation of the experimental procedure for cell extract analysis using the fluorescent biosensor. (B, C) Fluorescence readings for different concentrations of L929 (B) or HEK293T (C) cell extracts (DNA- or mock-stimulated). (D) Fluorescence readings for L929 cell extracts (DNA- or mock-stimulated) treated with active or denatured SVPD prior to biosensor addition. (E) Standard curve of fluorescence readings from mock-stimulated L929 cell extracts doped with known concentrations of 2′, 3′-cGAMP (black circles). Average fluorescence reading from DNA-stimulated L929 cell extracts is shown in open circle. Error bars indicate standard deviations for three (B, C) or two (D, E) independent biological replicates. P-values are calculated from student’s t-test comparison: **, P < 0.01; ***, P < 0.001, n.s, not significant, P > 0.05.
Interestingly, we observed higher background fluorescence signal for mock-stimulated L929 cells than HEK293T cells. We checked whether there was basal 2′, 3′-cGAMP in mock-stimulated L929 cells that was activating the fluorescent biosensor. Background fluorescence for the mock-stimulated sample did not change upon treatment with active snake venom phosphodiesterase (SVPD), which degrades 2′, 3′-cGAMP (Diner et al., 2013) (Fig. 4D, S4). In contrast, active SVPD did reduce the fluorescence signal of DNA-stimulated L929 cell lysates to the same levels as mock-stimulated samples. Therefore, we concluded that the observed difference in background fluorescence was due to distinct levels of auto-fluorescence between the cell types and includes contributions to background fluorescence from the presence of the dye DFHBI and the RNA biosensor (Fig. S4).
Finally, using our fluorescent biosensor we were able to determine the absolute concentration of 2′, 3′-cGAMP produced upon DNA stimulation in L929 cells. Mock-stimulated L929 cell lysate was doped with known concentrations of 2′, 3′-cGAMP and the corresponding biosensor fluorescence signals were measured to obtain a standard curve with a lower limit of detection of 0.95 μM (Fig. 4E). The fluorescence signal from L929 cells stimulated with DNA for 5 hours was compared to the standard curve, and showed that there was a concentration of 1.5 μM for 2′, 3′-cGAMP in L929 cell lysate. Assuming 100% extraction efficiency, it was calculated that DNA stimulation by transfection resulted in 60 attomoles of 2′, 3′-cGAMP produced on average per L929 cell, or 6 million molecules of 2′, 3′-cGAMP per cell (Fig. 4E, see SI for detailed calculations).
Discussion
Even though no natural riboswitches for 2′, 3′-cGAMP have been discovered, we were able to engineer RNA-based fluorescent biosensors for 2′, 3′-cGAMP by employing structure-based rational design. Ligand-bound riboswitch structures informed the initial choice of the GEMM-II scaffold and mutations to the ligand binding pocket. A modest screen of different phylogenetic and stem variants was guided by empirical design rules for functional biosensors that start with use of the natural P1 stem (Kellenberger and Hammond, 2015; Wang et al., 2016). Our results in this study again demonstrate that riboswitch-based biosensors can be reprogrammed to recognize other ligands via point mutations, although not necessarily with full selectivity (Kellenberger et al., 2013; Ren et al., 2015; Wu et al., 2015). The broader implication is that natural riboswitch-ligand pairs can serve as advanced starting points for the engineering of biosensors for related ligands not recognized by known riboswitches. In one case, this actually led to the rational discovery of natural riboswitches (Kellenberger et al., 2015).
Our biosensors exhibit fluorescence turn-on activity in direct response to 2′, 3′-cGAMP and thus are readily adapted for high-throughput screening assays of cGAS enzyme activity and inhibition. Given the emerging clinical relevance of the cGAS/STING innate immune signaling pathway for infectious diseases, cancer immunotherapy, and DNA-triggered autoimmune disorders, a fluorescent plate reader screen for small molecule modulators of cGAS activity is a timely and valuable resource for the academic and industrial research community. We evaluated the Z′ factor for the assay, which showed that high signal-to-background ratios gave statistically reliable results. The ability to use DFHBI analogs with different fluorescent emission wavelengths (Song et al., 2014) may be further beneficial to avoid false positive hits. We also showed that our assay provides a straightforward way to distinguish between compounds that target cGAS enzyme versus interact with the activating ligand, double-stranded DNA.
Our finding that intercalating compounds affect cGAS activation reveals that the mechanism of small molecule inhibition of cGAS enzyme should be carefully examined. In addition, since DNA intercalators are used as anti-cancer drugs (Wheate et al., 2007), it potentially raises interesting questions about the activation status of cGAS during drug treatment. There also are broader implications for the design of synthetically modified double-stranded nucleic acids for therapeutic or basic research purposes. In particular, we expect that backbone modifications that favor B-form-like helical conformations may cause the nucleic acid agent to bind and activate cGAS, thus triggering interferon response. This adds to the existing body of knowledge about the length and sequence requirements for cGAS activation (Gao et al., 2013b; Herzner et al., 2015; Li et al., 2013), and further implies that some synthetic modifications may be better at eluding cGAS surveillance and thus the cytosolic immune response. Our assay provides a rapid, non-radioactive, and low material cost method to screen nucleic acid agents for such activity.
Finally, we have employed a fluorescent biosensor to quantitate the levels of 2′, 3′-cGAMP in L929 cell lysates, which previously had been shown to be cGAS+ via LC-MS analysis (Sun et al., 2013), but not in a quantitative manner. Very recently, an LC-MS/MS method has been developed for quantitation of 2′, 3′-cGAMP (Paijo et al., 2016). The amount of 2′, 3′-cGAMP produced upon HCMV virus infection was measured in the range of 0.5 to 3 attomoles on average per cell, depending on the cell type (Paijo et al., 2016). As expected, we have found that transfection of DNA leads to a much stronger response, as we measured 60 attomoles of 2′, 3′-cGAMP on average per cell at an earlier time point (6 h post-transfection versus 24 h post-infection). Based on the standard curve in L929 lysates, the current lower limit of detection of our fluorescent biosensor is ~1 μM or 40 attomoles per cell. Thus, improving biosensor sensitivity by 10- to 100-fold would provide a high-throughput method to analyze viral-induced 2′, 3′-cGAMP levels in the future. Perhaps more excitingly, this analysis suggests that second-generation fluorescent biosensors with the requisite higher sensitivity, e.g. binding affinities in the 10 to 100 nM range, would be useful for single-cell analysis of 2′, 3′-cGAMP production upon virus infection. In addition, biosensors with improved sensitivity would enable high-throughput biochemical and/or cell-based assays for high affinity compounds that modulate cGAS activity. We recently described a phylogenetic search strategy for generating high affinity and high turn-on RNA-based biosensors, which resulted in a biosensor capable of detecting sub-nanomolar levels of cyclic di-GMP (Wang et al., 2016). Work on applying this methodology to the 2′, 3′-cGAMP sensor is ongoing in the lab.
Much in the way that antibody-based assays are indispensable for analysis of kinase signaling, fluorescent biosensor-based assays will be a valuable resource for the analysis of cyclic dinucleotide signaling. Measuring 2′, 3′-cGAMP levels is important for investigating the basic biology of the cGAS-STING signaling pathway and its mechanistic contribution to innate immune responses. A recent study showed that in a viral infection model, cGAS and STING expression levels do not necessarily correlate with 2′, 3′-cGAMP production, and furthermore 2′, 3′-cGAMP levels do not necessarily correlate with IFN production (Paijo et al., 2016). These findings underscore the importance of measuring cGAS enzyme activity, rather than using its expression level or induction of IFN as proxies. In the context of cancer immunology, it has been suggested that tumor-derived DNA activates cGAS activity in host immune cells (Woo et al., 2015b), but direct proof of this hypothesis is still lacking. Addressing these and other pressing questions about cGAS activity will be facilitated by a high-throughput fluorescent plate reader assay capable of quantitating 2′, 3′-cGAMP levels directly from lysates of different mammalian cell types. We also demonstrate use of the biosensor for both biochemical and cell-based analysis of cGAS activity, which provides two different high-throughput screening methods to identify chemical compounds or novel protein factors that affect cGAS activity.
Significance
To our knowledge, this study demonstrates the first fluorescent plate reader assay for detection of 2′, 3′-cGAMP, a signaling molecule central to the cGAS/STING innate immune pathway that has emerged as a promising new target for cancer immunotherapy and autoimmune diseases. The assay relies on a riboswitch-based fluorescent biosensor that was generated by structure-based rational design. Several intercalating compounds were found to affect cGAS activity, which has broader implications for related anti-cancer agents and the design of synthetically modified double-stranded nucleic acids for therapeutic or basic research purposes. Finally, as proof of principle, we demonstrated quantitation of 2′, 3′-cGAMP from DNA-stimulated cell lysates using the fluorescent biosensor.
Experimental Procedures
Reagents and oligonucleotides
DNA oligonucleotides for biosensor constructs were purchased as Ultramers from Integrated DNA Technologies (Coralville, IA) and other DNA oligonucleotides were purchased from Elim Biopharmaceuticals (Hayward, CA). DFHBI and DFHBI-1T were either purchased from Lucerna (New York, NY) or were synthesized following previously described protocols (Song et al., 2014) and were stored as 10–30 mM stocks in DMSO. C-di-GMP, 3′, 3′-cGAMP, 2′, 3′-cGAMP were purchased from Axxora (Farmingdale, NY). Commercially available reagents were used without further purification. T7 RNA polymerase was either purchased from New England Biolabs Inc (Ipswich, MA) or given as a generous gift by the laboratory of Terrence Oas at Duke University. Phusion DNA polymerase were purchased from New England Biolabs Inc (Ipswich, MA). Chemically competent BL21 (DE3) Star cells were purchased from Life Technologies (Carlsbad, CA). Quinacrine dihydrochloride, herring testes DNA (HT-DNA), and snake venom phosphodiesterase (SVPD) was purchased from Sigma-Aldrich (St Louis, MO). L929 and HEK293T cells were purchased from ATCC (Manassas, VA).
In vitro transcription
DNA templates for in vitro transcription were prepared through PCR amplification using Phusion DNA polymerase (NEB) from sequence-confirmed plasmids using primers that added the T7 polymerase promoter sequence. PCR products were purified by QIAquick PCR purification kit (Qiagen) for characterization and application experiments. Templates were then transcribed using T7 RNA polymerase in 40 mM Tris-HCl, pH 8.0, 6 mM MgCl2, 2 mM spermidine, and 10 mM DTT. RNAs were either purified by a 96-well format ZR-96 Clean & Concentrator (Zymo Research) for the candidate biosensor screen or by denaturing (7.5 M urea) 6% PAGE for biosensor characterization and use in quantitative assays. RNAs purified from PAGE were subsequently extracted from gel pieces using Crush Soak buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl and 1 mM EDTA, pH 8.0). RNAs were precipitated with ethanol, dried, and then resuspended in TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). Accurate measurement of RNA concentration was determined by measuring the absorbance at 260 nm after performing a hydrolysis assay to eliminate the hypochromic effect due to secondary structure in these RNAs (Wilson et al., 2014).
Expression and purification of cGAS enzyme
N-terminal 6xHis-MBP tagged full length cGAS encoding plasmid was donated by the Doudna lab (Kranzusch et al., 2013). Protein expression was induced in E. coli BL21-RIL DE3 along with pRARE2 human tRNA plasmid (Agilent) with addition of 0.5 M IPTG to LB media for 20 hr at 16 °C. Cells were lysed by sonication in 20 mM HEPES (pH 7.5), 400 mM NaCl, 10% glycerol, 30 mM imidazole, 1 mM PMSF, 1 mM TCEP. The lysate was treated with DNase (Worthington, NJ) for 30 min to remove residual DNA bound to cGAS. Clarified lysate was bound to Ni-NTA agarose (QIAGEN) and was washed with lysis buffer supplemented with 1 M NaCl. The bound protein was eluted using lysis buffer supplemented with 300 mM imidazole. The eluted protein was dialyzed overnight at 4 °C in buffer having 20 mM HEPES (pH 7.5), 150 mM KCl, 10% glycerol and 1 mM TCEP.
Thin-layer chromatography assay of cGAS activity
Enzyme activity of recombinant full-length cGAS was assayed in buffer containing 40 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 10 mM MgCl2 (Sigma-Aldrich, St Louis, MO). cGAS (estimated final concentration 1.5 μM) or equal volume of water was incubated with activating ligand HT-DNA (final concentration 0.1 mg/ml) in the presence of 250 μM ATP and 250 μM GTP and trace amounts of [α-32P] ATP or GTP (~ 30 nM) at 37 °C for 1.5 hr. Reactions were terminated with addition of 5 U of alkaline phosphatase (New England Biolabs) and further incubation at 37°C for 30 min. One microliter of each reaction was spotted onto a PEI-Cellulose F thin-layer chromatography plate (EMD Millipore), and reaction products were separated with the use of 1.5 M KH2PO4 (pH 3.8) as solvent. Plates were dried and radiolabeled products were detected by imaging the exposed phosphor screen using a Typhoon phosphorimager (GE Life Sciences).
General procedure for in vitro fluorescence assays
In vitro fluorescence assays were carried out in a buffer containing 40 mM HEPES, pH 7.5 and 125 mM KCl. Other conditions, including temperature, concentration of MgCl2, DFHBI (or DFHBI-1T), ligand (or cell extract) and RNA were varied in different experiments and are indicated in the figures. The RNA was renatured in buffer at 70 °C for 3 min and cooled to ambient temperature for 5 min prior to addition to the reaction solution containing DFHBI (or DFHBI-1T), buffer, and ligand. Binding reactions were performed either in 100 μL (96-well plate) or 30 μL (384-well plate) volumes and were incubated at the indicated temperature in either a Corning Costar 3915 96-well black plate or Greiner 781077 384-well black plate until equilibrium was reached, which typically takes 30 to 60 minutes. The fluorescence emission was measured using a Molecular Devices SpectraMax Paradigm Multi-Mode detection platform plate reader (Sunnyvale, CA) with the following instrument parameters: 448 nm excitation, 506 nm emission for DFHBI or 470 nm excitation, 510 nm emission for DFHBI-1T.
cGAS activity and inhibition assay
To initiate the enzyme reaction, 0.5 μL of HT-DNA was added to 2.5 μL of 6X enzyme reaction solution containing cGAS, GTP, ATP and reaction buffer. Generally, final concentrations are 100 μg/mL HT-DNA, 1.5 μM cGAS, 200 μM GTP, and 200 μM ATP in enzyme reaction buffer (40 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10 mM MgCl2). The enzyme reactions are incubated at 37 °C for 2 h. For the inhibition assay shown in Figure S2B, the reaction conditions are the same as described above except various concentrations of quinacrine was added.
To initiate the fluorescent biosensor reaction, 3 μL of the enzyme reaction was added to 27 μL of biosensor reaction solution containing renatured RNA, DFHBI (or DFHBI-1T), and biosensor binding buffer. Final concentrations are 200 nM RNA, 10 μM DFHBI (or DFHBI-1T), 40 mM HEPES, pH 7.5, 125 mM KCl, 10 mM MgCl2, and 0.1X enzyme reaction buffer (4 mM Tris-HCl (pH 7.5), 10 mM NaCl, 1 mM MgCl2). Fluorescence measurements were conducted as described above. For the inhibition assay, due to the intrinsic fluorescence of quinacrine, DFHBI-1T was used to decrease overlap in its excitation wavelength. Samples containing only quinacrine at various concentrations in the biosensor binding buffer were used for background fluorescence subtraction.
High-throughput screening (HTS) assay of cGAS activity
For HTS experiments, reaction component samples were dispensed into a Grenier 781077 384-well flat bottom black plate using an Eppendorf Repeater Xstream pipetter. Z′ factors were calculated from 16 well replicates in the 384-well plate. For the cGAS activity assay, 0.5 μL of HT-DNA or water was added to 2.5 μL of reaction solution containing cGAS, GTP, ATP and cGAS reaction buffer in each 0.2 mL PCR tube in two 8-tube strips (BioRad). Final concentrations are: 100 μg/mL HT-DNA, 1.5 μM cGAS, 200 μM GTP, 200 μM ATP, and enzyme reaction buffer (40 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10 mM MgCl2). After 2 h incubation at 37 °C, to initiate the fluorescent biosensor reaction, the total reaction solution (3 μL) was added in each well of the 384-well plate to 27 μL of biosensor assay solution containing renatured RNA, DFHBI, and biosensor binding buffer. Final concentrations are 200 nM RNA, 10 μM DFHBI, 40 mM HEPES, pH 7.5, 125 mM KCl, 10 mM MgCl2, and 0.1x cGAS reaction buffer (4 mM Tris-HCl (pH 7.5), 10 mM NaCl, 1 mM MgCl2). Fluorescence measurements were conducted as described above.
Biosensor-based determination of 2′, 3′-cGAMP concentration in mammalian cell extracts
L929 and HEK293T cells were maintained in DMEM medium with 10% FBS in a humidified incubator at 37°C and 5% CO2. To introduce cytosolic DNA, transfections were performed using lipofectamine2000 (Invitrogen) using the manufacturer’s protocol. Typically, cells were seeded in 175 cm flasks at 90% confluence (2 x 106 cells) in 25 mL media. The lipofectamine (125 μL) was mixed with 250 μg of HT-DNA (10 μg/ml) or no HT-DNA (mock) in OptiMEM to a final volume of 200 μL and incubated at room temperature for 30 min. The transfection mixture was added to the cell culture and incubated for 6 h, then cyclic dinucleotides were extracted from three 175 cm flasks of cells (~ 6 x 106 cells, quantified in a Neubauer chamber using bright field microscope) after brief trypsinization to detach from flask. The extraction procedure was performed as described in the section on E. coli cell lysates. Mammalian cell lysates were concentrated to a final volume of 30 μL using a vacuum concentrator and used immediately or stored at −20 °C.
To initiate the fluorescent biosensor reactions, 2 μL (1X), 5 μL (2.5X), or 10 μL (5X) of cell extracts (stimulated or non-stimulated) was added to a reaction solution adjusted to a final volume of 30 μL. Final concentrations are 200 nM RNA, 25 μM DFHBI-1T, 40 mM HEPES, pH 7.5, 125 mM KCl, and 10 mM MgCl2. Fluorescence measurements were conducted as described above. For experiments with SVPD, active (200 μU) or heat inactivated SVPD (95 °C for 5 mins) was added to 2 μL (1X cell lysate) of cell extract and incubated at 37 °C for 1 h. The reactions were stopped by heating at 95 °C for 5 mins, rapidly cooled on ice, and then analyzed in fluorescent biosensor reactions.
To generate a standard fluorescence response curve in L929 cell lysates, known concentrations of commercial 2′, 3′-cGAMP in water were mixed with 1 μL of non-stimulated cell extract, followed by addition to a reaction solution adjusted to a final volume of 25 μL. See SI for details on calculation.
Supplementary Material
Highlights.
A fluorescent biosensor that responds to 2′, 3′-cGAMP was designed
A biosensor-based fluorescence turn-on assay detects cGAS activity and inhibition
High-throughput analysis via fluorescence plate reader is enabled
2′,3′-cGAMP levels were quantified in DNA-stimulated L929 cells
Acknowledgments
We thank Philipp Kranzusch from the Doudna lab at University of California, Berkeley for providing the 6xHis-MBP-cGAS plasmid. This work was also supported by DP2 OD008677 (to M.C.H.), R01 AI113041 (to D.R.), and NIH Training Grant T32 GM066698 (for Y.S.)
Footnotes
Author Contributions
D.B., Y.S. and M.C.H. designed research; D.B. and Y.S. conducted experiments; A.M. contributed new reagents; D.B., Y.S., A.M., D.R., and M.C.H. analyzed data; and D.B., Y.S., and M.C.H. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Konno H, Barber GN. Diverse roles of STING-dependent signaling on the development of cancer. Oncogene. 2015;34:5302–5308. doi: 10.1038/onc.2014.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J Immunol. 2014;193:4634–4642. doi: 10.4049/jimmunol.1401337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Woodward JJ, Sasaki T, Minie M, Elkon KB. Cutting edge: Antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J Immunol. 2015;194:4089–4093. doi: 10.4049/jimmunol.1402793. [DOI] [PubMed] [Google Scholar]
- Baird JR, Friedman D, Cottam B, Dubensky TW, Jr, Kanne DB, Bambina S, Bahjat K, Crittenden MR, Gough MJ. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016;76:50–61. doi: 10.1158/0008-5472.CAN-14-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio. 2013;4:e00018–00013. doi: 10.1128/mBio.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer DR, Canals A, Coll M. DNA-binding drugs caught in action: the latest 3D pictures of drug-DNA complexes. Dalton Trans. 2009:399–414. doi: 10.1039/b809873p. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burhenne H, Kaever V. Quantification of cyclic dinucleotides by reversed-phase LC-MS/MS. Methods Mol Biol. 2013;1016:27–37. doi: 10.1007/978-1-62703-441-8_3. [DOI] [PubMed] [Google Scholar]
- Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901–910. doi: 10.1158/2326-6066.CIR-13-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Gajewski TF. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin Cancer Res. 2015;21:4774–4779. doi: 10.1158/1078-0432.CCR-15-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran E, Corrales L, Kline J. Targeting the innate immune system as immunotherapy for acute myeloid leukemia. Front Oncol. 2015;5:83. doi: 10.3389/fonc.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A. 2015;112:15408–15413. doi: 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey CM, Aghaei M, Schwendener RA, Jirik FR. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PLoS One. 2014;9:e99988. doi: 10.1371/journal.pone.0099988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, Chen ZJ. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. 2015;112:E5699–5705. doi: 10.1073/pnas.1516465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013a;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013b;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, Nielsen R, Leber JH, Decker T, Horan KA, et al. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014;33:1654–1666. doi: 10.15252/embj.201488029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kubler K, Wittmann S, Gramberg T, Andreeva L, Hopfner KP, Mertens C, et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol. 2015;16:1025–1033. doi: 10.1038/ni.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain M, Giri P, Kumar GS. DNA intercalation by quinacrine and methylene blue: a comparative binding and thermodynamic characterization study. DNA Cell Biol. 2008;27:81–90. doi: 10.1089/dna.2007.0652. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger CA, Hammond MC. In vitro analysis of riboswitch-Spinach aptamer fusions as metabolite-sensing fluorescent biosensors. Methods Enzymol. 2015;550:147–172. doi: 10.1016/bs.mie.2014.10.045. [DOI] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Hickey SF, Gonzalez TL, Su Y, Hallberg ZF, Brewer TF, Iavarone AT, Carlson HK, Hsieh YF, et al. GEMM-I riboswitches from Geobacter sense the bacterial second messenger cyclic AMP-GMP. Proc Natl Acad Sci U S A. 2015;112:5383–5388. doi: 10.1073/pnas.1419328112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC. RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J Am Chem Soc. 2013;135:4906–4909. doi: 10.1021/ja311960g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Kobayashi CI, Nakamura-Ishizu A, Karigane D, Haeno H, Yamamoto KN, Sato T, Ohteki T, Hayakawa Y, Barber GN, et al. Bacterial c-di-GMP affects hematopoietic stem/progenitors and their niches through STING. Cell Rep. 2015;11:71–84. doi: 10.1016/j.celrep.2015.02.066. [DOI] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013;3:1362–1368. doi: 10.1016/j.celrep.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ER, Baker JL, Weinberg Z, Sudarsan N, Breaker RR. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science. 2010;329:845–848. doi: 10.1126/science.1190713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LePecq JB, Paoletti C. A fluorescent complex between ethidium bromide and nucleic acids. Physical-chemical characterization. J Mol Biol. 1967;27:87–106. doi: 10.1016/0022-2836(67)90353-1. [DOI] [PubMed] [Google Scholar]
- Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, Xu P, Tan J, Rui Y, Li P, et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep. 2016;6:19049. doi: 10.1038/srep19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39:1019–1031. doi: 10.1016/j.immuni.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Damania B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe. 2016;19:150–158. doi: 10.1016/j.chom.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Miyabe H, Hyodo M, Sato Y, Hayakawa Y, Harashima H. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J Control Release. 2015;216:149–157. doi: 10.1016/j.jconrel.2015.08.026. [DOI] [PubMed] [Google Scholar]
- Paijo J, Doring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, Witte G, Messerle M, Hornung V, Kaever V, et al. cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog. 2016;12:e1005546. doi: 10.1371/journal.ppat.1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren A, Wang XC, Kellenberger CA, Rajashankar KR, Jones RA, Hammond MC, Patel DJ. Structural basis for molecular discrimination by a 3′,3′-cGAMP sensing riboswitch. Cell Rep. 2015;11:1–12. doi: 10.1016/j.celrep.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan CA, Gaffney BL, Jones RA, Strobel SA. Differential analogue binding by two classes of c-di-GMP riboswitches. J Am Chem Soc. 2011;133:15578–15592. doi: 10.1021/ja204650q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M, Niewold TB. Nucleic Acid sensors and type I interferon production in systemic lupus erythematosus. Front Immunol. 2013;4:319. doi: 10.3389/fimmu.2013.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Hossain M, Kumar GS. RNA targeting by DNA binding drugs: structural, conformational and energetic aspects of the binding of quinacrine and DAPI to A-form and H(L)-form of poly(rC).poly(rG) Biochim Biophys Acta. 2007;1770:1636–1650. doi: 10.1016/j.bbagen.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Smith KD, Shanahan CA, Moore EL, Simon AC, Strobel SA. Structural basis of differential ligand recognition by two classes of bis-(3′-5′)-cyclic dimeric guanosine monophosphate-binding riboswitches. Proc Natl Acad Sci U S A. 2011;108:7757–7762. doi: 10.1073/pnas.1018857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Strack RL, Svensen N, Jaffrey SR. Plug-and-play fluorophores extend the spectral properties of Spinach. J Am Chem Soc. 2014;136:1198–1201. doi: 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Hickey SF, Keyser SG, Hammond MC. In Vitro and In Vivo Enzyme Activity Screening via RNA-Based Fluorescent Biosensors for S-Adenosyl-l-homocysteine (SAH) J Am Chem Soc. 2016;138:7040–7047. doi: 10.1021/jacs.6b01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensen N, Jaffrey SR. Fluorescent RNA Aptamers as a Tool to Study RNA-Modifying Enzymes. Cell Chem Biol. 2016;23:415–425. doi: 10.1016/j.chembiol.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XC, Stephen CW, Hammond MC. Next-generation RNA-based fluorescent biosensors enable anaerobic detection of cyclic di-GMP. Nucleic Acids Research. 2016 doi: 10.1093/nar/gkw580. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CM. The second messenger cyclic Di-GMP. Washington, DC: ASM Press; 2010. Chapter6: Methods for Cyclic di-GMP Detection. [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe. 2015;17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheate NJ, Brodie CR, Collins JG, Kemp S, Aldrich-Wright JR. DNA intercalators in cancer therapy: organic and inorganic drugs and their spectroscopic tools of analysis. Mini Rev Med Chem. 2007;7:627–648. doi: 10.2174/138955707780859413. [DOI] [PubMed] [Google Scholar]
- White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SC, Cohen DT, Wang XC, Hammond MC. A neutral pH thermal hydrolysis method for quantification of structured RNAs. RNA. 2014;20:1153–1160. doi: 10.1261/rna.045856.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015a;33:445–474. doi: 10.1146/annurev-immunol-032414-112043. [DOI] [PubMed] [Google Scholar]
- Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015b;36:250–256. doi: 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Lowe PT, Robinson CJ, Vincent HA, Dixon N, Leigh J, Micklefield J. Rational Re-engineering of a Transcriptional Silencing PreQ1 Riboswitch. J Am Chem Soc. 2015;137:9015–9021. doi: 10.1021/jacs.5b03405. [DOI] [PubMed] [Google Scholar]
- Zhang H, Tang K, Zhang Y, Ma R, Ma J, Li Y, Luo S, Liang X, Ji T, Gu Z, et al. Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res. 2015;3:196–205. doi: 10.1158/2326-6066.CIR-14-0177. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.