

Abstract
Costello syndrome is part of the RASopathies, a group of neurocardiofaciocutaneous syndromes caused by deregulation of the RAS mitogen-activated protein kinase pathway. Heterozygous mutations in HRAS are responsible for Costello syndrome, with more than 80% of the patients harboring the specific p.Gly12Ser variant. These individuals show a homogeneous phenotype. The clinical characteristics of the Costello syndrome individuals harboring rarer HRAS mutations are less understood, due to the small number of reported cases. Here we describe the phenotypic spectrum of five additional individuals with HRAS c.38G>A; p ly13Asp, including one with somatic mosaicism, and review five previously described cases. The facial and hair abnormalities of the HRAS p.Gly13Asp individuals differ from the typical pattern observed in those showing the common HRAS (p.Gly12Ser) mutation, with less coarse facial features and slow growing, sparse hair with abnormal texture, the latter resembling the pattern observed in Noonan syndrome-like disorder with loose anagen hair and individuals harboring another amino acid substitution in HRAS (p.Gly13Cys). Although some individuals with HRAS p.Gly13Asp developed papillomata and vascular proliferation lesions, no malignant tumors occurred, similar to what was reported for individuals harboring the HRAS p.Gly13Cys. The fact that no malignant tumors were described in these individuals does not allow definitive conclusions about the risk for cancer development. It remains to be determined if substitutions of amino acid 13 in HRAS (p.Gly13Asp and p.Gly13Cys) increase the risk of tumor development.
Keywords: Costello syndrome, HRAS, mutation p.Gly13Asp, ectodermal, cancer
Introduction
A group of clinically overlapping neurocardiofaciocutaneous syndromes, known as RASopathies, are caused by heterozygous germline mutations in genes belonging to the RAS mitogen-activated protein kinase (MAPK) pathway, known for cellular proliferation, differentiation and survival [Aoki et al., 2016]. Among these disorders, Costello syndrome (CS) is a rare disorder caused by mainly de novo, heterozygous mutations in HRAS. It is characterized by failure-to-thrive in infancy, short stature, characteristic facial features, curly or sparse hair, papillomata, osteoporosis, cardiovascular malformations such as pulmonic stenosis and hypertrophic cardiomyopathy, and rhythm disturbances such as multifocal atrial tachycardia; neurological abnormalities including intellectual disability, a friendly outgoing personality, Chiari I malformation, syringomyelia and hydrocephalus, and a predisposition to malignancies, especially embryonal rhabdomyosarcoma [Gripp et al., 2006; Gripp et al., 2012; McCormick et al., 2013].
More than 90% of the mutations in CS patients are clustered in codons 12 and 13 (p.Gly12Ala/Ser/Val/Cys/Asp/Glu and p.G13Cys/Asp), constituting a mutational hotspot [Giannoulatou et al., 2013]. Phenotypic characterization has a bias towards individuals harboring the specific p.Gly12Ser mutation, present in approximately 85%. These individuals tend to show a homogeneous phenotype [Zampino et al., 2007]. Recently, Gripp et al. [2011] analyzed clinical data in 12 individuals showing the rare p.Gly13Cys HRAS mutation and observed lower rates of neurological abnormalities requiring surgery, lack of multifocal atrial tachycardia and papillomata, and long eyelashes requiring trimming, termed dolichocilia, when compared to individuals with the most frequent mutation (p.Gly12Ser). Interestingly, two of these 12 individuals showed loose anagen hair (LAH), an ectodermal condition characterized by easily pluckable, sparse, thin, and slow growing hair with abnormal hair bulb. This hair abnormality is considered a hallmark of another RASopathy – Noonan syndrome-like disorder with loose anagen hair (NSLAH) [Cordeddu et al., 2009].
We report on the clinical findings of five CS individuals harboring a rarer mutation in codon 13 (p.Gly13Asp) and review the phenotype described in another five individuals reported in the literature, in order to delineate the phenotypic spectrum.
Patients and Methods
Patient 1 was identified clinically and consent was obtained to share the information and images. To perform the molecular analysis (Sanger sequencing), the patient was enrolled in an ongoing clinical and molecular study of individuals with RASopathies, approved by the local institutional review board (Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo – CAPpesq # 0843/08). Patients 2-5 were enrolled in an IRB approved research study (Nemours #2005-051). Molecular studies were completed in a clinical diagnostic laboratory or performed as previously published [Gripp et al., 2006]. Clinical data were obtained through parent interview and documentation was obtained as possible. Signed consent was obtained in order to publish images.
We reviewed the phenotypic description of CS individuals with the HRAS p.Gly13Asp mutation reported in the English language literature, as well as available photographs.
Clinical Reports
Patient 1
This 13 year-old girl (Fig.1A-D) was the first child of healthy and non-consanguineous parents. She had a younger healthy sister. As a neonate, she developed respiratory distress requiring mechanical ventilation, and hypoglycemic episodes, which resolved with glucose infusion. She was released from the hospital after 13 days. An echocardiogram revealed pulmonary hypertension, patent ductus arteriosus and patent foramen ovale. She had swallowing difficulties and slow weight gain, requiring tube feeding for 5 months. At the age 1 year, her weight was 6 kg (well below the 5th centile). She had motor developmental delay with sitting unsupported at 1 year, walking independently and saying first words at 2 6/12 years. She attended a regular school, with learning difficulties only in Mathematics. She has been evaluated by a cardiologist and her most recent echocardiogram and electrocardiogram showed no abnormalities. She never had arrhythmias. At age 3 years, she had an abnormal increase of her OFC and cranial CT scan showed hydrocephaly, requiring ventriculostomy. At age 12, brain MRI disclosed Chiari I abnormality, syringomyelia and microgyria in the occipital region. No abnormal EEG discharges have been observed. Two other surgeries were performed: Achilles tendon release at 6 and correction of the palpebral ptosis at 7 years; the latter, without resolution of the ptosis. Ophthalmologic evaluation disclosed, besides palpebral ptosis, optic nerve hypoplasia, nystagmus and myopia. She had short stature, but never received growth hormone therapy. At age 10 she developed lower limb edema and a vascular evaluation showed greater saphenous vein insufficiency. She used compression stockings. At age 9 she developed a hemangioma in her neck, which was surgically excised and, at 12, she developed perinasal and external ear canal papillomata. A rapid, progressive scoliosis developed at adolescence. She always had slow growing hair, not requiring a haircut. Long eyelashes were evident in infancy and childhood, but never required trimming. She had a hoarse voice. Ophthalmologic evaluation disclosed myopia, nystagmus, and optic nerve atrophy.
Figure 1.
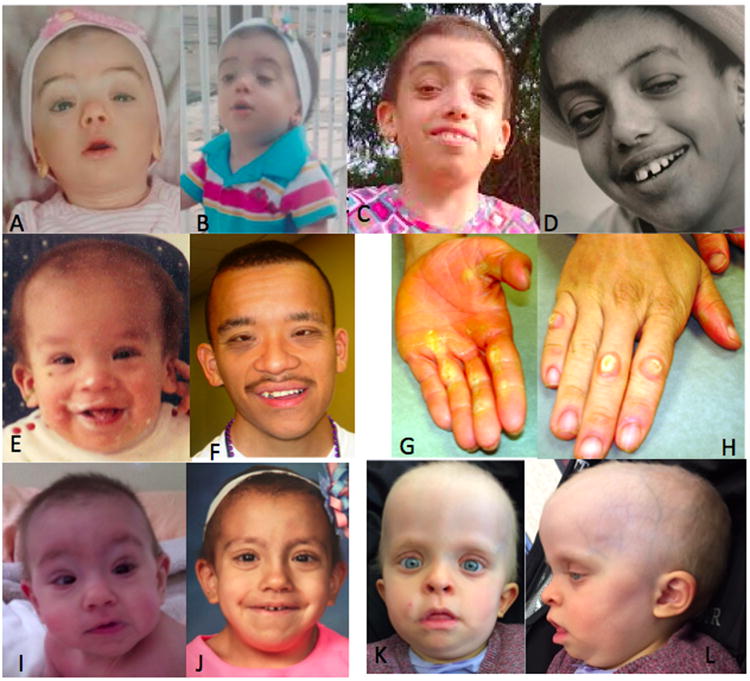
Clinical features of patients 1-4. Facial features at different ages observed in patients 1 (A – infancy, B – childhood and C and D – adolescence), 2 (E – infancy and F – adulthood) and 3 (I – infancy and J – childhood). Frontal and lateral view of patient 4 is depicted in K and L. Note the short, sparse hair with abnormal texture, especially in individuals 1 and 3, perinasal papilloma in patient 2(D), and the prominent hyperkeratosis in the hands of patient 2 (G,H).
A clinical diagnosis of Costello or cardiofaciocutaneous syndrome was made at 5 years. G-banded karyotype was normal.
At the age 13, when evaluated at our center, her clinical features were compatible with the diagnosis of a RASopathy. Among the distinct disorders within this group, the presence of slow growing hair, associated with hoarse voice and Chiari I in cranial MRI in this patient, favored the diagnosis of Noonan-like/LAH. Initially the recurrent p.Ser2Gly mutation in SHOC2 was tested and excluded. Following the investigation, the heterozygous variant c.38G>A (p.Gly13Asp) in HRAS was found and the diagnosis of Costello syndrome established.
Patient 2
This 28 year-old man (Fig. 1E, F) was born vaginally after an uncomplicated pregnancy to a 33-year-old mother of Japanese ancestry and a 32-year-old father of Croatian ancestry. His neonatal course was complicated by hypoglycemia requiring intravenous glucose supplementation for one week. Maternal hyperparathyroidism resulted in hypocalcemia and hypomagnesemia and patient 2 had two brief hypocalcemic seizures at days 8 and 9. No seizure occurred subsequently. Brain imaging studies including brain and spinal MRI had normal results. He had gastroesophageal reflux and feeding difficulties, but never required a feeding tube. He had failure-to-thrive and short stature. Echocardiograms showed mild pulmonary stenosis with mild right ventricular outflow tract obstruction, moderate right and left ventricular and septal hypertrophy, and a dilated aortic root measuring 3.5 cm at age 27 years. Progressive thoracic scoliosis was first noted at 3 years and resulted in spinal fusion at age 18 years and 19 years. His development was delayed and intellectual disability was diagnosed. He required special education and attended a life skills program in college. During his college years he lived in a dormitory with a resident advisor. Anxiety resulted in frequent emesis and need for medication. He was very social and enjoyed his work in a grocery store. This individual was included in the cohorts reported in Detweiler et al. [2013], McCormick et al. [2013] and Schwartz et al. [2013].
Patient 3
This 6 year-old girl (Fig. 1I, J) was born by repeat cesarean at 35 6/7 weeks gestation after a pregnancy complicated by polyhydramnios and non-immune fetal hydrops to a 45-year-old mother and a 47-year-old father. She required intubation with significant support including high frequency jet ventilation for respiratory distress and pleural effusions. Cardiac echography showed biventricular hypertrophy in the neonate and propranolol use resulted in improvement by age 18 months. Feeding difficulties resulted in placement of a gastric feeding tube, which was still used at age 6 years to supplement her fluid intake. Her motor development was delayed (Table I), but she was very social and had mild intellectual and learning disabilities. At age 6 years, foot position abnormalities and tight Achilles tendons were becoming more prominent. She had papillomata in her external ear canal and below her nose and hyperkeratosis on her soles. Hair remained very slow growing and short.
Table 1.
Clinical findings of the individuals presented here (patients 1 to 5) and those previously described in the literature with the HRAS p.Gly13Asp mutation, compared to findings in individuals with the HRAS p.Gly13Cys and p.Gly12Ser mutations
| Clinical Findings | Patient 1 Present report |
Patient 2 Present report |
Patient 3 Present report |
Patient 4 Present report |
Patient
5 Present report Note somatic mosaicism |
Aoki et al.,
2005 (COS30); Abe et al., 2012 (NS30) |
Aoki et al.,
2005 (COS44) |
Digilio et al.,
2008 (Case 3) |
Limongelli et al., 2008 |
Takahashi et
al., 2013 (patient 4) |
Total HRAS p.G13D (N=9) |
Gripp et al.,
2011 HRAS p.G13C (N=12) |
Gripp et al.,
2011 HRAS p.G12S |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Demographics | |||||||||||||
| Sex | F | M | F | F | F | F | M | M | F | F | 7F/3M | ||
| Age | 13y3m | 28y | 6y | 3y 8 mo | 3y 11mo | 8y3mo/18y | 3mo | 6mo | 15y | 7y | |||
| Origin | Brazil | Japan Europe | North Europe Turkey/Lebanon | Northern Europe | North Europe | Japan | Japan | Europe | Europe | Japan | |||
| Prenatal | |||||||||||||
| Polyhydramnios/fetal hydrops | - | - | + | + | + | + | 4/6 (66%) | 7/11 (64%) | 71/97 (73%)a | ||||
| Neonatal | |||||||||||||
| Gestational age (weeks) | 40 | 38 | 35 6/7 | 37 | 38 | 32 | 36 | Preterm 3/7 (43%) | Preterm 6/12 (50%) | preterma 19/48 (40%) | |||
| BW (g) | 2860 (10-25%) | 3170 (50-75%) | 4260 (>90%) | 3220 (75-90%) | 3690 (>90%) | 2500 (>90%) | 2970 (50-75%) | BW>75% 4/7 (57%) | BW>75% 10/12 (83%) | BW>90%a 28/48 (58%) | |||
| BL (cm) | 45.5 (<10%) | 50 (75%) | 48.3 (75-90%) | 48 (50%) | 51 (75-90%) | 43 (25-50%) | BL>75% 2/6 (33%) | BL>75% 5/12 (42%) | |||||
| BOFC (cm) | 35 (50-75%) | 35.5 (75-90%) | 37 (>90%) | ? | 34 (>90%) | BOFC >75% 3/4 (75%) | BOFC >75% 5/6 (83%) | BOFC>90% 25/48 (52%) | |||||
| Hypoglycemia | + | + | + | - | - | + | 4/6 (67%) | 1/? | 8/18* (44%) | ||||
| Feeding problems | + | + | + | + | - | + | 5/6 (83%) | 12/12 (100%) | 36/48 (75%)a | ||||
| Feeding tube NG/gastrostomy | + | - | + | + | + | 4/5 (80%) | 7/12 (58%) | ||||||
| Growth | |||||||||||||
| Failure to thrive | + | + | + | + | + | + | + | + | 8/8 (100%) | 11/12 (92%) | 33/33 (100%) | ||
| Height (cm) (Z-score) | 135.5 (13y5mo) (-3.38) | 153 (-3.15) | 83 (2y8mo) (-2.23) | 103 (3y11mo) (0.64) | 62 (-2.35) | 154 (-1.22) | (-2.7) | ||||||
| Short stature | + | + | + | + | - | + | - | + | 6/8 (75%) | 5/12 (42%) | 19/19 (100%) | ||
| GH deficiency | NA | - | Low normal | + | 1/3 (33%) | 2/8 (25%) | 14/30 (47%) | ||||||
| GH treatment | - | - | - | - | - | 0/5 (0%) | 1/12 (8%) | 10/30 (33%) | |||||
| Craniofacial | |||||||||||||
| Macrocephaly (absolute or relative) | absolute | + | relative | - | relative | relative | relative | absolute | OFC >98% 2/7 (28.5%) | OFC>98% 6/12 (50%) | OFC >98% 9/30 (30%) | ||
| Coarse facial appearance | - | - | - | - | - | + | + | + | - | distinct | 3/10 (30%) | Typical)b 29/29 (100%) | |
| Thick lips | + | + | + | - | - | + | + | + | + | + | 8/10 (80%) | ||
| Ophthalmologic abnormalities | + | + | + | + | - | - | - | 4/7 (57%) | |||||
| Refractory error/strabismus | myopia | Strabismus | Mild myopia | NA | |||||||||
| Optic nerve atrophy | + | NA | |||||||||||
| Nystagmus | + | + | + | - | 3/4 (75%) | 4/12 (33%) | 13/30 (43%) | ||||||
| Cardiac | |||||||||||||
| Structural anomaly | PDA, PFO, PH neonatal | mild PS; DAR | PDA | - | - | ASD | - | - | - | ASD 1/10 (10%) | ASD 3/12 (25%) | ASD 3/30 (10%) | |
| Hypertrophic cardiomyopathy | - | moderate | Mild, stable to improving | mild | - | + | - | - | Severe myectomy | + | 6/10 (60%) | 8/12 (67%) | 15/33 (45%) |
| Arrythmias | - | - | - | - | - | - | + | + | + | + | 4/10 (40%) | 1/12 (8%) | 15/33 (45%) |
| CNS/developmental milestones | |||||||||||||
| Hypotonia/delayed milestones | + | + | delayed | delayed | - | + | delayed | delayed | 7/8 (87.5%) | 12/12 (100%) | Hypotonia 22/30 (73%) c | ||
| Walked unassisted | 2y6m | 2y9m | 2y | 2y6m | 16m | Cane-assisted gait | |||||||
| First words | 2y6m | 2y | 18m | 2y | 10m; Sentences at 18 months | ||||||||
| Intellectual disability | - | + | mild | NA | - | severe | ? | moderate | + | 5/7 (71%) | 3/7 (43%) | 11/14 (78.5%) d | |
| Seizures | - | hypocalcemia | - | - | - | 1/5 (20%) | 2/12 (17%) | ||||||
| Hydrocephalus | + | - | - | NA | - | - | 1/5 (20%) | 1/12 (8%) | 3/30 (10%) | ||||
| Crowded posterior fossa | - | - | - | NA | - | - | 0/5 (0%) | 7/7 (100%) | 21/22 (95%) | ||||
| Chiari I | + | - | - | NA | - | - | 1/5 (20%) | 1/8 (12.5%) | 7/22 (32%) | ||||
| CNS surgical procedure | + | - | - | - | - | 1/5 (10%) | 1/12 (8%) | 11/22 (50%) | |||||
| Ectodermal | |||||||||||||
| Slow growing hair | + | + | + | + | + | 5/5 (100%) | 4/10 (40%) | ||||||
| Uncombable hair | + | + | + | NA | + | 4/5(80%) | 2/10 (20%) | ||||||
| Sparse hair/loose anagen hair | + | slighlty | + | + | + | + | + | - | + | 8/9 (89%) | 6/10 (60%) | Sparse 5/8 (62.5%) e | |
| Curly hair | - | + | - | - | - | + | + | - | - | 3/9 (33%) | 7/8(87.5%)e | ||
| Thin hair | + | + | + | 3/3 (100%) | |||||||||
| Dolichocilia | - | - | - | - | - | 0/5 (0%) | 8/10 (80%) | ||||||
| Freckles | + | + | 2/2 (100%) | ||||||||||
| Deep palmar/plantar creases | - | palms | - | + | ? | palms | 3/5(60%) | ||||||
| Palmo-plantar hyperkeratosis | + | + | + | + | - | + | 5/6 (83%) | 5/8 (62.5%) | |||||
| Skeletal | |||||||||||||
| Hyperextensible fingers | - | - | - | + | ? | - | 1/5 (20%) | ||||||
| Kyphosis/scoliosis | + severe | + spinal fusion | ? | ? | - | 2/3 (66%) | 2/12 (17%) | ||||||
| Limited extension of the elbows | + | ? | - | ? | - | - | 1/4 (25%) | ||||||
| Ulnar deviation | - | - | mild | - | - | 1/5 (20%) | 0/12 (0%) | 24/30 (80%) | |||||
| Abnormal foot position | - | ? | Developing age 6 years | ? | Toe walking | + | ? | + | 4/5 (80%) | ||||
| Tight Achilles tendon | + | ? | + | + | + | 4/4 (100%) | 2/12 (17%) | ||||||
| Osteoporosis | NA | osteopenia | + | 2/2 (100%) | 1/? | ||||||||
| Tumors | |||||||||||||
| Papillomata | + | - | + | - | - | + | - | - | + | 4/9 (44%) | 0/12 (0%) | 16/33 (48%) | |
| Vascular anomalies | hemangioma | - | - | hemangioma | - | Renal angioma | 3/6 (50%) | 2/10 (20%) | |||||
| Cancer | - | - | - | - | - | - | - | - | - | 0/9 (0%) | 0/12 (0%) | 4/33 (12%) | |
| Other | venous insufficiency | anxiety | ovarian cyst | Bifid uvula; somatic mosaicism | gallbladder; polyps | LM; EK; FLA; OSA | oral cavity anomalies |
NA: not assessed; ? unknown; PDA: patent duct arteriosus; PFO: patent foramen ovale; PH: pulmonary hypertension; PS: pulmonary stenosis; DAR: dilated aortic root; ASD: atrial septal defect; LM: laryngomalacia; EK: ectopic kidney; FLA: frontal lobe atrophy; OSA: obstructive sleep apnea; a. Myers et al., 2014; b. Kerr et al., 2006; c. Gripp et al., 2005; d. Axelrad et al., 2007; e. Morice-Picard et al., 2013
Patient 4
This girl (Fig. 1K, L) was born to 35-year-old mother and a 32-year old father of Scandinavian ancestry. The pregnancy was complicated by polyhydramnios and severe maternal pre-eclampsia, resulting in delivery by cesarean at 37 weeks gestation. Pleural effusion resolved without intervention. A facial hemangioma was treated with propranolol. Mild hypertrophic cardiomyopathy and a narrow aortic root were noted on the neonatal echocardiography but resolved without intervention. A weak suck and persistent feeding difficulties resulted in placement of a g-tube at age 13 months. Brain and spine imaging studies had normal results. Her development was delayed with standing at 22 months and walking independently at 2.5 years. At age 3 8/12 years she used short sentences with difficulties in pronunciation and remained almost exclusively G-tube fed.
Patient 5
This girl was born after a pregnancy complicated by late polyhydramnios to a 32-year-old mother with a history of childhood epilepsy and a 34-year-old father with rheumatoid arthritis, both parents were of North European ancestry. Two older and one younger sibling were in good health. Several tufts of darker hair were noted in the neonate and considered to present hyperpigmented moles. A weight loss of about 450 gr occurred postnatally. Otherwise, she had a normal perinatal course and fed without difficulties. A cardiac murmur led to a cardiac echography at age 2 months, with normal results. A re-evaluation through cardiology at age 13 months resulted in her being discharged from further re-evaluations. At age 4 months, failure-to-thrive with difficulty gaining weight developed. Endocrinology evaluation reportedly showed delayed bone age, and a low growth hormone level at age 1 year which normalized subsequently. She never received supplemental feeding through a feeding tube, growth hormone or other medication for her mild failure to gain weight. During adenoidectomy and pressure equalizing tube placement at age 2 years a bifid uvula was noted. Her motor and cognitive development was age appropriate and she was completely toilet trained at age 22 months. At age 3 years 11 months her OFC was 51.5 cm, height 103 cm and weight 14.6 kg.
Sparse scalp hair described as brittle and slow growing, with few interspersed regions of longer, straight and typical appearing brown hair (Fig. 2A,D,E), led to dermatology evaluations and the local use of minoxidil starting at age 3 years without significant improvement of scalp hair growth. Hyperkeratosis of knees, palms and soles (Fig 2 B, C) occurred at age 3 years. Areas of irregular skin hypo- or hyperpigmentation (Fig 2E) suggested the possibility of somatic mosaicism.
Figure 2.
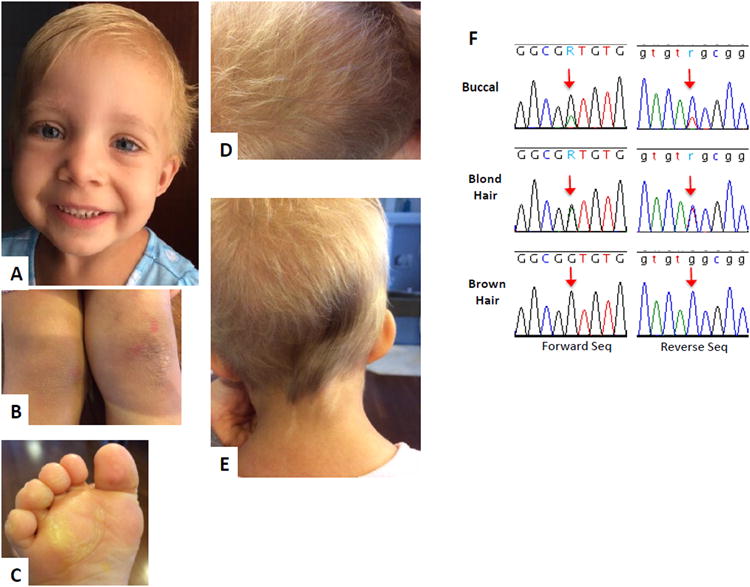
Clinical features of patient 5 and chromatogram of HRAS sequencing. A- Facial features at age 3 years 11 months, note high forehead and short blond hair, B- Hyperkeratosis on her knees, C-hyperkeratosis on her foot, D- scalp hair with a majority of short blond hairs and very few long brown hairs, E- scalp hair over her occiput showing the majority of blond hair with a patch of darker, longer and typical appearing hair, as well as streaky skin hypopigmentation compared to the relatively hyperpigmented surrounding skin of the neck. F- chromatogram of HRAS sequencing of the probands cheek swab derived DNA (labeled buccal) showing the more prominent wild type and the lesser mutant allele (arrow); the chromatogram for hair root cell derived DNA (labeled blond and brown, respectively) showing 50% mutant allele (blond hair, arrow) or only wild type allele (brown hair, arrow), respectively.
Laboratory evaluations performed clinically included a chromosomal microarray and karyotype, both were nondiagnostic. Whole exome analysis on blood derived DNA samples from the patient and her parents was performed through Baylor Genetics and showed a de novo HRAS c.38G>A; p.Gly13Asp variant, first reported as heterozygous. Upon a requested reanalysis, the variant was reported to be present in less than 50% of alleles (variant: reference read depth 39:166) and considered mosaic. After consenting into the above referenced research study, Sanger sequencing of cheek swab derived DNA on the patient and her parents and on the patient's hair root cells was performed. Blond and brown hair roots were collected and analyzed separately.
Molecular analysis of buccal and hair root samples
Buccal and hair root samples were collected in 600ul cell lysis and processed for DNA using the Puregene Tissue kit (Qiagen) following the buccal brush protocol. Hair samples were supplemented with an additional 10ul proteinase K and 30ul 1M DTT during lysis at 55°C. Sample DNA was PCR amplified for a 649bp discreet region of HRAS containing exon 2 with primers 5′-ACCTGTTCTGGAGGACGGTAA-3′ and 5′-CCTCTAGAGGAAGCAGGAGACA-3′, using an annealing temperature of 62°C. Sanger sequencing was performed in both directions using the BigDye Terminater v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) and analyzed on an ABI3130XL Genetic Analyzer.
Sequencing results confirmed the HRAS mutation reported by Baylor Genetics and was consistent with somatic mosaicism in the buccal sample, which showed more wild-type alleles than mutant (Fig. 2F). DNA isolated from blond hair was heterozygous for the mutation while DNA from brown hair was only wild-type (Fig. 2F). The mutation was not found in parental samples.
Literature review
We identified five individuals reported in the literature harboring the p.Gly13Asp [Aoki et al., 2005; Digilio et al., 2008; Limongelli et al., 2008; Takahashi et al., 2013]. The female patient described by Aoki et al. [2005] was reevaluated at age 18 years [Abe et al., 2012]. Photographs were available for the patients reported by Digilio et al., 2008 (age 3 months) and Limongelli et al., [2008] (age 15 years). The clinical features were considered present in these individuals only if mentioned by the authors or depicted by photographs.
Results
Phenotypic characteristics in our cohort and the five individuals reported in literature are detailed in Table I, displaying absolute and relative frequencies of each variable. Facial photographs of the four patients reported here in detail are depicted in figure 1 and patient 5, in figure 2.
Comparing the clinical findings in individuals with HRAS p.Gly13Asp to those with HRAS p.Gly13Cys and p.Gly12Ser (Table I), we observe that several cardinal characteristics of CS showed a frequency of 50% or more in all three groups, such as prenatal (polyhydramnios) and perinatal complications (feeding problems, often requiring tube feeding), high birth weight and OCF, failure to thrive, developmental delay/hypotonia and cardiac abnormality. On the other hand, the facial features, associated with ectodermal abnormalities, especially the hair pattern, seem to be more uniform in individuals harboring mutations in the HRAS residue 13 (p.Gly13Cys and pGly13Asp), although sparse hair is frequently observed in the three groups. Interestingly, no malignant tumors have been reported in these two groups of individuals, contrasting to the increased prevalence (12%) in individuals harboring the HRAS p.Gly12Ser.
Discussion
RASopathies constitute a group of disorders sharing phenotypic overlap. In some of these disorders, genetic heterogeneity is evident, as is the case of Noonan syndrome, the most prevalent RASopathy associated with more than ten different genes. On the other hand, CS and NSLAH are caused by mutations in HRAS and SHOC2, respectively [Aoki et al., 2016]. Before the employment of next-generation sequencing (NGS) in a diagnostic setting, the precise delineation of the phenotype was essential for a more directed molecular analysis. In the last decade, NGS allowed the simultaneous study of gene panels, such as genes responsible for the RASopathies, identifying the molecular basis of a specific RASopathy more easily and faster. Still, phenotypic delineation of each RASopathy and its genotype-phenotype correlation is important in order to most accurately tailor medical care, therapeutic intervention and genetic counseling to the individuals needs.
The mutational spectrum in CS is narrow. The vast majority of mutations affect the residue p.Gly12, mainly p.Gly12Ser (71%), followed by p.Gly12Ala (9%) and the residue p.Gly13, especially p.Gly13Cys (6%). Other mutations reported in more than two individuals in residue 12, includes p.Gly12Cys (2.5%), p.Gly12Asp (2%) and p.Gly12Val (2%) [Giannoulatou et al., 2013]. Rarer mutations in other residues have been described occasionally and the associated phenotype could differ from that observed in individuals harboring the p.Gly12Ser HRAS mutations. Amino acid substitutions in residue 12 other than the common p.12GlySer have been associated with a more severe phenotype, with lethal outcome caused by cardiac compromise, whereas mutations in other residues, such as p. Thr58 and p.Gly60, often cause an attenuated phenotype [Lorenz et al., 2012; Gripp et al., 2012; Gripp et al., 2015].
The small number of individuals harboring HRAS p.Gly13Asp mutation and lack of a systematically exploration of the clinical features in all reports preclude a robust analytical approach comparing our cohort and the groups of individuals harboring the other HRAS p.Gly13Cys and p.Gly12Ser mutations. Nevertheless, Table I shows that the patients with HRAS p.Gly13Asp frequently present with perinatal abnormalities (data from seven individuals, note somatic mosaicism in patient 5): Polyhydramnios and/or fetal hydrops (4/6), premature labor (3/7), high birth weight (4/7) and head circumference (3/4), hypoglycemic episodes (4/6), and feeding difficulties (5/6), commonly requiring nasogastric tube and/or gastrostomy (4/5). Perinatal abnormalities can be observed in all RASopathies, but are more prevalent in CS, with hypoglycemic episodes having the greatest specificity for CS [Myers et al., 2014]. Failure-to-thrive was universal and short stature (Z-score of >2SD) was observed in 6 out of 8 individuals, one of them with growth hormone deficiency. The craniofacial features described as typical in CS include relative macrocephaly, coarse facial features with curly and sparse hair, prominent epicanthal folds, long eyelashes, full nasal tip, fleshy ear lobes, and a wide mouth with full lips [Gripp et al., 2012]. We identified a different facial profile in the individuals harboring the HRAS p.Gly13Asp, which is most evident in individuals 1 and 3 (Fig.1), and recognizable in the photographs of the patient reported by Limongelli et al. [2008]. The craniofacial features include macrocephaly (absolute in 2/7 and of prenatal onset in 3/4); very short, uncombable, sparse hair; less coarseness of the facial features, although full lips and a wide mouth remain; wrinkled inferior palpebral skin, probably secondary to the skin laxity. The decreased coarseness of the facial features and the hair abnormality was also noted by Gripp et al. [2011] in the patients harboring the HRAS p.Gly13Cys mutation. Among the ectodermal findings, the hair in the typical CS individuals is usually sparse and curly. Although poor hair growth has been reported in these individuals, the extent observed here is not common [Siegel et al., 2012]. The effect of this mutation on hair is very clearly demonstrated through the analysis of the roots of the long brown “typical” appearing hair and its comparison to the short, blond and sparse appearing slow growing hair in patient 5 (Fig. 2). The typical brown hair with normal growth did not show the mutation in its roots, whereas the mutation accounts for approximately 50% of the alleles in the blond hair roots. This demonstrates the somatic mosaicism within the patient, as well the direct correlation between the hair structure and the mutation. Notably slow growing hair, not requiring haircuts, has been occasionally described in CS, mainly associated with HRAS p.Gly13Cys mutations [Gripp et al., 2011]. The hair was described as less curly than typical for most CS individuals, sparse and uncombable, similar to what was observed in some patients reported here. Recently, Gripp et al. [2016] described mutations in a novel gene (PPP1CB) associated with a Noonan-like phenotype also presenting slow growing hair, with unruly texture. Thus, slow growing hair, frequently not requiring haircuts, seems to be more widespread among the RASopathies, and not restricted to NSLAH. Interestingly, one of our patients (patient 1) was screened initially for mutations in SHOC2, as the clinical impression lead to the suspected diagnosis of NSLAH. Another ectodermal finding is palmo-plantar hyperkeratosis (Fig. 1G,H; Fig 2B,C), a characteristic shared by CS and cardiofaciocutaneous syndrome. No dolichocilia, as described in patients with p.Gly13Cys HRAS mutations, was observed in this cohort. The cardiac abnormalities were typical of CS, with a high prevalence of hypertrophic cardiomyopathy (6/10) and arrhythmias 4/10. Global developmental delay was present in all, but patient 5, with variable cognitive impairment (5/7), ranging from mild to severe. On the other hand, CNS abnormalities requiring surgical intervention were observed only in patient 1 in our cohort. Macrocephaly was prevalent in this group, but was not associated with crowded posterior fossa. As the number of individuals that had a formal CNS evaluation is small, it is too early to conclude that this group shows fewer CNS abnormalities. It is important to consider the somatic mosaicism in patient 5 in the context of her normal developmental milestones. Among the skeletal abnormalities, severe scoliosis was observed in the two older patients in our cohort, an orthopedic problem that should be closely monitored.
A serious medical concern in CS individuals is the increased risk for cancer. HRAS is a proto-oncogene, in which somatic mutations usually affecting codons 12 and 13, are responsible for several types of cancers. Among the RASopathies, CS shows the highest malignancy risk, especially for embryonal rhabdomyosarcoma, leading to the establishment of a tumor screening protocol [Gripp et al., 2005; Kratz et al., 2015]. Kerr et al. [2006] showed a higher tumor risk in individuals harboring HRAS p.Gly12Ala, rather the commonest p.Gly12Ser. Biological assays suggest that the transforming potential of HRAS is dependent of the location of the amino acid substitution [Fasano et al., 1984]. Benign tumors, such as papillomatas and/or vascular proliferative lesions, have been observed in five individuals harboring the p.Gly13Asp HRAS mutation. The prevalence of papillomatas is similar to the individuals with p.Gly12Ser HRAS. On the other hand, no patient reported or reviewed here with a mean age of approximately 9 years, presented a malignant neoplasm, similar to the individuals harboring the p.Gly13Cys mutations in HRAS [Gripp et al., 2011]. Although a preliminary impression favors the possibility that individuals with mutations in residue p.Gly13 have a lower cancer risk compared to those with mutations affecting the p.Gly12, the number of reported individuals (N=21) is too small and the cohort is young to draw a definitive conclusion. Description of further patients, with long-term follow-up, is required. In the meantime, we suggest that the screening tumor protocol proposed for CS should be applied to all individuals with this diagnosis, independently of the identified mutation.
Acknowledgments
This study was supported by FAPESP (CEPID 2013/08028-1) and CNPq (302605/2013-4). Research reported in this publication was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under award number grants P30GM114736 and P20GM103446.
Footnotes
The authors declare no conflict of interests.
References
- Abe Y, Aoki Y, Kuriyama S, Kawame H, Okamoto N, Kurosawa K, Ohashi H, Mizuno S, Ogata T, Kure S, Niihori T, Matsubara Y. Costello and CFC syndrome study group in Japan. Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan: findings from a nationwide epidemiological survey. Am J Med Genet A. 2012;158A:1083–1094. doi: 10.1002/ajmg.a.35292. [DOI] [PubMed] [Google Scholar]
- Axelrad ME, Nicholson L, Stabley DL, Sol-Church K, Gripp KW. Longitudinal assessment of cognitive characteristics in Costello syndrome. Am J Med Genet A. 2007;143A:3185–3193. doi: 10.1002/ajmg.a.31968. [DOI] [PubMed] [Google Scholar]
- Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016;61:33–39. doi: 10.1038/jhg.2015.114. [DOI] [PubMed] [Google Scholar]
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, Lipzen A, Zampino G, Mazzanti L, Digilio MC, Martinelli S, Flex E, Lepri F, Bartholdi D, Kutsche K, Ferrero GB, Anichini C, Selicorni A, Rossi C, Tenconi R, Zenker M, Merlo D, Dallapiccola B, Iyengar R, Bazzicalupo P, Gelb BD, Tartaglia M. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detweiler S, Thacker M, Hopkins E, Conway L, Gripp KW. Orthopedic manifestations and implications for individuals with Costello Syndrome. Am J Med Genet Part A. 2013;161A:1940–1949. doi: 10.1002/ajmg.a.36047. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Sarkozy A, Capolino R, Chiarini Testa MB, Esposito G, de Zorzi A, Cutrera R, Marino B, Dallapiccola B. Costello syndrome: clinical diagnosis in the first year of life. Eur J Pediatr. 2008;167:621–628. doi: 10.1007/s00431-007-0558-0. [DOI] [PubMed] [Google Scholar]
- Fasano O, Aldrich T, Tamanoi F, Taparowsky E, Furth M, Wigler M. Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc Natl Acad Sci U S A. 1984;81:4008–4012. doi: 10.1073/pnas.81.13.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoulatou E, McVean G, Taylor IB, McGowan SJ, Maher GJ, Iqbal Z, Pfeifer SP, Turner I, Burkitt Wright EM, Shorto J, Itani A, Turner K, Gregory L, Buck D, Rajpert-De Meyts E, Looijenga LH, Kerr B, Wilkie AO, Goriely A. Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS mutations in the paternal germline. Proc Natl Acad Sci U S A. 2013;110:20152–20157. doi: 10.1073/pnas.1311381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Aldinger KA, Bennett JT, Baker L, Tusi J, Powell-Hamilton N, Stabley D, Sol-Church K, Timms AE, Dobyns WB. A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am J Med Genet A. 2016 Jun 5; doi: 10.1002/ajmg.a.37781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Hopkins E, Serrano A, Leonard NJ, Stabley DL, Sol-Church K. Transmission of the rare HRAS mutation (c. 173C > T; p.T58I) further illustrates its attenuated phenotype. Am J Med Genet A. 2012;158A:1095–1101. doi: 10.1002/ajmg.a.35294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Hopkins E, Sol-Church K, Stabley DL, Axelrad ME, Doyle D, Dobyns WB, Hudson C, Johnson J, Tenconi R, Graham GE, Sousa AB, Heller R, Piccione M, Corsello G, Herman GE, Tartaglia M, Lin AE. Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am J Med Genet A. 2011;155A:706–716. doi: 10.1002/ajmg.a.33884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Lin AE. Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med. 2012;14:285–292. doi: 10.1038/gim.0b013e31822dd91f. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Lin AE, Stabley DL, Nicholson L, Scott CI, Jr, Doyle D, Aoki Y, Matsubara Y, Zackai EH, Lapunzina P, Gonzalez-Meneses A, Holbrook J, Agresta CA, Gonzalez IL, Sol-Church K. HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A. 2006;140:1–7. doi: 10.1002/ajmg.a.31047. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Sol-Church K, Smpokou P, Graham GE, Stevenson DA, Hanson H, Viskochil DH, Baker LC, Russo B, Gardner N, Stabley DL, Kolbe V, Rosenberger G. An attenuated phenotype of Costello syndrome in three unrelated individuals with a HRAS c.179G>A (p.Gly60Asp) mutation correlates with uncommon functional consequences. Am J Med Genet A. 2015;167A:2085–2097. doi: 10.1002/ajmg.a.37128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:72–77. doi: 10.1002/ajmg.c.30065. [DOI] [PubMed] [Google Scholar]
- Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, Stef M, Tang B, Eden OB, O'Sullivan J, De Sandre-Giovannoli A, Reardon W, Brewer C, Bennett C, Quarell O, M'Cann E, Donnai D, Stewart F, Hennekam R, Cavé H, Verloes A, Philip N, Lacombe D, Levy N, Arveiler B, Black G. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet. 2006;43:401–405. doi: 10.1136/jmg.2005.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, Bier A, Eichhorn B, Blank C, Kraus C, Kohlhase J, Pauli S, Wildhardt G, Kutsche K, Auber B, Christmann A, Bachmann N, Mitter D, Cremer FW, Mayer K, Daumer-Haas C, Nevinny-Stickel-Hinzpeter C, Oeffner F, Schlüter G, Gencik M, Überlacker B, Lissewski C, Schanze I, Greene MH, Spix C, Zenker M. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;112:1392–1397. doi: 10.1038/bjc.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limongelli G, Pacileo G, Digilio MC, Calabro' P, Di Salvo G, Rea A, Miele T, Frigiola A, Sarkozy A, Dallapiccola B, Marino B, Calabro' R. Severe, obstructive biventricular hypertrophy in a patient with Costello syndrome: Clinical impact and management. Int J Cardiol. 2008;130:e108–110. doi: 10.1016/j.ijcard.2007.06.107. [DOI] [PubMed] [Google Scholar]
- Lorenz S, Petersen C, Kordaß U, Seidel H, Zenker M, Kutsche K. Two cases with severe lethal course of Costello syndrome associated with HRAS p.G12C and p.G12D. Eur J Med Genet. 2012;55:615–619. doi: 10.1016/j.ejmg.2012.07.007. [DOI] [PubMed] [Google Scholar]
- McCormick EM, Hopkins E, Conway L, Catalano S, Hossain J, Sol-Church K, Stabley DL, Gripp KW. Assessing genotype-phenotype correlation in Costello syndrome using a severity score. Genet Med. 2013;15:554–557. doi: 10.1038/gim.2013.6. [DOI] [PubMed] [Google Scholar]
- Morice-Picard F, Ezzedine K, Delrue MA, Arveiler B, Fergelot P, Taïeb A, Lacombe D, Boralevi F. Cutaneous manifestations in Costello and cardiofaciocutaneous syndrome: report of 18 cases and literature review. Pediatr Dermatol. 2013;30:665–673. doi: 10.1111/pde.12171. [DOI] [PubMed] [Google Scholar]
- Myers A, Bernstein JA, Brennan ML, Curry C, Esplin ED, Fisher J, Homeyer M, Manning MA, Muller EA, Niemi AK, Seaver LH, Hintz SR, Hudgins L. Perinatal features of the RASopathies: Noonan syndrome, cardiofaciocutaneous syndrome and Costello syndrome. Am J Med Genet A. 2014;164A:2814–2821. doi: 10.1002/ajmg.a.36737. [DOI] [PubMed] [Google Scholar]
- Schwartz DD, Katzenstein JM, Hopkins E, Stabley DL, Sol-Church K, Gripp KW, Axelrad ME. Verbal memory functioning in adolescents and young adults with Costello syndrome: Evidence for relative preservation in recognition memory. Am J Med Genet Part A. 2013;161A:2258–2265. doi: 10.1002/ajmg.a.36078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel DH, Mann JA, Krol AL, Rauen KA. Dermatological phenotype in Costello syndrome: consequences of Ras dysregulation in development. Br J Dermatol. 2012;166:601–607. doi: 10.1111/j.1365-2133.2011.10744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Ohashi H. Craniofacial and dental malformations in Costello syndrome: A detailed evaluation using multi-detector row computed tomography. Congenit Anom (Kyoto) 2013;53:67–72. doi: 10.1111/cga.12004. [DOI] [PubMed] [Google Scholar]
- Zampino G, Pantaleoni F, Carta C, Cobellis G, Vasta I, Neri C, Pogna EA, De Feo E, Delogu A, Sarkozy A, Atzeri F, Selicorni A, Rauen KA, Cytrynbaum CS, Weksberg R, Dallapiccola B, Ballabio A, Gelb BD, Neri G, Tartaglia M. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat. 2007;28:265–272. doi: 10.1002/humu.20431. [DOI] [PubMed] [Google Scholar]