

Abstract
The many bystander macromolecules in the crowded cellular environments present both steric repulsion and weak attraction to proteins undergoing folding or binding and hence impact the thermodynamic and kinetic properties of these processes. The weak not nonrandom binding with bystander macromolecules may facilitate subcellular localization and biological function. Weak binding also leads to the emergence of a protein-rich, droplet phase, which has been implicated in regulating a variety of cellular functions. All these important problems can now be addressed by realistic modeling of intermolecular interactions. Configurational sampling of concentrated protein solutions is an ongoing challenge.
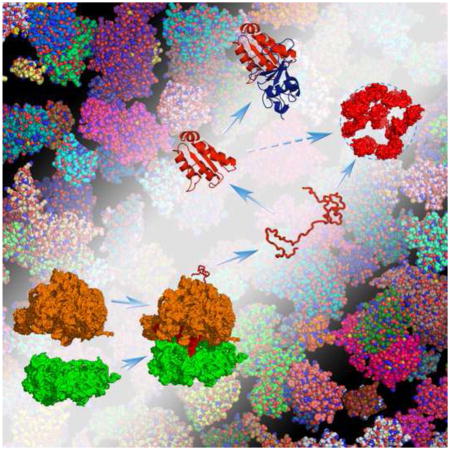
Graphical Abstract
Introduction
Proteins and other macromolecules are present at high total concentrations in all cells, a situation that is now known as macromolecular crowding [1]. Macromolecular crowding can affect protein folding and binding reactions (Fig. 1), typically studied in a dilute solution, in small and large ways. Even when the net effect of crowding is small, it does not mean that the bystander macromolecules, or crowders, exert no influence on direct participants, or test proteins, in the reactions. Instead, the crowders generate opposing effects that often cancel to a large extent. Accurate modeling of protein-crowder interactions and efficient computation are thus necessary to complements experiments in untangling the various effects of crowding.
Fig. 1.

Protein folding, binding, and droplet formation inside a cell. “Test” proteins are volume-excluded from but also weakly bind to bystander macromolecules in the cellular environment, and these interactions can steer folding and binding stability in complex ways. Test proteins (either unstructured or structured) can also weakly interact among themselves, and form a new, droplet phase in the cellular environment.
Recent years have seen continued growth of interest in protein folding and binding in cell-like conditions [2–11]. Injecting new interest in modeling cell-like conditions and posing new challenges to computation are experiments demonstrating intriguing or emergent behaviors that arise from nonspecific protein-crowder and protein-protein interactions. In particular, experimental evidence has indicated preference for specific sites on proteins in forming weak binding with crowders [12,13]. The implication is that proteins can bind weakly but nonrandomly with bystander macromolecules in their subcellular environments, and such nonrandom binding facilitates subcellular localization as well as biological function.
There is great excitement about a new phase, which is increasingly referred to as protein droplet (Fig. 1), that emerges in concentrated protein solutions under the right conditions of temperature, pH, salt concentration, etc. [14,15]. Cell biologists have for some time identified some of these membraneless intracellular “bodies” and associated them with regulating various cellular functions [16,17]. The physical nature of protein droplets has come into focus in recent years [18–21]. It is now known that these droplets represent a high-density phase of protein solutions, and their formation is similar to the condensation of water vapor into the liquid phase; the former corresponds to the ordinary low-density dissolved phase of protein solutions whereas the latter the droplet phase. Much like the vapor-liquid phase transition of water, the liquid-liquid phase separation of protein solutions is reversible and this reversibility is well suited for regulating cellular functions. The liquid-liquid phase boundaries are exquisitely sensitive to molecular details including phosphorylation, and can be significantly shifted by adding crowding agents [22]. Computational methods are now beginning to enable accurate calculations of liquid-liquid phase equilibria [23].
Below we review the major developments and challenges in modeling macromolecular crowding since the last time Curr. Opin. Struct. Biol. covered this topic [3]. Complementary coverage of the experimental literature can be found in two recent surveys in this journal [4,9,10].
Approaches to modeling protein folding and binding under crowding
Computation of crowding effects on protein folding and binding faces the twin challenges posed by the significant size of the protein-crowder systems and by the enormous amount of sampling over both the folding or binding reaction of the protein(s) and the reconfiguration of the crowders [5,8]. A number of groups have carried out direct simulations where test proteins are mixed with crowders [24–37] (horizontal paths in Fig. 2a). In most cases, a coarse-grained representation was used for the proteins and crowders to ensure adequate sampling. Others used an all-atom representation to study folding stability under crowding but whether an equilibrium conformational ensemble of the protein was generated and whether protein-crowder interactions were adequately sampled were not assessed [28,29].
Fig. 2.
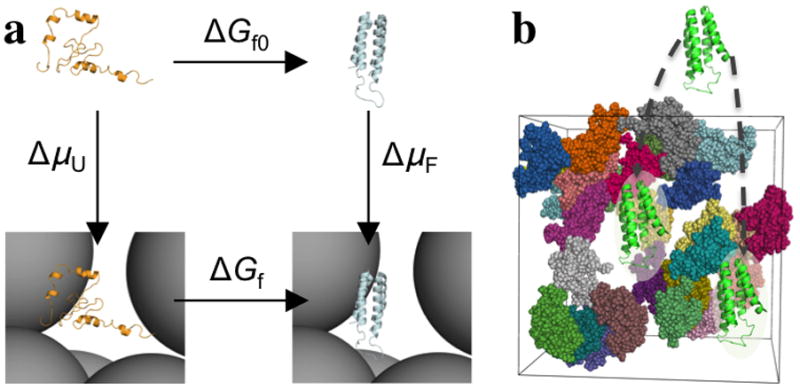
Direct simulation versus postprocessing approach, illustrated on the folding of cytochrome b562. (a) Direct simulation follows the vertical paths, whereas postprocessing follows the vertical paths. The former approach yields the folding free energies in the absence (ΔGf0) and presence (ΔGf) of crowders, whereas the latter approach yields the transfer free energies of the unfolded (ΔμU) and folded (ΔμF) states from a dilute solution to the crowder solution. By closing a thermodynamic cycle, they lead to the same effect of crowding on the folding free energy, ΔΔGf. Taken from ref [38]. (b) To calculate Δμ(X), one has to fictitiously place the protein with conformation X into different positions and evaluate the protein-crowder interaction energy at each position. Taken from ref [5].
To resolve the conflict between realistic representation and adequate sampling, we introduced the postprocessing approach [5,38,39] (vertical paths in Fig. 2a). Here the test protein and the crowders are separately simulated. Moreover, for the test protein, only the end states (e.g., folded and unfolded states) are simulated, not their transitions, which are rare events and hence hard to sample. Each protein conformation (denoted by X) is then weighted by the Boltzmann factor of the of the transfer free energy Δμ(X),
| (1) |
where Uint(X,R) is the protein-crowder interaction energy for protein conformation X placed at position R inside the crowder solution; β = 1/kB T in which kB is the Boltzmann constant and T the absolute temperature; and < … >R,c signifies averaging over R and crowder configuration. The calculation of Δμ(X) entails probing the test protein at different positions inside the crowder solution (Fig. 2b), according to the Widom insertion [40]. Further averaging over protein conformations in an end state then yields ΔμU or ΔμF in the case of protein folding. The difference, ΔμF − ΔμU, yields ΔΔGf, the effect of crowding on the folding free energy.
Because the simulations of the test protein are performed for the end state only and without crowders, adequate sampling of protein conformations can be achieved even with an all-atom representation. Separate simulations of the crowders are needed but, once done, can be reused for the study of many test proteins. Lastly placing the protein in many positions and averaging over many crowder configurations [eq (1)] assure exhaustive sampling of protein-crowder interactions.
The sampling over protein-crowder interactions prescribed in eq (1) can make the calculation of Δμ(X) quite expensive. A direct implementation of the Widom insertion indeed incurred “very significant computational expense” [41]. We were able to develop efficient methods for atomistic proteins interacting with hard-sphere crowders [38,42]. Applications to folding and binding stability of single-domain proteins predicted modest effects of crowding (up to ~1 kcal/mol) [5,38,42,43], in line with magnitudes observed in many experimental studies [44–65]. In contrast, using simple protein models, much more dramatic effects of crowding were predicted [66].
In theory the postprocessing approach is rigorous, but in practice its accuracy depends on the extent to which the conformational ensemble of the test protein in the absence of crowders overlaps with the counterpart in the presence of crowders. We have validated the postprocessing approach against direct simulations in cases where coarse-grained test proteins interacted with hard-sphere crowders. One validation study [67] was motivated by a paper by Mittal and Best [26], who used replica-exchange umbrella sampling to generate folding free energy surfaces of three small proteins in the absence and presence of crowders. By postprocessing the crowder-free protein conformations, we were able to closely reproduce the folding free energy surfaces over a range of crowder concentrations (up to a volume fraction of 35%). Consequently the postprocessing approach, using the crowder-free protein conformations alone, was able to accurately predict the effects of crowding on the folding stability. Importantly, with assumptions or information about how crowding affects motions along reaction coordinates, the postprocessing approach can also predict folding and binding kinetics [67–69].
In a second validation study [70], the conformational ensembles of an intrinsically disordered protein (IDP) represented at the coarse-grained level in the presence of hard-sphere crowders at a range of concentrations, obtained from direct simulations, were used for benchmarking predictions of the postprocessing approach. Up to a crowder volume fraction of 31%, the postprocessing approach faithfully predicted the crowder-present conformational ensembles from the crowder-free conformational ensemble. The contraction of the IDP (as measured, e.g., by the mean radius of gyration) with increasing concentration of a repulsive crowder obtained in this study presaged similar observations in subsequent experimental [71] and computational [34,36] studies. However, the predicted conformational ensemble of the IDP at an even higher crowder volume fraction of 49% was discernibly skewed due to an under-sampling of the most compact conformations in the crowder-free simulations. In addition, if the crowders are not purely repulsive and the protein-crowder attraction has a sufficient strength, postprocessing predictions can be erroneous even at intermediate crowder volume fractions (Qin and Zhou, unpublished). This is in line with all-atom simulations showing that a partially denatured protein in the presence of crowders had only partial conformational overlap with the urea denatured state [29]. On the other hand, all-atom simulations of protein-crowder systems presently can reach only sub-microsecond times, during which test proteins can easily be trapped in local crowder environments. Continued cross-validation between direct simulation and postprocessing approaches is thus called for.
Recently we cleared a remaining hurdle for realizing the full potential of the postprocessing approach, by developing a practical method for calculating Δμ(X) when both the test protein and the crowders are represented at the all-atom level and the protein-crowder interactions have both hard-core repulsion and soft components [72,73]. To do so, we expressed these interactions as correlation functions and evaluated them via fast Fourier transform (FFT). A 40,000-fold speedup was gained over brute-force Widom insertion, without losing accuracy.
This FFT-based method for Modeling Atomistic Proteins-crowder interactions, or FMAP, has the unique advantage that its computational cost remains the same whether the crowder solution consists of a single species of macromolecules or is a heterogeneous mixture of many different macromolecules, as in cellular compartments. That is because all the crowder molecules are mapped to a grid, which circumvents the need for atomic identities in the expensive calculations. With FMAP, the postprocessing approach is poised to make quantitatively predictions of crowding effects and pair with in vitro and in vivo experiments to uncover the physical basis of complex and emergent behaviors of biomacromolecules in cellular environments.
Varying effects of protein-crowder hard-core repulsion and soft attraction
Intermolecular interactions generally comprise both hard-core repulsion and “soft” components, which are attractive in the case of van der Waals and hydrophobic interactions and either attractive or repulsive depending on the signs of charges in the case of electrostatic interactions. The effects of protein-crowder hard-core repulsion, also termed excluded-volume, have long been recognized [2,74]. They favor more compact conformations of test proteins over more open or extended conformations, i.e., favor those presenting less excluded volume to crowders. As a consequence, both folding and binding stability would be enhanced, and compaction of IDPs would be expected.
It is now widely recognized that hard-core repulsion alone does not dictate the outcome of crowding effects. Soft attraction typically oppose and can reverse the qualitative trend expected from hard-core repulsion. For example, the more open conformations of a protein in the unfolded state are expected to allow it to experience stronger attractive interactions with crowders than in the folded state. Accordingly the soft attraction would favor the unfolded state over the folded state, thereby further moderating or even reversing the already modest effect on folding stability expected of hard-core repulsion. This expectation is supported by exquisite experimental data of Pielak’s laboratory using NMR-detected hydrogen/deuterium exchange [50,52–54,59] and by other studies [60,64]. In cells, soft attraction manifests as weak binding with cellular components (see below), and exerts complex effects on folding stability [57,58,62–65].
For an IDP in the presence of protein crowders, the compaction expected from hard-core repulsion was not supported by small-angle neutron scattering data [75]; soft attraction was offered as a possible counteracting factor [76]. Similar subtle effects of hard-core repulsion and soft attraction can be expected for protein binding stability under crowding. As for binding kinetics, these subtle effects on thermodynamics are further muddied by crowding effects on inter- and intra-protein dynamics [77]. All these complications highlight the importance of accurate modeling of protein-crowder interactions for capturing both the trends and the magnitudes of crowding effects on protein folding and binding.
The dependence of crowding effects on temperature potentially brings out another level of complexity. Surprisingly, a simple prediction regarding folding stability was made: for any kind of crowders there exists a crossover temperature, at which the effect of crowding switches from destabilizing to stabilizing [78]. This prediction is based on the assumption that crowding decreases both the unfolding entropy (due to conformational compaction of the unfolded state) and the unfolding entropy (due to stronger attractive interactions of the unfolded state, relative to the folded state, with the crowders). Reanalysis of the temperature-dependent data from the Pielak’s laboratory [53,54] provides support for the existence of a crossover temperature, and more such data will be required to settle this issue. If proven, the existence of a crossover temperature has broad implications. In particular, macromolecular crowding might have provided some of the stabilization to proteins in the very early cells in thermophilic environments, such that the pressure on evolution to produce stably folded proteins was lessened. One also wonders whether the dependences of crowding effects on solvent properties other than temperature, e.g., pH or salt concentration, have their own crossover points.
Blurring the divide between specific and nonspecific binding
The high concentration of bystander macromolecules in the cellular environment of any given protein provides ample opportunities for chance encounters. Researchers have long focused on “specific” binding, i.e., interactions between proteins and their partners that are direct participants of biochemical processes, such as interactions between enzymes and activators or inhibitors. Many of the resulting complexes are stable and can be captured for structure determination by X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy. In contrast, “nonspecific” binding with bystander macromolecules in the past was either ignored or viewed as harmful.
High-throughput techniques such as yeast two-hybrid and affinity purification-mass spectrometry have now enabled the identifications of many protein interaction partners [79–82]. The notion of a cardinal divide between specific and nonspecific interactions has given rise to the classification of protein pairs as binders and non-binders, and likely accounts for the branding of at least some portions of high-throughput results as false positives. Is there a physical basis for classifying proteins pairs into binders and non-binders? The physiochemical property that measures the strength of protein association is the binding constant. Statistical thermodynamic theory [83] predicts and numerous experimental measurements confirm that the magnitudes of binding constants span a wide, continuous range, and therefore there is no obvious demarcation for classifying proteins pairs into binders and non-binders. It can be stated that the divide between specific and nonspecific binding is a matter of degree not type. Instead of the binding constant, weak nonspecific binary interactions are often measured by the second virial coefficient, which appears in the expansion of the osmotic pressure of a protein solution in terms of the protein concentration. The second virial coefficient can be determined by techniques including static light scattering [84].
Recently, many in-cell NMR studies [13,63–65,85–92] have shown that, in both bacterial and eukaryotic cells, nearly all proteins participate in weak, nonspecific binding with cellular components, resulting in disappearance of NMR peaks. Interestingly, the nonspecific interactions observed are not random. In some cases, injected proteins were found to bind with particular cellular components. For examples, the neural protein tau when injected into X. laevis oocytes bound to microtubules [86]. In E. coli the MetJ repressor formed extensive nonspecific interactions with genomic DNA [87]. In other cases, there was evidence implicating a specific site of a protein for the nonspecific interactions. The Pin1 WW domain used the substrate recognition site for nonspecific interactions; nonspecific interactions were apparently abrogated when either the substrate recognition site was phosphorylated or a substrate peptide was bound [13]. Similarly, MBP formed nonspecific interactions with protein and polymer crowders, but this ability was weakened or lost when maltose was bound [12] (Fig. 3a).
Fig. 3.

Nonrandom weak binding of the maltose-binding protein (MBP) and the Pin1 WW domain with bystander macromolecules. (a) Competition of Ficoll and maltose for interaction with MBP, shown by NMR spectroscopy. In buffer, apo MBP shows well-resolved 1H-15N TROSY spectra. With 200 g/l Ficoll, most of the TROSY peaks are broadened beyond detection, indicating MBP-Ficoll binding. Upon further addition of 1 mM maltose, the peaks are recovered, indicating that the ligand has competed out the weakly bound Ficoll. (b) Shuttling of MBP in the E. coli periplasm for transport of maltose into the cytoplasm. The apo form may be weakly bound to the outer membrane-attached peptidoglycan; upon binding maltose, MBP is released from the peptidoglycan and diffuses toward the inner membrane, where it hands over the ligand to the ABC transporter for translocation into the cytoplasm. Red and black arrows indicate the flow of maltose and the shuttling of MBP, respectively. (a)and (b) taken from [12]. (c) Protein-crowder interaction energies calculated by FMAP. Top panel: the test protein (green) is the Pin1 WW protein, and the crowder is ovalbumin, with 8 copies present in a cubic box with a 157.4-Å side length (corresponding to a concentration of approximately 150 mg/mL). The crowder configuration was a snapshot taken from molecular dynamics simulation in explicit solvent. Note that the crowder molecules formed clusters. Bottom panel: in the FMAP calculation, both the protein and crowder molecules were represented at the all-atom level, and the energy function consisted of Lennard-Jones terms for modeling steric, van der Waals, and hydrophobic interactions and Debye-Hu ckel terms for modeling electrostatic interactions [73]. The energy map on a slice through the crowder box is shown according to a color scale from white to dark red; the gray regions are occupied by the crowder molecules. The placement of the test protein shown in the top panel has the minimum interaction energy, in which the substrate recognition site of the WW domain forms close contacts with one of the ovalbumin molecules (enlarged view on the left).
In many of the cases cited above, nonspecific interactions can be inferred to impart biological function. In particular, the binding of tau to microtubules is thought to be important for the latter’s stability. Nonspecific binding of the MetJ repressor to genomic DNA may facilitate the search for a specific site. Nonspecific binding with endogenous proteins via the substrate recognition site of the WW domain may contribute to Pin1’s subcellular localization. For MBP, it has been proposed that nonspecific binding with the outer membrane-attached peptidoglycan primes the protein for receiving maltose; binding of maltose releases the protein, allowing it to diffuse to the inner membrane-bound ABC transporter and hand over the ligand for translocation into the cytoplasm [12] (Fig. 3b).
It is remarkable that nonspecific binding can be tuned out by phosphorylation or substrate binding [13], or by ligand binding [12]. Apparently, nonspecific binding can be regulated by some of the same biochemical signals, e.g., phosphorylation or ligand or substrate binding, as those for specific binding. So in many respects the divide between specific and nonspecific binding is becoming blurred.
The nonrandom nature of weak protein-crowder binding and its sensitivity to biochemical signals can only be recapitulated by realistic models of protein-crowder interaction energies. An atomistic energy function consisting of Lennard-Jones and Debye-Hu ckel terms exhibit the desired features (Fig. 3c). The energy map is highly non-uniform, with multiple minima corresponding to particular orientations and positions of the test protein (the Pin1 WW domain) relative to the crowder (ovalbumin) molecules. In many of these minima, the substrate recognition site of the WW domain faces the crowder molecules, consistent with experimental observations [13]. Such nonrandom protein-crowder weak binding has also been observed in direct simulations of trp-cage crowded by bovine pancreatic trypsin inhibitor [33]. The crowder molecules also weakly bind among themselves, forming various transient clusters (Fig. 3c). Cluster formation reduces the magnitude of volume exclusion to and affects the soft attraction for test proteins, and thereby impact their folding and binding.
Protein droplet formation and regulation of cellular functions
At sufficiently high concentrations and under appropriate solvent conditions, weak interactions of protein molecules result in the co-existence of the droplet phase with the low-density dissolved phase. Experimentally, many purified proteins, mostly components of intracellular bodies but also engineered constructs, have been shown to undergo the liquid-liquid phase separation [19,22,93–101]. These proteins typically contain disordered regions and/or bind RNA. Droplet formation can facilitate the assembly of multi-component complexes for biochemical reactions, but the concentration of disordered proteins is also inductive to fibrillization and degenerative diseases.
Liquid-liquid phase separation of globular protein solutions has been studied theoretically and computationally in the past by representing proteins as spheres (with either centrosymmetric or site-specific interactions) or by other simple shapes [102–110]. FMAP, the FFT-based method for modeling atomistic intermolecular interactions, has now opened the door to accurate calculation of liquid-liquid phase equilibria for protein(/RNA) mixtures in cell-like conditions [23]. The co-existence of two phases requires equality in chemical potential. Using FMAP, we can calculate chemical potentials over a range of protein concentration (Fig. 4a). The concentration dependence of the chemical potential can then be used to identify the concentrations of the dissolved and droplet phases (Fig. 4b). The first such results, for γII-crystallin (a globular protein in the eye lens), are shown in Fig. 4c. Compared to the experimental data [111], the broadness of the phase diagram on the high-concentration side is underestimated. This discrepancy was attributed to under-sampling of cluster formation [23]. While FMAP enables accurate calculation of the chemical potential once the configurations of a protein solution at given concentrations are properly sampled, achieving this configurational sampling at high protein concentrations is an ongoing challenge [112].
Fig. 4.

Calculation of liquid-liquid coexistence curves [23]. (a) By the Widom insertion, such as implemented by FMAP, the chemical potentials of a protein over a range of concentrations are obtained. (b) Left: the phase co-existence condition is located by applying the Maxwell equal-area rule on the isotherm in the chemical potential (μ) – concentration (ρ) plane. The blue horizontal dash (at μ = μCO) crosses the isotherm with equal areas enclosed above and below. The concentrations (ρ1 and ρ2) at the low and high crossing points are those of the dissolved and droplet phases, respectively. Right: by repeating the process over a range of temperature, the full phase diagram is constructed. (c) Liquid-liquid phase diagram for γII-crystallin calculated by FMAP, compared to the experimental data. In the calculation, γII-crystallin molecules were represented at the all-atom level, and their interactions were modeled by Lennard-Jones and Debye-Hu ckel potentials. Snapshots of protein configurations at 123 and 307 mg/mL in an 81-Å thick slab are shown to the left and right, respectively.
In regulating cellular functions, cells apparently use a variety of means, including pH change and phosphorylation/dephosphorylation, to modify the liquid-liquid phase boundary and thereby control droplet formation. An in vitro study has shown that the phase boundary can be significantly shifted by adding crowding agents [22]. Some protein components are selected into the droplet phase while others are excluded. These critical issues can now be addressed by computational methods.
Highlights.
Proteins and other macromolecules weakly bind in cell-like conditions
Weak binding has many consequences, including the formation of protein droplets
Computation is positioned to accurately capture the many effects of weak binding
Acknowledgments
This work was supported in part by Grants GM88187 and GM118091 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Minton AP, Wilf J. Effect of macromolecular crowding upon the structure and function of an enzyme: glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 1981;20:4821–4826. doi: 10.1021/bi00520a003. [DOI] [PubMed] [Google Scholar]
- 2.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elcock AH. Models of macromolecular crowding effects and the need for quantitative comparisons with experiment. Curr Opin Struct Biol. 2010;20:196–206. doi: 10.1016/j.sbi.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gershenson A, Gierasch LM. Protein folding in the cell: challenges and progress. Curr Opin Struct Biol. 2011;21:32–41. doi: 10.1016/j.sbi.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou HX, Qin S. Simulation and modeling of crowding effects on the thermodynamic and kinetic properties of proteins with atomic details. Biophys Rev. 2013;5:207–215. doi: 10.1007/s12551-013-0101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phillip Y, Schreiber G. Formation of protein complexes in crowded environments--from in vitro to in vivo. FEBS Lett. 2013;587:1046–1052. doi: 10.1016/j.febslet.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou HX. Influence of crowded cellular environments on protein folding, binding, and oligomerization: biological consequences and potentials of atomistic modeling. FEBS Lett. 2013;587:1053–1061. doi: 10.1016/j.febslet.2013.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feig M, Sugita Y. Reaching new levels of realism in modeling biological macromolecules in cellular environments. J Mol Graph Model. 2013;45:144–156. doi: 10.1016/j.jmgm.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hingorani KS, Gierasch LM. Comparing protein folding in vitro and in vivo: foldability meets the fitness challenge. Curr Opin Struct Biol. 2014;24:81–90. doi: 10.1016/j.sbi.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith AE, Zhang ZT, Pielak GJ, Li CG. NMR studies of protein folding and binding in cells and cell-like environments. Curr Opin Struct Biol. 2015;30:7–16. doi: 10.1016/j.sbi.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Im W, Liang J, Olson A, Zhou HX, Vajda S, Vakser IA. Challenges in structural approaches to cell modeling. J Mol Biol. 2016 doi: 10.1016/j.jmb.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Miklos AC, Sumpter M, Zhou HX. Competitive interactions of ligands and macromolecular crowders with maltose binding protein. PLoS One. 2013;8:e74969. doi: 10.1371/journal.pone.0074969. NMR data showing that ligand binding competed out weak protein-crowder binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13**.Luh LM, Hansel R, Lohr F, Kirchner DK, Krauskopf K, Pitzius S, Schafer B, Tufar P, Corbeski I, Guntert P, et al. Molecular crowding drives active Pin1 into nonspecific complexes with endogenous proteins prior to substrate recognition. J Am Chem Soc. 2013;135:13796–13803. doi: 10.1021/ja405244v. NMR data showing that both phosphrylation and substrate binding aborgated weak protein-crowder binding. [DOI] [PubMed] [Google Scholar]
- 14.Garber K. CELL BIOLOGY. Protein 'drops' may seed brain disease. Science. 2015;350:366–367. doi: 10.1126/science.350.6259.366. [DOI] [PubMed] [Google Scholar]
- 15.Strzyz P. Molecular networks: Protein droplets in the spotlight. Nat Rev Mol Cell Biol. 2015;16:639. doi: 10.1038/nrm4070. [DOI] [PubMed] [Google Scholar]
- 16.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spector DL. SnapShot: Cellular bodies. Cell. 2006;127:1071. doi: 10.1016/j.cell.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 18.Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, Hyman AA. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 19.Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- 21.Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys. 2015;11:899–904. [Google Scholar]
- 22**.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. Experimental determination of the liquid-liquid phase boundary of a purifed component protein in stress granules, showing that the phase boundary was significantly shifted by adding a crowidng agent Ficoll. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23**.Qin S, Zhou HX. Fast method for computing chemical potentials and liquid-liquid phase equilibria of macromolecular solutions. J Phys Chem B. 2016 doi: 10.1021/acs.jpcb.6b01607. Method for determing liquid-liquid phase boundaries by equilibirum calculations on atomistically represented protein molecules. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheung MS, Klimov D, Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc Natl Acad Sci U S A. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minh DD, Chang CE, Trylska J, Tozzini V, McCammon JA. The influence of macromolecular crowding on HIV-1 protease internal dynamics. J Am Chem Soc. 2006;128:6006–6007. doi: 10.1021/ja060483s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mittal J, Best RB. Dependence of protein folding stability and dynamics on the density and composition of macromolecular crowders. Biophys J. 2010;98:315–320. doi: 10.1016/j.bpj.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dhar A, Samiotakis A, Ebbinghaus S, Nienhaus L, Homouz D, Gruebele M, Cheung MS. Structure, function, and folding of phosphoglycerate kinase are strongly perturbed by macromolecular crowding. Proc Natl Acad Sci U S A. 2010;107:17586–17591. doi: 10.1073/pnas.1006760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feig M, Sugita Y. Variable interactions between protein crowders and biomolecular solutes are important in understanding cellular crowding. J Phys Chem B. 2012;116:599–605. doi: 10.1021/jp209302e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Harada R, Tochio N, Kigawa T, Sugita Y, Feig M. Reduced native state stability in crowded cellular environment due to protein-protein interactions. J Am Chem Soc. 2013;135:3696–3701. doi: 10.1021/ja3126992. All-atom molecular dynamics simulations suggesting that weak binding with protein crowders may shift the unfold-state ensemble of a test protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YC, Mittal J. Crowding induced entropy-enthalpy compensation in protein association equilibria. Phys Rev Lett. 2013;110:208102. doi: 10.1103/PhysRevLett.110.208102. [DOI] [PubMed] [Google Scholar]
- 31.Kim YC, Bhattacharya A, Mittal J. Macromolecular crowding effects on coupled folding and binding. J Phys Chem B. 2014;118:12621–12629. doi: 10.1021/jp508046y. [DOI] [PubMed] [Google Scholar]
- 32.Latshaw DC, Cheon M, Hall CK. Effects of macromolecular crowding on amyloid beta (16–22) aggregation using coarse-grained simulations. J Phys Chem B. 2014;118:13513–13526. doi: 10.1021/jp508970q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Bille A, Linse B, Mohanty S, Irback A. Equilibrium simulation of trp-cage in the presence of protein crowders. J Chem Phys. 2015;143:175102. doi: 10.1063/1.4934997. Direct simulations of the folding a small protein in the presence of protein crowders, showing that weak protein-crowder interactions are nonrandom. [DOI] [PubMed] [Google Scholar]
- 34.Kang H, Pincus PA, Hyeon C, Thirumalai D. Effects of macromolecular crowding on the collapse of biopolymers. Phys Rev Lett. 2015;114:068303. doi: 10.1103/PhysRevLett.114.068303. [DOI] [PubMed] [Google Scholar]
- 35.Macdonald B, McCarley S, Noeen S, van Giessen AE. Protein-protein interactions affect alpha helix stability in crowded environments. J Phys Chem B. 2015;119:2956–2967. doi: 10.1021/jp512630s. [DOI] [PubMed] [Google Scholar]
- 36.Miller CM, Kim YC, Mittal J. Protein composition determines the effect of crowding on the properties of disordered proteins. Biophys J. 2016;111:28–37. doi: 10.1016/j.bpj.2016.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bille A, Mohanty S, Irback A. Peptide folding in the presence of interacting protein crowders. J Chem Phys. 2016;144:175105. doi: 10.1063/1.4948462. [DOI] [PubMed] [Google Scholar]
- 38.Qin S, Zhou HX. Atomistic modeling of macromolecular crowding predicts modest increases in protein folding and binding stability. Biophys J. 2009;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin S, Minh DD, McCammon JA, Zhou HX. Method to predict crowding effects by postprocessing molecular dynamics trajectories: application to the flap dynamics of HIV-1 protease. J Phys Chem Lett. 2010;1:107–110. doi: 10.1021/jz900023w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Widom B. Some topics in theory of fluids. J Chem Phys. 1963;39:2808–2812. [Google Scholar]
- 41.McGuffee SR, Elcock AH. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput Biol. 2010;6:e1000694. doi: 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin S, Zhou HX. Generalized fundamental measure theory for atomistic modeling of macromolecular crowding. Phys Rev E. 2010;81:031919. doi: 10.1103/PhysRevE.81.031919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tjong H, Zhou HX. The folding transition-state ensemble of a four-helix bundle protein: helix propensity as a determinant and macromolecular crowding as a probe. Biophys J. 2010;98:2273–2280. doi: 10.1016/j.bpj.2010.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spencer DS, Xu K, Logan TM, Zhou HX. Effects of pH, salt, and macromolecular crowding on the stability of FK506-binding protein: an integrated experimental and theoretical study. J Mol Biol. 2005;351:219–232. doi: 10.1016/j.jmb.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 45.Ai X, Zhou Z, Bai Y, Choy WY. 15N NMR spin relaxation dispersion study of the molecular crowding effects on protein folding under native conditions. J Am Chem Soc. 2006;128:3916–3917. doi: 10.1021/ja057832n. [DOI] [PubMed] [Google Scholar]
- 46.Roberts A, Jackson SE. Destabilised mutants of ubiquitin gain equal stability in crowded solutions. Biophys Chem. 2007;128:140–149. doi: 10.1016/j.bpc.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 47.Batra J, Xu K, Zhou HX. Nonadditive effects of mixed crowding on protein stability. Proteins. 2009;77:133–138. doi: 10.1002/prot.22425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batra J, Xu K, Qin S, Zhou HX. Effect of macromolecular crowding on protein binding stability: modest stabilization and significant biological consequences. Biophys J. 2009;97:906–911. doi: 10.1016/j.bpj.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillip Y, Sherman E, Haran G, Schreiber G. Common crowding agents have only a small effect on protein-protein interactions. Biophys J. 2009;97:875–885. doi: 10.1016/j.bpj.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miklos AC, Li CG, Sharaf NG, Pielak GJ. Volume exclusion and soft interaction effects on protein stability under crowded conditions. Biochemistry. 2010;49:6984–6991. doi: 10.1021/bi100727y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong J, Gierasch LM. Macromolecular crowding remodels the energy landscape of a protein by favoring a more compact unfolded state. J Am Chem Soc. 2010;132:10445–10452. doi: 10.1021/ja103166y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miklos AC, Sarkar M, Wang Y, Pielak GJ. Protein crowding tunes protein stability. J Am Chem Soc. 2011;133:7116–7120. doi: 10.1021/ja200067p. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Sarkar M, Smith AE, Krois AS, Pielak GJ. Macromolecular crowding and protein stability. J Am Chem Soc. 2012;134:16614–16618. doi: 10.1021/ja305300m. [DOI] [PubMed] [Google Scholar]
- 54.Benton LA, Smith AE, Young GB, Pielak GJ. Unexpected effects of macromolecular crowding on protein stability. Biochemistry. 2012;51:9773–9775. doi: 10.1021/bi300909q. [DOI] [PubMed] [Google Scholar]
- 55.Denos S, Dhar A, Gruebele M. Crowding effects on the small, fast-folding protein λ6–85. Faraday Discuss. 2012;157:451–462. doi: 10.1039/c2fd20009k. discussion 475–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christiansen A, Wittung-Stafshede P. Quantification of excluded volume effects on the folding landscape of Pseudomonas aeruginosa apoazurin in vitro. Biophys J. 2013;105:1689–1699. doi: 10.1016/j.bpj.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sarkar M, Smith AE, Pielak GJ. Impact of reconstituted cytosol on protein stability. Proc Natl Acad Sci U S A. 2013;110:19342–19347. doi: 10.1073/pnas.1312678110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guzman I, Gelman H, Tai J, Gruebele M. The extracellular protein VlsE is destabilized inside cells. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sarkar M, Lu J, Pielak GJ. Protein crowder charge and protein stability. Biochemistry. 2014;53:1601–1606. doi: 10.1021/bi4016346. [DOI] [PubMed] [Google Scholar]
- 60.Senske M, Törk L, Born B, Havenith M, Herrmann C, Ebbinghaus S. Protein stabilization by macromolecular crowding through enthalpy rather than entropy. J Am Chem Soc. 2014;136:9036–9041. doi: 10.1021/ja503205y. [DOI] [PubMed] [Google Scholar]
- 61.Ådén J, Wittung-Stafshede P. Folding of an unfolded protein by macromolecular crowding in vitro. Biochemistry. 2014;53:2271–2277. doi: 10.1021/bi500222g. [DOI] [PubMed] [Google Scholar]
- 62.Monteith WB, Pielak GJ. Residue level quantification of protein stability in living cells. Proc Natl Acad Sci U S A. 2014;111:11335–11340. doi: 10.1073/pnas.1406845111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monteith WB, Cohen RD, Smith AE, Guzman-Cisneros E, Pielak GJ. Quinary structure modulates protein stability in cells. Proc Natl Acad Sci U S A. 2015;112:1739–1742. doi: 10.1073/pnas.1417415112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64*.Danielsson J, Mu X, Lang L, Wang HB, Binolfi A, Theillet FX, Bekei B, Logan DT, Selenko P, Wennerstrom H, et al. Thermodynamics of protein destabilization in live cells. Proc Natl Acad Sci U S A. 2015;112:12402–12407. doi: 10.1073/pnas.1511308112. In-cell NMR study of protein folding stability implicating weak, sequence-specific interactions with cellular components, acting preferentially on the unfolded state. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65*.Smith AE, Zhou LZ, Gorensek AH, Senske M, Pielak GJ. In-cell thermodynamics and a new role for protein surfaces. Proc Natl Acad Sci U S A. 2016;113:1725–1730. doi: 10.1073/pnas.1518620113. In-cell NMR study of protein folding stability demonstrating importance of protein surface charges in interacting with cellular components. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Minton AP. Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: macromolecular crowding and protein stability revisited. Biophys J. 2005;88:971–985. doi: 10.1529/biophysj.104.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qin S, Mittal J, Zhou HX. Folding free energy surfaces of three small proteins under crowding: validation of the postprocessing method by direct simulation. Phys Biol. 2013;10:045001. doi: 10.1088/1478-3975/10/4/045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yuan JM, Chyan CL, Zhou HX, Chung TY, Peng H, Ping G, Yang G. The effects of macromolecular crowding on the mechanical stability of protein molecules. Protein Sci. 2008;17:2156–2166. doi: 10.1110/ps.037325.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qin S, Cai L, Zhou HX. A method for computing association rate constants of atomistically represented proteins under macromolecular crowding. Phys Biol. 2012;9:066008. doi: 10.1088/1478-3975/9/6/066008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qin S, Zhou H-X. Effects of macromolecular crowding on the conformational ensembles of disordered proteins. J Phys Chem Lett. 2013;4:3429–3434. doi: 10.1021/jz401817x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71*.Soranno A, Koenig I, Borgia MB, Hofmann H, Zosel F, Nettels D, Schuler B. Single-molecule spectroscopy reveals polymer effects of disordered proteins in crowded environments. Proc Natl Acad Sci U S A. 2014;111:4874–4879. doi: 10.1073/pnas.1322611111. FRET data demonstrating compaction of IDPs by polymer crowders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qin S, Zhou HX. FFT-based method for modeling protein folding and binding under crowding: benchmarking on ellipsoidal and all-atom crowders. J Chem Theory Comput. 2013;9:4633–4643. doi: 10.1021/ct4005195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73*.Qin S, Zhou HX. Further development of the FFT-based method for atomistic modeling of protein folding and binding under crowding: Optimization of accuracy and speed. J Chem Theory Comput. 2014;10:2824–2835. doi: 10.1021/ct5001878. FFT-based method that enables efficient modeling of atomistic protein-crowder interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Minton AP. Excluded volume as a determinant of macromolecular structure and reactivity. Biopolymers. 1981;20:2093–2120. [Google Scholar]
- 75.Goldenberg DP, Argyle B. Minimal effects of macromolecular crowding on an intrinsically disordered protein: a small-angle neutron scattering study. Biophys J. 2014;106:905–914. doi: 10.1016/j.bpj.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou HX, Bilsel O. SAXS/SANS probe of intermolecular interactions in concentrated protein solutions. Biophys J. 2014;106:771–773. doi: 10.1016/j.bpj.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Phillip Y, Harel M, Khait R, Qin S, Zhou HX, Schreiber G. Contrasting factors on the kinetic path to protein complex formation diminish the effects of crowding agents. Biophys J. 2012;103:1011–1019. doi: 10.1016/j.bpj.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou HX. Polymer crowders and protein crowders act similarly on protein folding stability. FEBS Lett. 2013;587:394–397. doi: 10.1016/j.febslet.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu HY, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, et al. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322:104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273–299. doi: 10.1146/annurev-biochem-061308-093216. [DOI] [PubMed] [Google Scholar]
- 81.Stark C, Breitkreutz BJ, Chatr-aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang XD, Shi XQ, et al. The BioGRID Interaction Database: 2011 update. Nucl Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucl Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou HX, Gilson MK. Theory of free energy and entropy in noncovalent binding. Chem Rev. 2009;109:4092–4107. doi: 10.1021/cr800551w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu D, Minton AP. Quantitative characterization of nonspecific self- and hetero-interactions of proteins in nonideal solutions via static light scattering. J Phys Chem B. 2015;119:1891–1898. doi: 10.1021/jp510918d. [DOI] [PubMed] [Google Scholar]
- 85.Sakai T, Tochio H, Tenno T, Ito Y, Kokubo T, Hiroaki H, Shirakawa M. In-cell NMR spectroscopy of proteins inside Xenopus laevis oocytes. J Biomol NMR. 2006;36:179–188. doi: 10.1007/s10858-006-9079-9. [DOI] [PubMed] [Google Scholar]
- 86.Bodart JF, Wieruszeski JM, Amniai L, Leroy A, Landrieu I, Rousseau-Lescuyer A, Vilain JP, Lippens G. NMR observation of Tau in Xenopus oocytes. J Magn Reson. 2008;192:252–257. doi: 10.1016/j.jmr.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 87.Augustus AM, Reardon PN, Spicer LD. MetJ repressor interactions with DNA probed by in-cell NMR. Proc Natl Acad Sci U S A. 2009;106:5065–5069. doi: 10.1073/pnas.0811130106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li C, Wang GF, Wang Y, Creager-Allen R, Lutz EA, Scronce H, Slade KM, Ruf RA, Mehl RA, Pielak GJ. Protein 19F NMR in Escherichia coli. J Am Chem Soc. 2010;132:321–327. doi: 10.1021/ja907966n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Q, Zhuravleva A, Gierasch LM. Exploring weak, transient protein--protein interactions in crowded in vivo environments by in-cell nuclear magnetic resonance spectroscopy. Biochemistry. 2011;50:9225–9236. doi: 10.1021/bi201287e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crowley PB, Chow E, Papkovskaia T. Protein interactions in the Escherichia coli cytosol: an impediment to in-cell NMR spectroscopy. Chembiochem. 2011;12:1043–1048. doi: 10.1002/cbic.201100063. [DOI] [PubMed] [Google Scholar]
- 91.Waudby CA, Camilloni C, Fitzpatrick AW, Cabrita LD, Dobson CM, Vendruscolo M, Christodoulou J. In-cell NMR characterization of the secondary structure populations of a disordered conformation of alpha-synuclein within E coli cells. PLoS One. 2013;8:e72286. doi: 10.1371/journal.pone.0072286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 93.Elbaum-Garfinkle S, Kim Y, Szczepaniak K, Chen CC-H, Eckmann CR, Myong S, Brangwynne CP. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci U S A. 2015;112:7189–7194. doi: 10.1073/pnas.1504822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 95.Jiang H, Wang S, Huang Y, He X, Cui H, Zhu X, Zheng Y. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell. 2015;163:108–122. doi: 10.1016/j.cell.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xiang S, Kato M, Wu LC, Lin Y, Ding M, Zhang Y, Yu Y, McKnight SL. The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei. Cell. 2015;163:829–839. doi: 10.1016/j.cell.2015.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015;57:936–947. doi: 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin Y, Protter DSW, Rosen MK, Parker R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell. 2015;60:208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang H, Elbaum-Garfinkle S, Langdon EM, Taylor N, Occhipinti P, Bridges AA, Brangwynne CP, Gladfelter AS. RNA controls PolyQ protein phase transitions. Mol Cell. 2015;60:220–230. doi: 10.1016/j.molcel.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue-by-residue view of In vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol Cell. 2015;60:231–241. doi: 10.1016/j.molcel.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murakami T, Qamar S, Lin JQ, Schierle GSK, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FTS, Michel CH, et al. ALS/FTD mutation-induced phase transition of FUS liquid proplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lomakin A, Asherie N, Benedek GB. Monte Carlo study of phase separation in aqueous protein solutions. J Chem Phys. 1996;104:1646–1656. [Google Scholar]
- 103.Kern N, Frenkel D. Fluid-fluid coexistence in colloidal systems with short-ranged strongly directional attraction. J Chem Phys. 2003;118:9882–9889. [Google Scholar]
- 104.Rosch TW, Errington JR. Investigation of the phase behavior of an embedded charge protein model through molecular simulation. J Phys Chem B. 2007;111:12591–12598. doi: 10.1021/jp075455q. [DOI] [PubMed] [Google Scholar]
- 105.Gogelein C, Nagele G, Tuinier R, Gibaud T, Stradner A, Schurtenberger P. A simple patchy colloid model for the phase behavior of lysozyme dispersions. J Chem Phys. 2008;129:085102. doi: 10.1063/1.2951987. [DOI] [PubMed] [Google Scholar]
- 106.Bianchi E, Tartaglia P, Zaccarelli E, Sciortino F. Theoretical and numerical study of the phase diagram of patchy colloids: ordered and disordered patch arrangements. J Chem Phys. 2008;128:144504. doi: 10.1063/1.2888997. [DOI] [PubMed] [Google Scholar]
- 107.Cho EJ, Kim JS. Crowding effects on the formation and maintenance of nuclear bodies: insights from molecular-dynamics simulations of simple spherical model particles. Biophys J. 2012;103:424–433. doi: 10.1016/j.bpj.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kurut A, Åkesson T, Forsman J, Lund M. Anisotropic interactions in protein mixtures: self assembly and phase behavior in aqueous solution. J Phys Chem Lett. 2012;3:731–734. doi: 10.1021/jz201680m. [DOI] [PubMed] [Google Scholar]
- 109*.Kastelic M, Kalyuzhnyi YV, Hribar-Lee B, Dill KA, Vlachy V. Protein aggregation in salt solutions. Proc Natl Acad Sci U S A. 2015;112:6766–6770. doi: 10.1073/pnas.1507303112. Theoretical prediction of liquid-liquid co-existence curves for two proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baumketner A, Melnyk R, Holovko MF, Cai W, Costa D, Caccamo C. Softness and non-spherical shape define the phase behavior and the structural properties of lysozyme in aqueous solutions. J Chem Phys. 2016;144:015103. doi: 10.1063/1.4939637. [DOI] [PubMed] [Google Scholar]
- 111.Broide ML, Berland CR, Pande J, Ogun OO, Benedek GB. Binary-liquid phase separation of lens protein solutions. Proc Natl Acad Sci U S A. 1991;88:5660–5664. doi: 10.1073/pnas.88.13.5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feig M, Harada R, Mori T, Yu I, Takahashi K, Sugita Y. Complete atomistic model of a bacterial cytoplasm for integrating physics, biochemistry, and systems biology. J Mol Graph Model. 2015;58:1–9. doi: 10.1016/j.jmgm.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]