

Abstract
Comprehensive understanding of the structure–function relationship of RNA both in real time and at atomic level will have a profound impact in advancing our understanding of RNA functions in biology. Here, we describe the first example of a multifunctional nucleoside probe, containing a conformation‐sensitive fluorophore and an anomalous X‐ray diffraction label (5‐selenophene uracil), which enables the correlation of RNA conformation and recognition under equilibrium and in 3D. The probe incorporated into the bacterial ribosomal RNA decoding site, fluorescently reports antibiotic binding and provides diffraction information in determining the structure without distorting native RNA fold. Further, by comparing solution binding data and crystal structure, we gained insight on how the probe senses ligand‐induced conformational change in RNA. Taken together, our nucleoside probe represents a new class of biophysical tool that would complement available tools for functional RNA investigations.
Keywords: antibiotics, fluorescent nucleosides, ribosomal RNA, selenium, X-ray diffraction
Biophysical techniques including fluorescence labeling, NMR, EPR and X‐ray crystallography have provided valuable information on RNA folding, molecular recognition and function.1, 2, 3, 4 Such investigations mostly involve RNA sequences labeled with appropriate reporters as natural nucleosides are practically non‐fluorescent and do not provide intrinsic labels that are suitable for analysis by spectroscopic and diffraction techniques.5 Minimally perturbing fluorescent nucleoside analogs,6 isotope‐ (e.g., 13C, 15N),7 spin‐ (e.g., nitroxide)8 and heavy‐atom‐labeled (e.g., Br, Se)9 nucleosides incorporated into oligonucleotides (ONs) have been applied in assays to study structure, dynamics and function of RNA. However, given the multitude and diversity of RNA transcripts, probing structure–function relationships of RNA sequences using established biochemical and biophysical methods remains a major challenge.10 This is largely due to the lack of chemical probes that can be efficiently deployed in multiple biophysical techniques as almost all studies use the traditional approach of “one label‐one technique”, wherein a custom labeled RNA suitable for a particular technique is used. Therefore, development of smart chemical tools that can 1) be readily incorporated into RNA ONs and 2) enable the direct correlation of RNA structure and function in real time and in 3D is highly desired. We concluded that ribonucleoside probes containing multiple labels (e.g., a fluorophore and an anomalous X‐ray scattering label) would be advantageous as they would serve as common probes to analyze RNA conformation and function in solution in real time by fluorescence and, concurrently, RNA structure in the solid‐state by X‐ray crystallography (Figure 1 and Figure S1).
Figure 1.
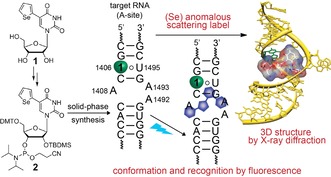
Chemical structure of 5‐selenophene‐modified nucleoside probe (1 or SeU) and corresponding phosphoramidite 2.11 Incorporation of SeU into the RNA target (e.g., A‐site) would enable simultaneous biophysical investigation of the same RNA construct in solution by fluorescence techniques and in the solid‐state by X‐ray crystallography.
Here, we describe the first example of a dual‐purpose ribonucleoside probe (SeU), based on the 5‐(selenophen‐2‐yl)uracil scaffold, which provides a tool for comprehensive investigations of RNA–drug interaction by fluorescence and X‐ray crystallography techniques (Figure 1). The nucleoside probe was chemically incorporated into the bacterial ribosomal decoding site (A‐site), and the environmental sensitivity of the fluorophore was used for monitoring the binding of aminoglycoside antibiotics. X‐ray diffraction analysis of A‐site RNA crystals containing the modified uridine revealed a prominent diffraction signal from the selenium atom and also provided insight into the structural basis on how the nucleoside probe senses conformational changes in RNA during ligand binding.
We recently introduced a 5‐selenophene‐modified uridine analog (1), which is composed of an environment‐sensitive base‐modified fluorescent nucleobase and an X‐ray crystallography phasing agent (Se atom, Figure 1).12 Selenium provides key advantages over halogen modification. First, Se gives rise to a strong anomalous scattering signal, which has been widely used in protein crystallography and more recently in nucleic acid analysis.13 Second, halogen‐modified ONs are known to undergo dehalogenation when exposed to X‐ray radiation causing failures in phasing.14 Ribonucleoside 1 has an emission maximum in the visible region (λ em=452 nm in water), and exhibits probe‐like properties (e.g., solvatochromism and viscochromism). The corresponding ribonucleotide can be effectively incorporated into ONs by enzymatic in vitro transcription12 and used to monitor RNA‐ligand binding by fluorescence. Based on these observations we sought to explore the fluorescent and anomalous X‐ray scattering properties of the uridine analog 1 as a common RNA probe in complementing solution and solid‐state techniques, namely fluorescence and X‐ray crystallography. We chose bacterial A‐site RNA as the test system, which is a well‐studied target for aminoglycoside antibiotics.15 In this system, the effect of selenophene modification on the RNA structure and its ability to faithfully report drug binding to A‐site in solution and solid state can be validated by direct comparison with previously reported data on A‐site aminoglycoside complexes.16
The A‐site motif in 16S rRNA serves as the decoding site for protein translation by screening cognate paring between the mRNA codon and tRNA anticodon.17 Biochemical and structural studies revealed that natural aminoglycoside antibiotics bind to the RNA decoding site at an internal loop which contains two conformationally flexible adenosine residues (A1492 and A1493, Figure 2).18 A‐site‐aminoglycoside complexes are further stabilized by direct and water‐bridged H‐bonding interactions with the U1406‐U1495 noncanonical base pair. Aminoglycoside binding induces a conformational change analogous to cognate tRNA bound to mRNA in the A‐site wherein A1492 and A1493 are pushed out of the internal loop. This conformational change reduces the ability of the decoding site to discriminate between cognate and near‐cognate tRNAs and ultimately leads to mistranslation. Short duplexes and stem‐loop RNA ONs faithfully representing the wild‐type decoding site of rRNA have been used as models to investigate the structure and recognition properties of the A‐site.18a,18b A‐site RNA models containing fluorescent nucleoside analogs (e.g., 2‐aminopurine; 2‐AP) at positions A1492/A1493/U1406 have been used in assays to monitor aminoglycoside binding.19 Based on these studies, we devised a labeled A‐site model suitable for both fluorescence and X‐ray studies by replacing U1406 with the nucleoside SeU.
Figure 2.

Secondary structure of selenophene‐modified (3⋅4) and native (5⋅4) bacterial A‐site model RNA.11 The decoding site internal loop that binds aminoglycosides is highlighted in bold letters. On the right, aminoglycosides used in this study are shown.
We have used bipartite ONs 3 and 4 comprising the bacterial decoding site sequence to assemble the A‐site RNA motif 3⋅4 (Figure 2). Preparation of the phosphoramidite building block 2 required for the synthesis of SeU‐modified RNA ON 3 is outlined in Scheme S1 in the Supporting Information.11 The phosphoramidite 2 was site‐specifically incorporated by solid‐phase ON synthesis. Fully deprotected ON 3 was purified by polyacrylamide gel electrophoresis (PAGE, Figure S2). The integrity of SeU‐modified ON 3 was confirmed by MALDI‐TOF mass analysis (Figure S2). The labeled A‐site construct (3⋅4) was assembled by hybridizing equimolar amounts of ON 3 and 4 in cacodylate buffer. The impact of the selenophene modification on the A‐site structure and stability was studied by circular dichroism (CD) and thermal melting experiments. Both, control unmodified 5⋅4 and modified A‐site 3⋅4 RNAs displayed similar CD spectra and Tm values indicating that the selenophene modification had only a minor effect on the native structure and stability of the A‐site RNA (Figure S3).
The ability of the nucleoside probe to photophysically report a ligand‐induced conformational change in the A‐site was evaluated by titrating RNA 3⋅4 with aminoglycoside antibiotics derived from 2‐deoxystreptamine (2‐DOS). We chose 4,5‐disubstituted 2‐DOS (paromomycin and neomycin B) and 4,6‐disubstituted 2‐DOS (tobramycin) aminoglycosides as they are known to reduce translation fidelity in bacteria by interacting with residues present in the internal loop of the decoding site (Figure 2).17, 18 A‐site model RNA was excited at 330 nm, and changes in emission intensity at λ em=450 nm were monitored as a function of increasing concentration of aminoglycosides. Titration with paromomycin and neomycin resulted in an increase in fluorescence intensity, corresponding to an apparent KD of 3.80±0.20 μm and 2.10±0.22 μm, respectively (Figure 3 and Figure S4). The binding constant and higher affinity exhibited by neomycin compared to paromomycin are consistent with literature reports (KD≈10−6 m).19a–19c It is important to note that 2‐AP, the most widely used conformation‐sensitive nucleoside probe, fails to report the binding of neomycin to A‐site RNA despite that both paromomycin and neomycin are known to bind the RNA target in a similar fashion and result in ribosomal miscoding during translation.16, 19 Next, we studied the binding of tobramycin, which binds A‐site RNA with lower affinity compared to 4,5‐disubstituted 2‐DOS aminoglycosides.18b SeU reported the binding of tobramycin with discernible enhancement in fluorescence intensity, albeit with reduced affinity compared to the native decoding site (66.3±5.9 μm and 9.5±2.0 μm, respectively, Figure S5).19a
Figure 3.
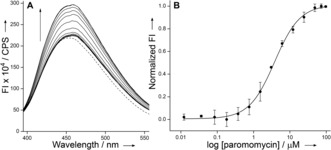
A) Emission spectra (solid lines) of A‐site RNA 3⋅4 as a function of increasing concentration of paromomycin. The dashed line represents an emission profile in the absence of aminoglycoside. B) Curve fitted for the titration of A‐site with paromomycin. Normalized fluorescence intensity (FI) at λ em=450 nm is plotted against log[aminoglycoside].
In order to elucidate the structural basis of conformation sensitivity of the nucleoside analog and allow a direct comparison between solution binding data and crystallographic structure, selenophene‐modified A‐site model RNA 3⋅4 was crystallized and its 3D structure determined by X‐ray diffraction at 2.14 Å resolution (PDB: 5T3K). Hanging‐drop vapor diffusion resulted in well‐diffracting crystals, which contained two unique A‐site RNA molecules with 50 % occupancy in the unit cell (Figure S6, Table S1). In one of the RNA copies, both A1492 and A1493 residues were flipped‐out of the loop, closely resembling the structure of the decoding site bound to aminoglycosides (Figure 4, top). In the second RNA copy, only A1493 was flipped‐out of the loop, while A1492 was found to be inside, base pairing and stacking with A1408 and G1491, respectively (Figure 4, bottom and Table S2). Other than these differences the two A‐site copies were nearly identical. Importantly, the electron density of the heavy Se atom was clearly visible, establishing the orientation of the 5‐membered ring as coplanar with the pyrimidine and Se facing the carbonyl O4. Like U1406 in the native decoding site, SeU1406 also formed a noncanonical Watson–Crick pair with U1495 (Figure S7, Table S2). The characteristically strong diffraction signal of the Se atom in the SeU analog suggests that anomalous dispersion from Se can be used in direct RNA structure determination by SAD or MAD phasing techniques.9, 13
Figure 4.
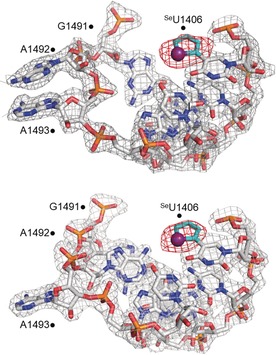
Electron density map around the SeU‐modified A‐site RNA 3⋅4 with A1492 flipped out (top) or in (bottom) the RNA helix, as is seen in a previous A‐site model RNA structure.19a The selenophene ring is shown in cyan with the selenium atom shown as an indigo sphere. The 2 F o−F c electron density map, contoured at 1.0σ, is shown in gray, and the F o−F c electron density map, contoured at 3.0σ, is shown in red (PDB ID: 5T3K).11.
The selenophene ring of the base‐paired fluorophore (SeU1406) is projected into the major groove and stacked with the imidazole ring of G1405 (3.72 Å, Figure S8). The uracil ring is stacked with the pyrimidine rings of G1405 (3.70 Å) and C1407 (3.71 Å). It is well documented that stacking interaction with neighboring bases and proximity to guanine residue can reduce the fluorescence efficiency of fluorophores incorporated into ONs.20 Hence, the stacked conformation of SeU in the unbound state of the A‐site RNA exhibits lower fluorescence intensity (Figure 3). However, upon titrating A‐site with aminoglycosides, the nucleoside probe reported the binding event by enhancement of fluorescence intensity. To gain further insight on the effect of modification on the A‐site structure and responsiveness of the label, the SeU‐A‐site crystal structure was superimposed onto structures of native A‐site RNA and aminoglycosides bound to the native A‐site RNA.
Superimposition of the selenophene‐modified and native decoding site structures reveal that the structures are almost identical with a RMS distance of 0.367 Å for 1305 identical atoms out of 1362 total atoms (Figure 5 A). The 2′‐OH group of the G1491 sugar is slightly drawn into the interior of the decoding site in the A1492‐out conformation of the modified RNA, whereas this is not seen in the structure of the native A‐site model. This 2′‐OH group interacts with a bridging water molecule to A1408 and with a chain of water molecules beneath the selenophene ring and above a Mg2+ ion (Figure S9). In addition to thermal melting and CD studies, the crystal structure confirms that selenophene‐modification has only a minor impact on the native A‐site RNA structure.
Figure 5.
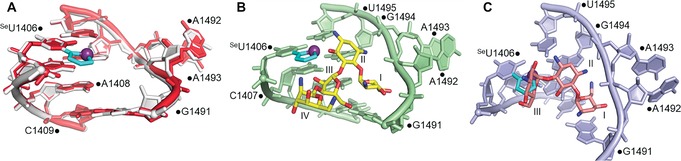
A) Superimposition of the SeU‐modified (white) and native (red; PDB: 1T0D)19a A‐site RNA structures. Superimposition of the SeU‐modified A‐site RNA and native A‐site RNA bound to B) paromomycin (yellow; PDB: 1J7T),21 and C) tobramycin (salmon; PDB: 1LC4).23 For clarity, superimposed selenophene‐modified uridine (cyan) is only shown.
In the superimposed structures of paromomycin and neomycin there was no apparent steric clash between the aminoglycosides and selenophene modification (Figure 5 B and Figure S10).21, 22 Other than the conserved interactions between the ligands and decoding site, 6‐ and 2′′‐hydroxyl groups of aminoglycoside rings II and III, respectively, were found to be in close proximity with the Se atom (2.2/2.4 Å and 3.4/2.6 Å, respectively). These superimposed models indicate that the SeU modification should not affect binding of paromomycin and neomycin to the decoding site. This notion is in consensus with the fluorescence binding experiments wherein SeU‐labeled A‐site exhibited binding affinity comparable to literature reports.19 In case of tobramycin, which binds differently to the decoding site as compared to 4,5‐disubstituted 2‐DOS aminoglycosides,23 the superimposed structure revealed a steric clash between ring III and the selenophene moiety (Figure 5 C). This would explain the observed lower affinity of tobramycin for the SeU‐labeled A‐site. Not surprisingly, mutations of U1406, predicted to confer antibiotic resistance, are known to reduce the binding of tobramycin due to a comparable steric clash.24 For example, replacing U1406 with adenine residue results in reduction in affinity of tobramycin (EC50 values, wt: 9.5 μm; mut: 110 μm).19a However, mutation of U1406 does not affect the binding of 4,5‐disubstituted 2‐DOS aminoglycosides to A‐site. In this context, SeU‐labeled A‐site can potentially serve as a mutant model because the nucleoside probe faithfully distinguishes the binding of aminoglycosides based on affinity and 2‐DOS class. Such labeled RNA constructs could be used for screening of small molecule ligands against resistant bacterial strains.25
Further, we compared the conformation of U1406 and its neighboring bases in the structures of native A‐site RNA in the absence and presence of aminoglycosides and SeU‐labeled A‐site to understand the photophysical behavior of SeU in solution binding experiments. The stacking interaction between U1406 and G1405 in the unbound state (3.77 Å) and aminoglycoside‐bound state (3.72–3.86 Å) is similar. However, U1406, which is completely stacked with C1407 (3.89 Å) in the free A‐site structure, is found to be only partially stacked with C1407 (3.96–4.27 Å) in all three aminoglycoside‐A‐site complexes (Figure S11, Table S2). Based on these structural observations, it is suggested that SeU placed at the 1406 position is likely to sense a similar environment as U1406 upon ligand binding. Hence, enhanced fluorescence intensity displayed by SeU in ligand binding experiments can be attributed to a reduced stacking interaction between the fluorophore and neighboring bases.
In summary, we have introduced a multipurpose nucleoside probe functioning both as an environment‐sensitive fluorophore and an anomalous X‐ray diffraction label, which facilitated the direct correlation of RNA structure and ligand recognition under equilibrium conditions and in 3D. The new bifunctional nucleoside probe will be a powerful tool for combined fluorescence and crystallography studies, including time‐resolved experiments, for example with femtosecond X‐ray laser pulses, on dynamic RNA systems such as riboswitches.26 The responsiveness of the fluorophore to subtle conformational changes and the strong diffraction signal from the Se atom as demonstrated by using decoding site RNA suggest that such a probe, when judiciously placed, would not only allow the concurrent investigation of RNA structure and function but also could support integrated approaches to antibiotics discovery against resistant bacterial strains.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
A.N. thanks UGC, India for a graduate research fellowship. M.A.B. was supported by a US Department of Education GAANN fellowship. Instrumentation at the UC San Diego Biomolecule Crystallography Facility was acquired with funding from the National Institutes of Health, grant OD011957. S.G.S thanks DST, India (EMR/2014/000419) and Wellcome Trust‐DBT India Alliance (IA/S/16/1/502360) for research grants.
A. Nuthanakanti, M. A. Boerneke, T. Hermann, S. G. Srivatsan, Angew. Chem. Int. Ed. 2017, 56, 2640.
Contributor Information
Prof. Dr. Thomas Hermann, Email: tch@ad.ucsd.edu
Prof. Dr. Seergazhi G. Srivatsan, Email: srivatsan@iiserpune.ac.in
References
- 1.
- 1a. Sinkeldam R. W., Greco N. J., Tor Y., Chem. Rev. 2010, 110, 2579–2619; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Phelps K., Morris A., Beal P. A., ACS Chem. Biol. 2012, 7, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Dethoff E. A., Chugh J., Mustoe A. M., Al-Hashimi H. M., Nature 2012, 482, 322–330; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. M. F. Bardaro Jr , Varani G., WIREs RNA 2012, 3, 122–132. [DOI] [PubMed] [Google Scholar]
- 3. Nguyen P., Qin P. Z., WIREs RNA 2012, 3, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Serganov A., Patel D. J., Curr. Opin. Struct. Biol. 2012, 22, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Wachowius F., Höbartner C., ChemBioChem 2010, 11, 469–480; [DOI] [PubMed] [Google Scholar]
- 5b. Khakshoor O., Kool E. T., Chem. Commun. 2011, 47, 7018–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanpure A. A., Pawar M. G., Srivatsan S. G., Isr. J. Chem. 2013, 53, 366–378. [Google Scholar]
- 7.
- 7a. Latham M. P., Brown D. J., McCallum S. A., Pardi A., ChemBioChem 2005, 6, 1492–1505; [DOI] [PubMed] [Google Scholar]
- 7b. Hänsel R., Luh L. M., Corbeski I., Trantirek L., Dötsch V., Angew. Chem. Int. Ed. 2014, 53, 10300–10314; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10466–10480; [Google Scholar]
- 7c. Salgado G. F., Cazenave C., Kerkour A., Mergny J.-L., Chem. Sci. 2015, 6, 3314–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Shelke S. A., Sigurdsson S. T., Angew. Chem. Int. Ed. 2010, 49, 7984–7986; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8156–8158; [Google Scholar]
- 8b. Helmling C., Bessi I., Wacker A., Schnorr K. A., Jonker H. R. A., Richter C., Wagner D., Kreibich M., Schwalbe H., ACS Chem. Biol. 2014, 9, 1330–1339. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Zhang W., Szostak J. W., Huang Z., Front. Chem. Sci. Eng. 2016, 10, 196–202; [Google Scholar]
- 9b. Westhof E., RNA 2015, 21, 486–487; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Egli M., Pallan P. S., Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 281–305. [DOI] [PubMed] [Google Scholar]
- 10. Mortimer S. A., Kidwell M. A., Doudna J. A., Nat. Rev. Genet. 2014, 15, 469–479. [DOI] [PubMed] [Google Scholar]
- 11.See Supporting Information for details.
- 12. Pawar M. G., Nuthanakanti A., Srivatsan S. G., Bioconjugate Chem. 2013, 24, 1367–1377. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Du Q., Carrasco N., Teplova M., Wilds C. J., Egli M., Huang Z., J. Am. Chem. Soc. 2002, 124, 24–25; [DOI] [PubMed] [Google Scholar]
- 13b. Serganov A., et al., Nat. Struct. Mol. Biol. 2005, 12, 218–224; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Höbartner C., Rieder R., Kreutz C., Puffer B., Lang K., Polonskaia A., Serganov A., Micura R., J. Am. Chem. Soc. 2005, 127, 12035–12045; [DOI] [PubMed] [Google Scholar]
- 13d. Freisz S., Lang K., Micura R., Dumas P., Ennifar E., Angew. Chem. Int. Ed. 2008, 47, 4110–4113; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4178–4181; [Google Scholar]
- 13e. Sheng J., Gan J., Soares A. S., Salon J., Huang Z., Nucleic Acids Res. 2013, 41, 10476–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ennifar E., Carpentier P., Ferrer J.-L., Walter P., Dumas P., Acta Crystallogr. Sect. D 2002, 58, 1262–1268. [DOI] [PubMed] [Google Scholar]
- 15. Moazed D., Noller H. F., Nature 1987, 327, 389–394. [DOI] [PubMed] [Google Scholar]
- 16. François B., Russell R. J. M., Murray J. B., Aboul-ela F., Masquida B., Vicens Q., Westhof E., Nucleic Acids Res. 2005, 33, 5677–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ogle J. M., Carter A. P., Ramakrishnan V., Trends Biochem. Sci. 2003, 28, 259–266. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Fourmy D., Recht M. I., Blanchard S. C., Puglisi J. D., Science 1996, 274, 1367–1371; [DOI] [PubMed] [Google Scholar]
- 18b. Wong C.-H., Hendrix M., Priestley E. S., Greenberg W. A., Chem. Biol. 1998, 5, 397–406; [DOI] [PubMed] [Google Scholar]
- 18c. Carter A. P., Clemons W. M., Brodersen D. E., Morgan-Warren R. J., Wimberly B. T., Ramakrishnan V., Nature 2000, 407, 340–348; [DOI] [PubMed] [Google Scholar]
- 18d. Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G., Nature 2012, 484, 256–259. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Shandrick S., Zhao Q., Han Q., Ayida B. K., Takahashi M., Winters G. C., Simonsen K. B., Vourloumis D., Hermann T., Angew. Chem. Int. Ed. 2004, 43, 3177–3182; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3239–3244; [Google Scholar]
- 19b. Kaul M., Barbieri C. M., Pilch D. S., J. Am. Chem. Soc. 2004, 126, 3447–3453; [DOI] [PubMed] [Google Scholar]
- 19c. Srivatsan S. G., Tor Y., J. Am. Chem. Soc. 2007, 129, 2044–2053; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19d. Parsons J., Hermann T., Tetrahedron 2007, 63, 3548–3552; [Google Scholar]
- 19e. Chao P.-W., Chow C. S., Bioorg. Med. Chem. 2007, 15, 3825–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Jean J. M., Hall K. B., Proc. Natl. Acad. Sci. USA 2001, 98, 37–41; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Doose S., Neuweiler H., Sauer M., ChemPhysChem 2009, 10, 1389–1398. [DOI] [PubMed] [Google Scholar]
- 21. Vicens Q., Westhof E., Structure 2001, 9, 647–658. [DOI] [PubMed] [Google Scholar]
- 22. Zhao F., Zhao Q., Blount K. F., Han Q., Tor Y., Hermann T., Angew. Chem. Int. Ed. 2005, 44, 5329–5334; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5463–5468. [Google Scholar]
- 23. Vicens Q., Westhof E., Chem. Biol. 2002, 9, 747–755. [DOI] [PubMed] [Google Scholar]
- 24. Pfister P., Hobbie S., Vicens Q., Böttger E. C., Westhof E., ChemBioChem 2003, 4, 1078–1088. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Perez-Fernandez D., et al., Nat. Commun. 2014, 5, 3112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Kondo J., Angew. Chem. Int. Ed. 2012, 51, 465–468; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 480–483. [Google Scholar]
- 26.J. R. Stagno, et al. Nature 2017, DOI: 10.1038/nature20599.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary