

Abstract
Pathogenic variants in PHOX2B lead to congenital central hypoventilation syndrome (CCHS), a rare disorder of the nervous system characterized by autonomic dysregulation and hypoventilation typically presenting in the neonatal period, although a milder late-onset (LO) presentation has been reported. More than 90% of cases are caused by polyalanine repeat mutations (PARMs) in the C-terminus of the protein; however non-polyalanine repeat mutations (NPARMs) have been reported. Most NPARMs are located in exon 3 of PHOX2B and result in a more severe clinical presentation including Hirschsprung disease (HSCR) and/or peripheral neuroblastic tumors (PNTs). A previously reported nonsense pathogenic variant in exon 1 of a patient with LO-CCHS and no HSCR or PNTs leads to translational reinitiation at a downstream AUG codon producing an N-terminally truncated protein. Here we report additional individuals with nonsense pathogenic variants in exon 1 of PHOX2B. In vitro analyses were used to determine if these and other reported nonsense variants in PHOX2B exon 1 produced N-terminally truncated proteins. We found that all tested nonsense variants in PHOX2B exon 1 produced a truncated protein of the same size. This truncated protein localized to the nucleus and transactivated a target promoter. These data suggest that nonsense pathogenic variants in the first exon of PHOX2B likely escape nonsense mediated decay (NMD) and produce N-terminally truncated proteins functionally distinct from those produced by the more common PARMs.
Keywords: CCHS, PHOX2B, mRNA reinitiation
INTRODUCTION
Congenital central hypoventilation syndrome (CCHS) is a rare disorder of the autonomic nervous system characterized by decreased sensitivity to hypercarbia (elevated carbon dioxide (CO2) levels in the blood) and hypoxia, (tissues depleted of oxygen) in the brainstem leading to an impaired respiratory response. Overt clinical signs of respiratory distress such as tachypnea, nasal flaring, and subcostal retractions are absent. CCHS is typically diagnosed in infancy, however diagnoses have been made as late as adulthood [Antic et al., 2006; Bygarski et al., 2013; Low et al., 2014; Rand et al., 2014; Trang et al., 2004; Weese-Mayer et al., 2013; Weese-Mayer et al., 2005]. Infants with CCHS increase their ventilation while awake, but have severely elevated levels of pCO2 during non-REM sleep [Rand et al., 2014]. During sleep, the infant’s oxygen levels fall and carbon dioxide levels rise with no compensatory rise in respiratory rate or signs of agitation. If untreated, infants may develop fixed pulmonary hypertension and right-sided heart failure.
CCHS is classified as a neurocristopathy, a diverse group of conditions arising from defects in the migration, division and differentiation of neural crest cells which leads to autonomic nervous system dysfunction (ANSD) [Bolande 1997]. Patients with CCHS can have additional manifestations of ANSD including abnormalities of temperature regulation, sweating, blood pressure, and cardiac rhythm [Gronli et al., 2008; Rand et al., 2014; Saiyed et al., 2016]. Infants with CCHS are at risk for abnormalities in organs of neural crest origin, such as altered gut motility or Hirschsprung disease (HSCR) and peripheral neuroblastic tumors (PNTs): extracranial tumors of neural crest origin including neuroblastomas, ganglioneuromas and ganglioneuroblastomas [Weese-Mayer et al., 2010].
More than 90% of CCHS causing pathogenic variants are located in a polyalanine repeat region in the third exon of PHOX2B. PHOX2B encodes a highly conserved homeobox domain transcription factor expressed in the peripheral and central autonomic nervous systems during embryonic development. PHOX2B is critical for the development of autonomic neural crest cell derivatives and controls hindbrain motor neuron development [Pattyn et al., 2000; Pattyn et al., 1999]. Unaffected individuals have 20 alanines or fewer [Amiel et al., 2003; Weese-Mayer et al., 2010]. In contrast, those heterozygous for 24 or 25 repeats (e.g., genotype 20/24) can have a mild phenotype only manifesting during illness or exposure to respiratory depressants, [Repetto et al., 2009], and those heterozygous for 26 to 33 alanine repeats are fully affected [Weese-Mayer et al., 2010; Weese-Mayer et al., 2003].
Non-polyalanine repeat pathogenic variants (NPARMs) have been reported in fewer than 100 patients [Weese-Mayer et al., 2010]. A majority of these pathogenic variants are frameshifts in PHOX2B exon 3, affect the polyalanine repeat region, and result in a severe phenotype with extensive gut involvement, increased risk of peripheral neuroblastic tumors, cardiac arrhythmia and need for continuous ventilator support (CVS) [Trochet et al., 2005; Weese-Mayer et al., 2010]. However, frameshift variants in the 3′ region of exon 2 and the 5′ region of exon 3 are associated with a milder form of CCHS that is variably penetrant [Berry-Kravis et al., 2006; Bygarski et al., 2013; Low et al., 2014]. It has been postulated that frameshift variants in this region result in a phenotype similar to smaller polyalanine repeat expansions and are more likely to result in incomplete penetrance or adult-onset disease [Berry-Kravis et al., 2006; Weese-Mayer et al., 2010].
Missense pathogenic variants primarily affect the homeo-domain of the protein and have a variable clinical presentation from only PNTs to PNTs plus CCHS and HSCR [Trochet et al., 2005; Trochet et al., 2009; Weese-Mayer et al., 2010]. Pathogenic variants primarily arise de novo, although inheritance from unaffected parents with incomplete penetrance or germline mosaicism has been reported [Parodi et al., 2008; Trochet et al., 2008].
To our knowledge, four nonsense pathogenic variants in PHOX2B have been reported. A c.463A>T (p.Lys155*) variant in exon 3 was reported in a patient with CCHS, HSCR and neuroblastoma [Weese-Mayer et al., 2003]. In vitro studies showed that the p.Lys155* variant produces a stable, C-terminally truncated protein that does not appropriately localize to the nucleus and is unable to transactivate the dopamine-β-hydroxylate (DBH) and PHOX2A promoters [Trochet et al., 2009]. Three nonsense pathogenic variants have been reported in exon 1: c.18T>C (p.Tyr6*), c.23delA (p.Tyr8*) and c.42C>A (p.Tyr14*) [Magalhaes et al., 2015; Parodi et al., 2008; Trochet et al., 2009]. Detailed clinical information was not available for the patient with the p.Tyr6* variant. Magalhaes et. al. [2015] reported a de novo p.Tyr8* variant in a patient who presented at nine, eleven, and thirteen months of age with severe respiratory failure during viral infections. Once the diagnosis of CCHS was made, bilevel positive airway pressure (BiPAP) non-invasive ventilation was initiated and the patient improved. At four years of age she required non-invasive ventilation during sleep and had no other systemic problems [Magalhaes et al., 2015]. The c.42C>T (p.Tyr14*) variant was reported in a patient who presented with LO-CCHS at 5 weeks of age without HSCR or PNTs [Trochet et al., 2009]. The p.Tyr14* variant leads to translational reinitiation at an alternate ATG start site and produces an N-terminally truncated protein missing the first 18 or 21 amino acids. Besides the differing clinical presentations, the p.Lys155* and p.Tyr14* variants have different subcellular localization and protein activity. The p.Lys155* variant has decreased nuclear localization and is unable to activate target promoters, whereas the p.Tyr14* variant localizes to the nucleus and transactivates the DBH and PHOX2A promoters similar to wild-type.
Here, we report a term infant with a novel NPARM, c.13G>T (p.Glu5*), who presented with cyanosis and hypoventilation at birth, and a ten year old child with c.18T>C (p.Tyr6*). Neither had detectable HSCR or PNTs or required waking ventilator support via tracheostomy, although both needed assisted ventilation during sleep. We found that the p.Glu5*, p.Tyr6*, and p.Tyr8* variants produce N-terminally truncated proteins that localize to the nucleus and activate the DBH target promoter similar to p.Tyr14*. These data suggest a genotype-phenotype correlation between nonsense variants in exon 1 of PHOX2B and CCHS, and that the location of nonsense variants within the gene should be taken into consideration when managing patients with this disorder.
CLINICAL ASSESSMENT
Patient 1 was a two-year-old male child born at 39 weeks to a 26-year-old gravida 1 mother by cesarean for failure to progress. His pregnancy was uncomplicated and he had normal ultrasounds at 19 and 22 weeks gestation. Maternal medications used during pregnancy included prenatal vitamins and docosahexaenoic acid. Following delivery, he was cyanotic and required treatment with nasal continuous positive airway pressure ventilation (NCPAP). He was admitted to the neonatal intensive care unit (NICU) with a respiratory rate of 38 breaths per minute and no respiratory distress. After a chest radiograph confirmed clear lung fields and no evidence of pneumothorax, he was weaned to nasal cannula; the initial capillary blood gas obtained while awake was normal (pH of 7.35; pCO2 54; pO2 37; base deficit +3 (normal capillary pCO2 45–55 mm Hg)). Despite the supplemental oxygen during sleep, he had compensated respiratory acidosis with hypoxemia (pH 7.4; pCO2 62; pO2 45; base deficit +13). Because of his respiratory acidosis ventilatory failure, he was placed on non-invasive positive pressure ventilation (NIPPV), and this normalized his pCO2 levels. His supplemental oxygen needs were minimal when awake, but as high as FiO2 0.45 when asleep.
Further medical evaluation included chest radiographs, cardiac echocardiogram, esophageal pH probe, magnetic resonance imaging of the brain, nasopharyngoscopy, and metabolic studies. All results were normal. A 12-hour oxypneumogram revealed 31 episodes of central apnea ranging in length from 12–33 seconds and excessive central hypopnea (shallow breathing) associated with oxygen desaturations without compensatory tachypnea or increased respiratory effort. A loading dose of caffeine citrate was administered, along with a daily dose, but this had no effect on respirations and did not permit weaning of supplemental oxygen.
The patient was unable to wean completely from supplemental oxygen. The lack of increased respiratory effort in the face of respiratory failure and the central apnea and hypopnea suggested impaired respiratory drive. The brain/brainstem MRI excluded gross structural abnormalities and the failure of caffeine therapy suggested against apnea of prematurity. Full gene sequencing of PHOX2B identified a c.13G>T (p.Glu5*) likely pathogenic variant confirming a diagnosis of CCHS. The mother tested negative for the pathogenic variant, the father was not tested. At two years, he required non-invasive ventilation only during sleep and has no reported HSCR or PNTs.
Patient 2 was a ten-year-old male who was a product of an uncomplicated term gestation. He was admitted at one month with cyanosis and hypotonia. He required intubation and mechanical ventilation for respiratory failure, but was successfully weaned and extubated to supplemental oxygen via nasal cannula. After a prolonged hospital stay, he was discharged without a clear diagnosis and on supplemental oxygen via nasal cannula. He continued to have cyanotic spells at home and did not achieve normal milestones. He was readmitted at 6–7 months with a severe cyanotic episode and was intubated. He failed extubation due to persistent hypercapnea. PHOX2B variant analysis was reported negative; however, he was given a clinical diagnosis of CCHS. Since age two years, he has been ventilated only during sleep. He has required changes to his ventilator settings every one to two years, but has otherwise remained stable. During the day he wears a Passy Muir valve. He intermittently struggles with constipation, but this is effectively managed with Miralax. EKG’s and Holter monitoring have not revealed arrhythmia. At age nine, repeat PHOX2B testing detected a heterozygous c.18T>C (p.Tyr6*) variant. His parents did not pursue familial testing.
METHODS
Plasmid generation
Human PHOX2B (NM_003924.3), with a C-terminal Myc tag was synthesized using the GeneArt Gene Synthesis service (ThermoFisher Scientific). The PHOX2B mutants were generated from the wild-type template by PCR using forward primers containing the mutation and the In-Fusion Cloning kit (Clontech). The DBH luciferase reporter construct was a kind gift from Dr. Kuixing Zhang.
Cell culture and transfection
HeLa cells were maintained in 5.0% CO2 at 37 °C in DMEM (SH3024301; HyClone) supplemented with 10% (vol/vol) FBS. For transient expression of the Phox2B constructs, HeLa cells were transfected via Lipofectamine 3000 (Life Technologies) using the manufacturer’s recommended protocols.
Luciferase Assay
HeLa cells were grown in DMEM (HyClone) supplemented with 10% Fetal Bovine Serum (VWR) and 1× Penicillin/Streptomycin (HyClone) in 24 well plates. When the cells reached 80% confluence, co-transfection of PHOX2B-pcDNA3.1 (245ng), DBH-pGL3-basic (245ng, courtesy of Dr. Sucheta Vaingankar University of California, San Diego), and pRL-TK (24.5ng, Promega) was performed using Lipofectamine 3000 (ThermoFisher) following the manufacturer’s suggested protocol. Forty eight hours after transfection, cell lysates were collected in 100ul of 1× Passive Lysis Buffer (Promega). Firefly and Renilla luciferase activity was detected using the Dual-Luciferase Reporter Assay System (E1910, Promega) following the manufacturer’s suggested protocol. Luciferase activity was read using a Promega GlowMax 96 well luminometer (Promega).
Western Blots
Cell Lysates were collected in 200ul of Laemmli Buffer (BioRad), heated to 98C for five minutes, and sonicated. Ten ul of lysate was ran on precast gradient gels, 4–20% (Mini-PROTEAN® TGX™, BioI-Rad), and then transferred to a nitrocellulose membrane using the BioRad Turbo Transfer System. Primary antibody incubation was performed with rabbit anti-myc antbodies (ab9106; Abcam) or mouse anti-tubulin (T9026; Sigma), followed by a secondary antibody incubation using goat-anti-rabbit-HRP antibodies (G21234; Life Technologies) or anti-mouse-HRP antibodies (F21453; Life Technologies). Bands were then visualized with Luminata Forte Western HRP substrate (Millipore) and imaged using UVP-gel documentation system.
Immunofluorescence
Cells transfected with the Phox2B constructs were fixed in 3% (wt/vol) paraformaldehyde/phosphate buffered saline (PBS) for 10 min followed by permeablization using 0.4% (wt/vol) Triton X-100/PBS for 15 min. Expressed Phox2B was labeled with rabbit polyclonal anti-myc (ab9106; Abcam) followed by a secondary antibody incubation using Alexa-Fluor 488 conjugated goat anti-rabbit (Life Technologies). DNA was detected with Hoechst dye 33258. Coverslips were mounted using 10% (wt/vol) Mowiol 4–88 (Polysciences). Images were obtained using Nikon Ni-E microscope (40×/0.75 Plan Fluor; Nikon) with a CCD camera (CoolSNAP Myo; Photometrics) linked to a workstation running NES-Element software (Nikon) was used.
RESULTS
PHOX2B nonsense variants in exon 1 produce N-terminally truncated proteins
Trochet et al. [2009] showed that the PHOX2B p.Tyr14* variant leads to an N-terminally truncated protein through translational reinitiation at either p.Met18 or p.Met21. Given their close proximity within the gene and similar clinical presentation, we hypothesized that the p.Glu5* and p.Tyr6* variants detected in our patients and the previously published p.Tyr8* also produce an N-terminally truncated PHOX2B protein (Figure 1). Mutant and wild-type myc-tagged cDNA constructs were expressed in HeLa cells, since they do not express endogenous PHOX2B. Immunoblot analysis using anti-myc antibodies demonstrated that p.Glu5*, p.Tyr6* and p.Tyr8* constructs all expressed a truncated protein with similar size as the previously reported p.Tyr14* mutant (Figure 2). Interestingly, the wild-type construct also expressed a minor band at the same size as the truncated proteins and the p.Tyr8* construct expressed a minor band at the same size as wild-type.
Figure 1. Location of nonsense pathogenic variants within PHOX2B.
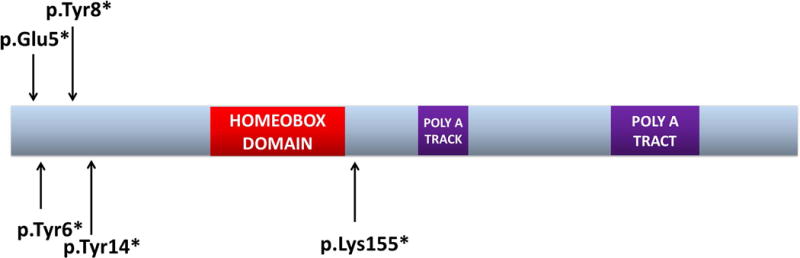
To date, five nonsense variants have been reported in PHOX2B. The p.Lys155* variant is predicted to lead to a C-terminally truncated protein, and was reported in a patient with CCHS, HSCR and neuroblastoma. In contrast, the other four nonsense variants are located in the first exon of PHOX2B and have been reported in patients who do not require daytime ventilation. HSCR and PNTs have not been reported in these patients.
Figure 2. Expression of PHOX2B transcripts in Hela cells.
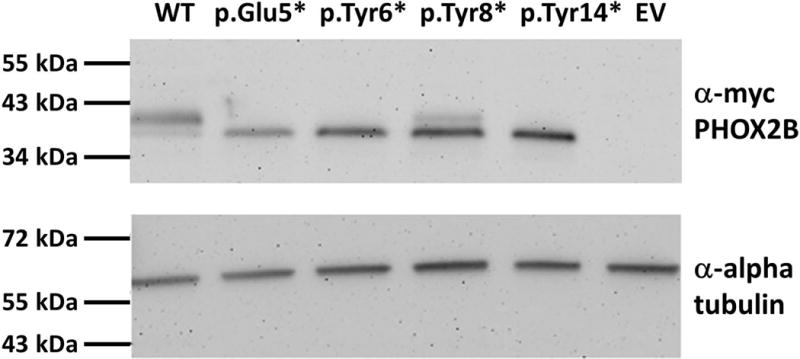
Expression constructs containing wild-type (WT) PHOX2B, N-terminal nonsense pathogenic variants, or empty vector (EV) were transiently transfected into Hela cells. Alpha tubulin was used as a loading control. All of the N-terminal nonsense variants produce proteins that are smaller in size than the WT protein. The WT construct expressed a minor band at the same size as the truncated proteins and the p.Tyr8* construct expressed a minor band the same size as the WT suggesting a small amount of nonspecific expression from the in vitro constructs.
To determine if p.Met18 or p.Met21 was the secondary ATG start site, the overexpressed proteins were isolated, digested with trypsin to cleave into smaller fragments and analyzed by mass spectrometry. An N-terminal peptide containing either start site was not detected (data not shown). Additional studies are required to determine which methionine is the true novel start site.
PHOX2B N-terminally truncated proteins localize to the nucleus
As a transcription factor, PHOX2B localization is almost exclusively nuclear. Studies have shown that expansion of the polyalanine repeat region causes protein aggregation and mislocalization to the cytoplasm [Bachetti et al., 2005; Trochet et al., 2005]. The p.Lys155* variant produces a C-terminally truncated protein that shows substantially decreased, but not completely abolished, nuclear localization. In contrast, the p.Tyr14* pathogenic variant has normal nuclear localization [Trochet et al., 2009]. To determine if the p.Glu5*, p.Tyr6*, and p.Tyr8* variants encoded proteins localizing to the nucleus, we overexpressed cDNAs containing these alleles as well as the wild-type and p.Tyr14* variant in HeLa cells and performed immunolabeling. All mutant proteins were expressed and localized correctly to the nucleus (Figure 3).
Figure 3. Localization of PHOX2B transcripts in Hela cells.
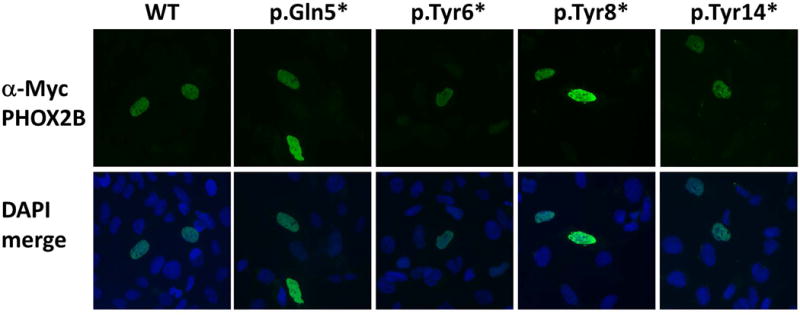
Expression constructs containing WT PHOX2B, N-terminal nonsense pathogenic variants, or empty vector were transiently transfected into Hela cells. All constructs properly localize to the nucleus in a pattern consistent with wild-type.
PHOX2B N-terminally truncated proteins transactivate a target promoter
Polyalanine expansions in PHOX2B result in a marked reduction in the ability of the protein to activate target gene promoters, such as DBH, PHOX2A, and TLX2 [Adachi et al., 2000; Di Lascio et al., 2013]. However, the truncated protein produced by the p.Tyr14* variant dimerizes like wild-type and transactivates the PHXO2A and DBH promoters [Trochet et al., 2009]. We therefore tested the ability of the additional variants to activate the DBH target promoter. Transactivation by all variants was similar to the wild-type protein (Figure 4), suggesting loss of the extreme N-terminus of the protein does not affect its ability to activate target promoters.
Figure 4. Luciferase activity resulting from transactivation of the DBH target promoter.
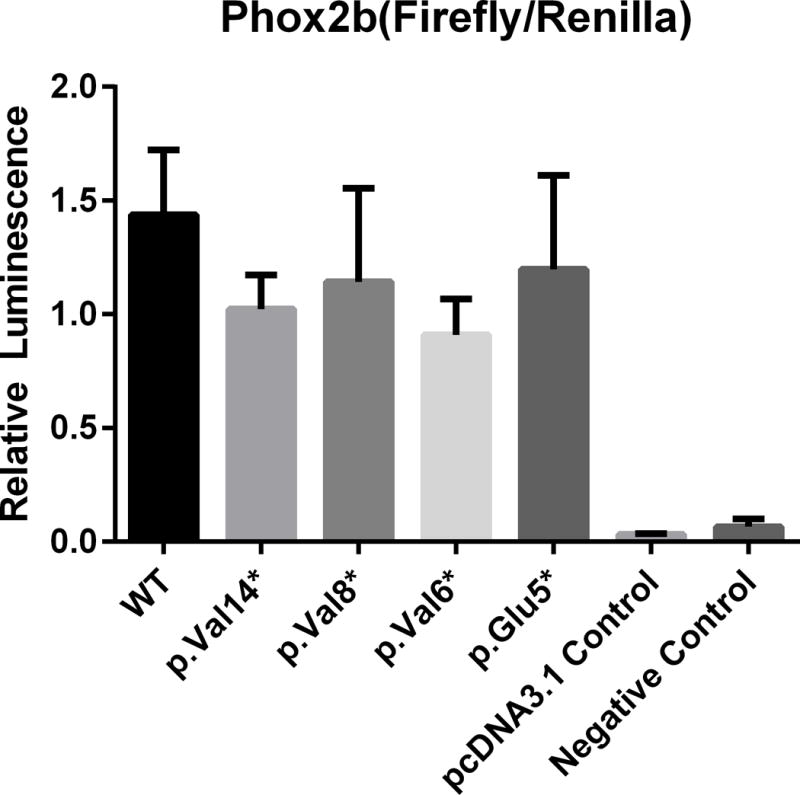
The bars indicate the relative transcriptional activity of the DBH reporter promoter construct upon co-transfection with plasmids containing the PHOX2B nonsense variants. The results are mean values ± SD (error bars) of experiments performed in triplicate. No significant differences between the WT and variant constructs were observed.
DISCUSSION
We report two individuals with CCHS and nonsense variants encoded in the first exon of PHOX2B. Expression of PHOX2B cDNAs encoding these variants resulted in production of a smaller protein, that we hypothesize results from reinitiation of translation downstream of the premature stop codon. We found that these smaller proteins localize to the nucleus and activate transcription of the same target genes as the wild type protein. We hypothesize that a similar phenomenon occurs in vivo, and that these truncated proteins have a distinct function compared to wild-type proteins or those containing altered polyalanine repeats.
Term infants displaying cyanosis immediately following birth is not uncommon. Often such infants require a brief course of respiratory support and/or antibiotics and rarely need additional intervention. Occasionally, depending upon complicating factors, the infants will require administration of surfactant or assisted ventilation. The two patients reported here presented at birth and at age one month, without complicating factors such as meconium-stained amniotic fluid, retained fetal lung fluid, pneumothorax, or maternal gestational diabetes.
The classic presentation of CCHS includes ANSD and hypoventilation with monotonous respiratory rates and shallow breathing during sleep, and occasionally during waking. Identification of a PHOX2B pathogenic variant establishes the diagnosis of CCHS. Additional evaluations for management of patients with CCHS include: 1) a full assessment of the ANS including tilt table testing and assessment for temperature dysregulation, 2) a barium enema and possibly a rectal biopsy to evaluate for Hirschsprung disease, 3) imaging for tumors of neural crest origin including chest radiograph/abdominal ultrasonography in infancy and later thoracic/abdominal magnetic resonance imaging or computed topography, 4) a neurocognitive evaluation, 5) an evaluation for cardiac arrhythmia, 6) cardiac echocardiograms to assess for cor pulmonale secondary to sustained hypoxemia. If molecular testing does not confirm a diagnosis of CCHS, other causes of hypoventilation such as cardiac disease, primary lung disease, inborn errors of metabolism, and ventilatory muscle weakness should be evaluated [Weese-Mayer et al., 2010].
Correlations between PHOX2B genotype and CCHS phenotype have been suggested. The respiratory phenotype appears to be more severe in patients with longer PARMs, requiring CVS [Berry-Kravis et al., 2006; Matera et al., 2004; Weese-Mayer et al., 2003]. NPARMs in PHOX2B have been associated with either a severe clinical presentation including CVS, HSCR and PNTs, or a variable milder clinical presentation often not detected until adulthood, depending on the type and location of the variant [Berry-Kravis et al., 2006; Bygarski et al., 2013; Low et al., 2014; Trochet et al., 2005; Weese-Mayer et al., 2010].
Nonsense variants are generally assumed to disrupt gene function by leading to a complete absence of the gene product, by lack of transcription or NMD of an abnormal transcript. Therefore, a more severe clinical presentation would be expected in patients carrying nonsense variants in PHOX2B. Indeed, the p.Lys155* variant in exon 3 was reported in a patient with classic CCHS including HSCR and neuroblastoma [Weese-Mayer et al., 2003]. However, no patients with a nonsense mutation in exon 1 for whom clinical information is available required waking ventilation support, or had HSCR or PNTs. PNTs, such as neuroblastoma, generally present in children younger than two years, with 90% of patients under five [Howman-Giles et al., 2007]. Therefore, while it is possible that these patients will develop tumors, the likelihood decreases greatly with age. We hypothesize that the clinical presentation observed in patients with nonsense mutations in PHOX2B exon 1 is the result of translational reinitiation at p.Met18 or p.Met21, and constitutes a distinctive group of patients with CCHS.
The finding of translational reinitiation following an early premature termination (PT) is increasingly observed as more variants are studied. Frequently these N-terminally truncated proteins are hypomorphic and associate with milder phenotypes. For example, pathogenic variants in ARX cause a range of clinical presentations: Lissencephaly with abnormal genitalia (XLAG), infantile spasms (ISSX), syndromic intellectual disability, and non-syndromic intellectual disability. The phenotype associating with these ARX variants depends on the type and location within the gene. Generally, patients with PT variants leading to total loss-of-function (LOF) have more severe XLAG, whereas patients with polyalanine expansions generally have ISSX or a form of intellectual disability [Kato et al., 2004]. Recently, cousins with c.81C>G (p.Tyr27*) and two brothers with c.34G>T (p.Glu12*) in ARX were reported with ISSX [Fullston et al., 2010; Moey et al., 2016]; none had the clinical features of XLAG as would be expected from a total loss of functional protein. Both, the p.Tyr27* and the p.Glu12* pathogenic variants, reinitiated translation at p.M41 suggesting that an N-terminally truncated ARX partially rescued the more severe brain malformation phenotype. Similar observations have been made for mutations of DMD. Encoded in exon 1, the c.9G>A (p.Tyr3*) variant is expected to cause a severe Duchenne muscular dystrophy (DMD); however, the clinical presentation is that of the milder Becker muscular dystrophy [Gurvich et al., 2009]. Immunoblot analysis of dystrophin detected a protein of reduced size consistent with translational reinitiation within exon 6. In contrast, the c.355C>T (p.Gln199*) variant in exon 5 associates with a DMD phenotype, and thus highlights that not every PT variant upstream from a putative translational reinitiation site functions similarly.
Often mRNAs encoding PT variants are degraded through nonsense-mediated decay (NMD), a post-transcriptional surveillance mechanism. This mechanism prevents synthesis of truncated proteins with potentially toxic effects. Transcripts with PT variants near the AUG start codon can escape NMD through juxtaposition of the PABPC1 protein to the mutation through mRNA circularization [Inacio et al., 2004; Silva et al., 2008], the ‘AUG-proximity effect’, and through the capacity to reinitiate translation at a downstream in-frame methionine [Neu-Yilik et al., 2011]. Consequently, only some transcripts with PT variants close to the AUG start site will be degraded by NMD and evaluation must be performed on a gene-by-gene and mutation-by-mutation basis. The variable activation of NMD versus reinitiation of translation likely contributes to phenotypic heterogeneity associated with proximal premature termination mutations.
PHOX2B forms homodimers through the homeobox domain, and in vitro also forms heterodimers with its paralogue PHOX2A. Expansion of the polyalanine track impairs dimerization by altering protein folding, leading to aggregation in the cytosol and impeding nuclear import [Bachetti et al., 2005; Di Lascio et al., 2013; Di Lascio et al., 2016; Trochet et al., 2005]. Consequently, even if the PHOX2B protein with expansion of the polyalanine track can escape cytosolic aggregation and move into the nucleus, its poor dimerization impedes transactivation of target promoters [Bachetti et al., 2005; Cargnin et al., 2005; Trochet et al., 2005]. These observations suggest that PHOX2B proteins with expansions of the polyalanine track have antimorphic and neomorphic properties, as well as loss of original function.
Consistent with this, mice with PHOX2B polyalanine track expansions have a different phenotype than those with a loss of the whole gene. Mice heterozygous for a +7 alanine expansion resemble a severe presentation of CCHS and die of respiratory failure in the first hours [Goridis et al., 2010]. In contrast, heterozygous PHOX2B knockout mice are viable and fertile, and have no detectable differences in apnea and ventilation during waking compared to wild-type mice [Durand et al., 2005], although they have an altered response to hypoxia and hypercarbia and disordered breathing and longer apnea events during sleep [Dauger et al., 2003; Durand et al., 2005]. It is difficult to compare the knockout mouse model to humans with CCHS, since only three patients with genomic deletions encompassing the whole PHOX2B have been reported and those deletions encompass additional genes [Jennings et al., 2012]. The identification of additional human deletions of PHOX2B will eventually clarify this.
In the cases in which translational reinitiation occurs, a PHOX2B protein presumably missing the first 18–21 amino acids appears to have normal nuclear localization, dimerization, DNA binding, and target promoter activation. When the clinical presentation is taken into account, it appears that the truncated protein is more likely to represent a haploinsufficiency model, similar to a whole gene deletion. Since PHOX2B regulates its own expression by an autoregulatory method and binds to multiple sites in its promoter region [Cargnin et al., 2005], it is possible that a threshold is required to reach full expression. Thus, reduction by either loss of the entire gene or a requisite function leads to a decrease in PHOX2B expression.
In conclusion, we have shown that four different nonsense pathogenic variants upstream from p.Met18/Met21 in PHOX2B are associated with a particular clinical presentation of CCHS. Patients did not require ventilation support during waking hours, and have not presented with HSCR or PNTs. Care should be taken when interpreting genetic test results as to the location of nonsense mutations and the correlation to the severity of the clinical presentation. Future studies on the function of the putative N-terminally truncated PHOX2B protein will further elucidate its mechanism in CCHS.
Acknowledgments
We thank the patients and their families who participated in this study. M. Landsverk would like to especially thank Dr. Darren Boehning for providing the motivation to pursue this project and Dr. Cornelius Boerkoel for his critical review.
FUNDING
This work is supported in party by funding from the Sanford Foundation and an Institutional Development Award from the NIH to support the Sanford Research Imaging Core (supported by P20 GM103620 and P20 GM103548).
Footnotes
CONTRIBUTIONS
Study concept and design: ML, AM, JW and KR. Collection of clinical information: AM, CM, SR, and MP. Design and performed experiments: JC, DK, MQ, WS, RP. Analysis and interpretation of data: ML, JW, KR. Critical review of the manuscript: JC, DK, MQ, WS, RP, CM, SR, MP, AM, JW, KR, ML. Study supervision: ML.
COMPETITING INTERESTS
The authors have no competing interests to report.
References
- Adachi M, Browne D, Lewis EJ. Paired-like homeodomain proteins Phox2a/Arix and Phox2b/NBPhox have similar genetic organization and independently regulate dopamine beta-hydroxylase gene transcription. DNA and cell biology. 2000;19(9):539–554. doi: 10.1089/104454900439773. [DOI] [PubMed] [Google Scholar]
- Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, Vekemans M, Munnich A, Gaultier C, Lyonnet S. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nature genetics. 2003;33(4):459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- Antic NA, Malow BA, Lange N, McEvoy RD, Olson AL, Turkington P, Windisch W, Samuels M, Stevens CA, Berry-Kravis EM, Weese-Mayer DE. PHOX2B mutation-confirmed congenital central hypoventilation syndrome: presentation in adulthood. American journal of respiratory and critical care medicine. 2006;174(8):923–927. doi: 10.1164/rccm.200605-607CR. [DOI] [PubMed] [Google Scholar]
- Bachetti T, Matera I, Borghini S, Di Duca M, Ravazzolo R, Ceccherini I. Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Human molecular genetics. 2005;14(13):1815–1824. doi: 10.1093/hmg/ddi188. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. American journal of respiratory and critical care medicine. 2006;174(10):1139–1144. doi: 10.1164/rccm.200602-305OC. [DOI] [PubMed] [Google Scholar]
- Bolande RP. Neurocristopathy: its growth and development in 20 years. Pediatric pathology & laboratory medicine: journal of the Society for Pediatric Pathology, affiliated with the International Paediatric Pathology Association. 1997;17(1):1–25. [PubMed] [Google Scholar]
- Bygarski E, Paterson M, Lemire EG. Extreme intra-familial variability of congenital central hypoventilation syndrome: a case series. Journal of medical case reports. 2013;7:117. doi: 10.1186/1752-1947-7-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnin F, Flora A, Di Lascio S, Battaglioli E, Longhi R, Clementi F, Fornasari D. PHOX2B regulates its own expression by a transcriptional auto-regulatory mechanism. The Journal of biological chemistry. 2005;280(45):37439–37448. doi: 10.1074/jbc.M508368200. [DOI] [PubMed] [Google Scholar]
- Dauger S, Pattyn A, Lofaso F, Gaultier C, Goridis C, Gallego J, Brunet JF. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development. 2003;130(26):6635–6642. doi: 10.1242/dev.00866. [DOI] [PubMed] [Google Scholar]
- Di Lascio S, Bachetti T, Saba E, Ceccherini I, Benfante R, Fornasari D. Transcriptional dysregulation and impairment of PHOX2B auto-regulatory mechanism induced by polyalanine expansion mutations associated with congenital central hypoventilation syndrome. Neurobiology of disease. 2013;50:187–200. doi: 10.1016/j.nbd.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Di Lascio S, Belperio D, Benfante R, Fornasari D. Alanine Expansions Associated With Congenital Central Hypoventilation Syndrome Impair PHOX2B Homeodomain-Mediated Dimerisation And Nuclear Import. The Journal of biological chemistry. 2016 doi: 10.1074/jbc.M115.679027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand E, Dauger S, Pattyn A, Gaultier C, Goridis C, Gallego J. Sleep-disordered breathing in newborn mice heterozygous for the transcription factor Phox2b. American journal of respiratory and critical care medicine. 2005;172(2):238–243. doi: 10.1164/rccm.200411-1528OC. [DOI] [PubMed] [Google Scholar]
- Fullston T, Brueton L, Willis T, Philip S, MacPherson L, Finnis M, Gecz J, Morton J. Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X) European journal of human genetics: EJHG. 2010;18(2):157–162. doi: 10.1038/ejhg.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goridis C, Dubreuil V, Thoby-Brisson M, Fortin G, Brunet JF. Phox2b, congenital central hypoventilation syndrome and the control of respiration. Seminars in cell & developmental biology. 2010;21(8):814–822. doi: 10.1016/j.semcdb.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Gronli JO, Santucci BA, Leurgans SE, Berry-Kravis EM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B genotype determines risk for sudden death. Pediatric pulmonology. 2008;43(1):77–86. doi: 10.1002/ppul.20744. [DOI] [PubMed] [Google Scholar]
- Gurvich OL, Maiti B, Weiss RB, Aggarwal G, Howard MT, Flanigan KM. DMD exon 1 truncating point mutations: amelioration of phenotype by alternative translation initiation in exon 6. Human mutation. 2009;30(4):633–640. doi: 10.1002/humu.20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howman-Giles R, Shaw PJ, Uren RF, Chung DK. Neuroblastoma and other neuroendocrine tumors. Seminars in nuclear medicine. 2007;37(4):286–302. doi: 10.1053/j.semnuclmed.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Inacio A, Silva AL, Pinto J, Ji X, Morgado A, Almeida F, Faustino P, Lavinha J, Liebhaber SA, Romao L. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. The Journal of biological chemistry. 2004;279(31):32170–32180. doi: 10.1074/jbc.M405024200. [DOI] [PubMed] [Google Scholar]
- Jennings LJ, Yu M, Rand CM, Kravis N, Berry-Kravis EM, Patwari PP, Weese-Mayer DE. Variable human phenotype associated with novel deletions of the PHOX2B gene. Pediatric pulmonology. 2012;47(2):153–161. doi: 10.1002/ppul.21527. [DOI] [PubMed] [Google Scholar]
- Kato M, Das S, Petras K, Kitamura K, Morohashi K, Abuelo DN, Barr M, Bonneau D, Brady AF, Carpenter NJ, Cipero KL, Frisone F, Fukuda T, Guerrini R, Iida E, Itoh M, Lewanda AF, Nanba Y, Oka A, Proud VK, Saugier-Veber P, Schelley SL, Selicorni A, Shaner R, Silengo M, Stewart F, Sugiyama N, Toyama J, Toutain A, Vargas AL, Yanazawa M, Zackai EH, Dobyns WB. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Human mutation. 2004;23(2):147–159. doi: 10.1002/humu.10310. [DOI] [PubMed] [Google Scholar]
- Low KJ, Turnbull AR, Smith KR, Hilliard TN, Hole LJ, Meecham Jones DJ, Williams MM, Donaldson A. A case of congenital central hypoventilation syndrome in a three-generation family with non-polyalanine repeat PHOX2B mutation. Pediatric pulmonology. 2014;49(10):E140–143. doi: 10.1002/ppul.23051. [DOI] [PubMed] [Google Scholar]
- Magalhaes J, Madureira N, Medeiros R, Fernandes PC, Oufadem M, Amiel J, Estevao MH, Reis MG. Late-onset congenital central hypoventilation syndrome and a rare PHOX2B gene mutation. Sleep & breathing = Schlaf & Atmung. 2015;19(1):55–60. doi: 10.1007/s11325-014-0996-7. [DOI] [PubMed] [Google Scholar]
- Matera I, Bachetti T, Puppo F, Di Duca M, Morandi F, Casiraghi GM, Cilio MR, Hennekam R, Hofstra R, Schober JG, Ravazzolo R, Ottonello G, Ceccherini I. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset Central Hypoventilation syndrome. Journal of medical genetics. 2004;41(5):373–380. doi: 10.1136/jmg.2003.015412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moey C, Topper S, Karn M, Johnson AK, Das S, Vidaurre J, Shoubridge C. Reinitiation of mRNA translation in a patient with X-linked infantile spasms with a protein-truncating variant in ARX. European journal of human genetics: EJHG. 2016;24(5):681–689. doi: 10.1038/ejhg.2015.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, Kulozik AE. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. Rna. 2011;17(5):843–854. doi: 10.1261/rna.2401811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi S, Bachetti T, Lantieri F, Di Duca M, Santamaria G, Ottonello G, Matera I, Ravazzolo R, Ceccherini I. Parental origin and somatic mosaicism of PHOX2B mutations in Congenital Central Hypoventilation Syndrome. Human mutation. 2008;29(1):206. doi: 10.1002/humu.9516. [DOI] [PubMed] [Google Scholar]
- Pattyn A, Hirsch M, Goridis C, Brunet JF. Control of hindbrain motor neuron differentiation by the homeobox gene Phox2b. Development. 2000;127(7):1349–1358. doi: 10.1242/dev.127.7.1349. [DOI] [PubMed] [Google Scholar]
- Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399(6734):366–370. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- Rand CM, Carroll MS, Weese-Mayer DE. Congenital central hypoventilation syndrome: a neurocristopathy with disordered respiratory control and autonomic regulation. Clinics in chest medicine. 2014;35(3):535–545. doi: 10.1016/j.ccm.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Repetto GM, Corrales RJ, Abara SG, Zhou L, Berry-Kravis EM, Rand CM, Weese-Mayer DE. Later-onset congenital central hypoventilation syndrome due to a heterozygous 24-polyalanine repeat expansion mutation in the PHOX2B gene. Acta paediatrica. 2009;98(1):192–195. doi: 10.1111/j.1651-2227.2008.01039.x. [DOI] [PubMed] [Google Scholar]
- Saiyed R, Rand CM, Carroll MS, Koliboski CM, Stewart TM, Brogadir CD, Kenny AS, Petersen EK, Carley DW, Weese-Mayer DE. Congenital central hypoventilation syndrome (CCHS): Circadian temperature variation. Pediatric pulmonology. 2016;51(3):300–307. doi: 10.1002/ppul.23236. [DOI] [PubMed] [Google Scholar]
- Silva AL, Ribeiro P, Inacio A, Liebhaber SA, Romao L. Proximity of the poly(A)-binding protein to a premature termination codon inhibits mammalian nonsense-mediated mRNA decay. Rna. 2008;14(3):563–576. doi: 10.1261/rna.815108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang H, Laudier B, Trochet D, Munnich A, Lyonnet S, Gaultier C, Amiel J. PHOX2B gene mutation in a patient with late-onset central hypoventilation. Pediatric pulmonology. 2004;38(4):349–351. doi: 10.1002/ppul.20074. [DOI] [PubMed] [Google Scholar]
- Trochet D, de Pontual L, Straus C, Gozal D, Trang H, Landrieu P, Munnich A, Lyonnet S, Gaultier C, Amiel J. PHOX2B germline and somatic mutations in late-onset central hypoventilation syndrome. American journal of respiratory and critical care medicine. 2008;177(8):906–911. doi: 10.1164/rccm.200707-1079OC. [DOI] [PubMed] [Google Scholar]
- Trochet D, Hong SJ, Lim JK, Brunet JF, Munnich A, Kim KS, Lyonnet S, Goridis C, Amiel J. Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Human molecular genetics. 2005;14(23):3697–3708. doi: 10.1093/hmg/ddi401. [DOI] [PubMed] [Google Scholar]
- Trochet D, Mathieu Y, Pontual L, Savarirayan R, Munnich A, Brunet JF, Lyonnet S, Goridis C, Amiel J. In Vitro studies of non poly alanine PHOX2B mutations argue against a loss-of-function mechanism for congenital central hypoventilation. Human mutation. 2009;30(2):E421–431. doi: 10.1002/humu.20923. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H, sous-commission de l’American Thoracic S ATS clinical policy statement: congenital central hypoventilation syndrome. Genetic basis, diagnosis and management. Revue des maladies respiratoires. 2013;30(8):706–733. doi: 10.1016/j.rmr.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H, Subcommittee ATSCCHS An official ATS clinical policy statement: Congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. American journal of respiratory and critical care medicine. 2010;181(6):626–644. doi: 10.1164/rccm.200807-1069ST. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Zhou L. Adult identified with congenital central hypoventilation syndrome–mutation in PHOX2b gene and late-onset CHS. American journal of respiratory and critical care medicine. 2005;171(1):88. doi: 10.1164/ajrccm.171.1.950. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, Marazita ML. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. American journal of medical genetics Part A. 2003;123A(3):267–278. doi: 10.1002/ajmg.a.20527. [DOI] [PubMed] [Google Scholar]