

Abstract
A principal product of the reaction between a protein cysteinyl thiol and hydrogen peroxide is a protein sulfenic acid. Because protein sulfenic acid formation is reversible, it provides a mechanism whereby changes in cellular hydrogen peroxide concentration may directly control protein function. We have developed methods for the detection and purification of proteins oxidized in this way. The methodology is based on the arsenite-specific reduction of protein sulfenic acid under denaturing conditions and their subsequent labeling with biotin–maleimide. Arsenite-dependent signal generation was fully blocked by pretreatment with dimedone, consistent with its reactivity with sulfenic acids to form a covalent adduct that is nonreducible by thiols. The biotin tag facilitates the detection of protein sulfenic acids on Western blots probed with streptavidin–horseradish peroxidase and also their purification by streptavidin–agarose. We have characterized protein sulfenic acid formation in isolated hearts subjected to hydrogen peroxide treatment. We have also purified and identified a number of the proteins that are oxidized in this way by using a proteomic approach. Using Western immunoblotting we demonstrated that a highly significant proportion of some individual proteins (68% of total in one case) form the sulfenic derivative. We conclude that protein sulfenic acids are widespread physiologically relevant posttranslational oxidative modifications that can be detected at basal levels in healthy tissue, and are elevated in response to hydrogen peroxide. These approaches may find widespread utility in the study of oxidative stress, particularly because hydrogen peroxide is used extensively in models of disease or redox signaling.
Keywords: heart, oxidative stress, myocardium, redox signaling
Redox imbalance leading to oxidative stress has been widely investigated as a causative mechanism in the pathogenesis of many diseases, although a definitive role for this mode of damage remains to be established. Overproduction of oxidants, including hydrogen peroxide (H2O2), may lead to the aberrant oxidation of cellular components resulting in dysfunction. Because H2O2 is produced so widely in cells and tissues, it is often used as the archetypal oxidant when studying redox cell biology. Thus, H2O2 is utilized in in vitro, cellular, organ, and animal models of a variety of diseases. For example, H2O2 treatment has been used to investigate mechanisms of cardiovascular diseases (including atherosclerosis, hypertrophy, apoptosis, ischemia and reperfusion) (1–5), cancer (6, 7), neurodegeneration (8), aging (9), and autoimmunity (10).
In addition to potential roles in disease and dysfunction, H2O2 is also produced in healthy cells and tissues. For example, peptide growth factors, cytokines, and agonists of heterotrimeric G protein-coupled receptors couple to H2O2 production, as reviewed by Rhee et al. (11). Therefore, in addition to a deleterious oxidant, H2O2 can be classified as a small, diffusible molecule that is rapidly produced at defined cellular locations and undergoes controlled breakdown by reaction with specific cellular targets, antioxidant molecules, and peroxidase enzymes. These characteristics of H2O2 allow it to act as a signaling molecule (11–13).
A major question in the redox control of proteins concerns how oxidants are sensed and transduced into a biological effect. When H2O2 is elevated, reactive protein thiols within the cell can become oxidized. A number of protein thiol oxidation states can be formed, including the S-thiolated and the S-nitrosylated form (14), which obviously require the presence of other chemical species.
A principal product of the reaction between a protein thiol and H2O2 is the protein sulfenic acid (PSOH), although protein sulfinic acid (PSO2H) or protein sulfonic acid (PSO3H) can also be formed (12, 15). Many protein sulfenic acid forms are unstable and preferentially react with local thiols to form disulphides, or may become internally stabilized sulfenyl-amide derivatives (16). However, at this time, the full extent of protein sulfenic acid formation in cellular control remains to be established.
In this study, we describe recently developed approaches for the study of protein sulfenic acids. Considering the number of studies where H2O2 is used as an archetypal oxidant or in models of disease, we believe these methods may find widespread utility. The method is based on the specific reduction of sulfenic acids by arsenite, which has allowed us to develop a biotin-switch method for labeling protein sulfenic acids. We have applied these methods to identify proteins susceptible to sulfenic acid modification(s) in cardiac tissue treated with physiologically relevant concentrations of H2O2.
Materials and Methods
Chemicals. These were obtained from Sigma or BDH unless stated otherwise and were of AnalaR grade or above.
Animals. Male Wistar rats (200–250 g) were used throughout this study and were obtained from B & K Universal (Hull, U.K.). The animals were maintained humanely in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and “Guide for Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (publication no. 85-23).
Isolated Heart Preparations and Perfusion Protocols. Isolated buffer-perfused heart preparations were as before (17). Controls were aerobically perfused for 30 min. The effect of H2O2 concentration on protein sulfenic acid formation was investigated by using 25-min aerobic perfusion followed by 5-min of the oxidant at 10, 100, 1,000, or 10,000 μM. The effect of time was investigated by using aerobic perfusion for 25 min followed by H2O2 (100 μM) administration for 5, 15, 30, or 60 min. In additional studies, after 20-min aerobic perfusion, the heart was exposed to N-acetylcysteine (NAC) (5 mM) for 5 min, followed by H2O2 (100 μM) for 5 min. At the end of each protocol, hearts were snap-frozen and stored in liquid nitrogen.
Labeling Protein Sulfenic Acids with Biotin. Fig. 1 shows the basic strategy used to label protein sulfenic acids with biotin, as a procedure that would allow their detection, purification, and identification. Frozen hearts (≅1 g) were ground to a powder under liquid nitrogen by using a tight fitting, metal pestle and mortar and then transferred to an argon-gassed buffer (100 mM Tris·HCl/100 mM maleimide, pH 7.0). The volume of buffer used was adjusted depending on the heart mass, producing a 20% wt/vol homogenate. SDS was then added (to a final concentration of 1% wt/vol), and the mixture was left at room temperature on a rotary shaker for 2 h. The SDS was added to denature proteins, allowing full access of the alkylation reagent and efficient blocking of protein thiols. Soluble protein was then collected by centrifugation (20,000 × g for 10 min) and applied to a calibrated HiLoad 16/60 prepgrade Superdex 200 gel filtration column (Amersham Pharmacia) powered by a Bio-Rad BioLogic DuoFlow chromatograph pumping a mobile phase of 100 mM Tris·HCl plus 1% SDS wt/vol. One large 50-ml fraction >5 kDa was collected, removing free maleimide from the bulk of the protein. The sample was then treated with sodium arsenite (20 mM) and biotin–maleimide (0.1 mM) for 30 min, allowing the reduction of protein sulfenic acids and their subsequent biotinylation. Preliminary studies confirmed that initial treatment with maleimide resulted in the full blockade of protein thiols, because when biotin–maleimide was used alone (without sodium arsenite), biotin labeling was not facilitated (results not shown). In additional experiments, the selectivity of arsenite reduction for sulfenic acids was tested by using 5,5-dimethyl-1,3-cyclohexanedione (dimedone), which specifically reacts with sulfenic acids (see Fig. 1) (18–22). In these experiments, the samples from hearts treated with 100 μM H2O2 for 5 min were treated with 5 mM dimedone for 30 min before adding biotin–maleimide.
Fig. 1.

Outline of the methodology used to detect and identify proteins that form sulfenic acids in the heart following hydrogen peroxide treatment. Protein thiols were blocked with maleimide in the presence of SDS, which facilitates access of the alkylation agent. Unincorporated maleimide was removed by using gel filtration, and the resulting protein fraction was reduced with sodium arsenite in the presence of biotin–maleimide. Protein sulfenic acids were visualized on nonreducing SDS/PAGE and Western blots probed with streptavidin–HRP. Protein sulfenic acids were purified by using streptavidin agarose, which involved first removing free biotin–maleimide by using desalting columns.
Protein Sulfenic Acid Detection and Purification. Samples that had undergone the labeling protocol described above were reconstituted in sample buffer without a reducing agent. SDS/PAGE was carried out by using the Bio-Rad mini protean II system, loading equal amount of protein (20 μg) per well. After electrophoresis, samples were transferred to poly(vinylidene difluoride) by using a Bio-Rad semidry blotter. Protein sulfenic acids were detected by virtue of their biotin tag using streptavidin–HRP and the enhanced chemiluminescence reagent (both Amersham Pharmacia). Images were digitized by using a flatbed scanner (Epson Perfection 1260) and quantitatively analyzed for total protein sulfenic acid in each lane by using image software (National Institutes of Health). In purification studies, free biotin–maleimide was first removed from the sample by using PD-10 Sephadex (G-25M) desalting columns (Amersham Pharmacia). The sample was then diluted 10-fold with 100 mM Tris·HCl/0.1% Triton X-100 wt/vol plus protease inhibitors (Roche Diagnostics, one tablet per 20 ml of buffer), to reduce the concentration of SDS. The sample was then rotated with 1 ml of streptavidin–agarose (Merck) overnight at 4°C. The matrix was then emptied into a disposable plastic column and washed with 200 ml of 100 mM Tris·HCl, pH 7.0/0.1% wt/vol SDS/0.1% wt/vol Triton X-100/100 mM NaCl. Protein sulfenic acids were then eluted by incubating the matrix with 2× SDS sample buffer at 50°C for 5 min. The samples were resolved by SDS/PAGE (large format Hoeffer system) and stained with Coomassie blue dye (Simply Blue, Invitrogen).
Determining the Sensitivity of the Protein Sulfenic Acid Detection Method. The sensitivity of the protein sulfenic acid method was determined by spiking aerobic control homogenates with known amounts of recombinant OxyR protein in the sulfenic acid state. An OxyR overexpressing E. coli strain (OxyR++ D1215/pAQ25, a gift from J. S. Stamler, Duke University Medical Center, Durham, NC) was used to generate OxyR protein, which was purified on a heparin HPLC column as described (21). This protein was reduced with 100 mM DTT and further purified on a Superdex 200 size exclusion column (as above). The reduced OxyR was either stored at 4°C in an argon environment or oxidized to sulfenic acid form by gassing with oxygen for 1 h. Full conversion to the oxidized form was achieved, as assessed by using spectral shift analysis of the reduced and oxidized forms with 7-chloro-4-nitrobenz-2-oxa-1,3-diazole (NBD-Cl), after removal of free NBD-Cl by using a PD-10 Sephadex (G-25M) desalting column (21, 22). The purified sulfenic acid OxyR protein was added to aerobic heart preparations and processed for detection of sulfenic acids as described above. By using this approach, we were able to detect 0.1 ng of sulfenic OxyR on Western blots (data not shown) by using the biotin switch method described. Several strategies could be used to enhance the sensitivity of this detection procedure, such as loading more total protein, longer incubation times with a more concentrated solution of streptavidin–HRP, or by utilising enhanced chemiluminescence procedures that generate more intense signals along with more sensitive detection systems. The detection of protein sulfenic acids in this system is essentially dependent on the visualization of a biotinylated protein on Western blot, a procedure that is highly sensitive and applicable to the study of proteins in cell systems.
Identification of Unknown Sulfenated Proteins by MS. Protein bands were cut out from the gel and transferred into a microSPE plate (Montage In-Gel DigestZP kit, Millipore). Destaining, digestion with MS grade trypsin (Promega), and desalting was accomplished according to the manufacturer's recommendation. For MALDI analysis, a sample preparation protocol using target plates with a 400-μm hydrophilic anchor (AnchorChip 400, Bruker Daltonics, Billerica, MA) was used. Briefly, a 1-μl aliquot of a mixture containing the MALDI matrix 2,5-dihydroxybenzoic acid (3 mg/ml, DHB, MassPREP, Waters) and the peptide calibrants angiotensin II, ACTH clip (18–39), and Vasoactive Intestinal Peptide clip (6–28) at concentrations of 50, 150, and 200 fmol/μl, respectively, was deposited on a target spot. A 1-μl aliquot of an in-gel digest was added to this droplet. The sample surfaces were crystallized in air before insertion into the mass spectrometer. MALDI-TOF mass spectra were acquired on an Autoflex mass spectrometer (Bruker Daltonics) operated in the positive ion mode by using an ion reflector. The mass spectra of 600 laser shots were averaged and internally calibrated in flexanalysis (version 2.0). A list of monoisotopic peptide masses was obtained for each sample and searched against the swissprot database by using the Mascot algorithm (Matrix Science). A mass accuracy of 40 ppm and the mammalian taxonomy was specified.
Determining the Proportion of a Protein That Is Oxidized to Sulfenic Acid. Protein sulfenic acids were isolated as described above, starting with 1 ml of a 20% (wt/vol) cardiac homogenate. After affinity purifications with excess streptavidin–agarose (0.5 ml), the purified proteins were resuspended in a final volume of 1 ml of SDS sample buffer. Twenty microliters of the affinity-purified proteins were resolved by SDS/PAGE adjacent to 20-, 10-, 5-, 2-, and 1-μl samples of the starting cardiac homogenate reconstituted in an equal volume of 2× SDS sample buffer. After blotting to poly(vinylidene difluoride), the samples were probed with antibodies to some of the proteins identified as forming sulfenic acids. If 100% of a protein formed sulfenic acid, the immunoblot signal present in the 10-μl sample of affinity-purified proteins would exactly match the amount present in a 20-μl sample of the starting sample, because it was diluted 2-fold in sample buffer. The extent to which a protein is modified to sulfenic acid can thus be calculated after quantitatively comparing the signal in the affinity-purified fraction with those in the various volumes of unfractionated samples. Three affinity preparations from different hearts treated with 100 μM H2O2 for 5 min were analyzed in this way. Antibodies used in these analyses were to actin (Hybridoma Bank, University of Iowa, Ames), myosin heavy chain (Hybridoma Bank), lactate dehydrogenase β-chain (Abcam, Cambridge, U.K.), tropomyosin (Abcam), aspartate aminotransferase (Research Diagnostics), malate dehydrogenase (Europa Bioproducts, Cambridge, U.K.), ATP synthase α (BD Transduction Laboratories), and ADP/ATP carrier protein (gift from A. Halestrap, University of Bristol, Bristol, U.K.).
Results
The dose dependency of protein sulfenic acid formation is shown in Fig. 2A. These Western blot data show protein sulfenic acids can be detected in control tissue under basal conditions and that lower concentrations (10 μM or 100 μM) of H2O2 investigated in these studies induced significant, widespread protein sulfenic acid formation. When higher concentrations (1 or 10 mM) were used, there was a progressive loss in the signals detected. Fig. 2B is a quantitative analysis of the amount of protein sulfenic acid formed in each treatment group after three separate experiments.
Fig. 2.

Western blot (A) and quantitative analysis (B) of multiple experiments showing that H2O2 induced protein sulfenic acid formation. However, as the H2O2 concentration was increased, there was a marked loss in the detected levels of sulfenic acid, which decreased below that observed in control tissue. This biphasic concentration-dependent effect is consistent with thiol oxidation to sulfinic or sulfonic forms at higher levels of H2O2, which are not reduced by arsenite treatment.
The time course of protein sulfenic acid generation is shown in Fig. 3A. The Western blot shows that the amount of protein sulfenic acid detected in the heart after exposure to 100 μM H2O2 is time dependent. A quantitative analysis of three different experiments is shown in Fig. 3B. At the concentration used and over the time scale investigated, protein sulfenic acid formation was maximal from 5 to 15 min. However, administration of H2O2 for longer periods decreased the amount of oxidized protein detected.
Fig. 3.

Western blot (A) and quantitative analysis (B) of multiple experiments showing that the duration of H2O2 (100 μM) administration influences the amount of protein sulfenic acid detected in the heart. Although exposure to the oxidant initially increases the amount of protein sulfenic acid detected, this decreases with prolonged exposure.
Fig. 4A is a Western blot showing that dimedone treatment abolished the arsenite-induced sulfenic acid signal generated when H2O2 is administered to the heart. This occurs because dimedone reacts with protein sulfenic acid to form a product that cannot be reduced by arsenite, as highlighted in Fig. 1. Therefore, the proteins have no reduced cysteine residues to react with biotin–maleimide, and consequently no signal is generated. The fact that dimedone blocks the signals generated in these experiments confirms that the arsenite-dependent signal is specific for protein sulfenic acids. The quantitative analysis of total protein sulfenic acid formation in each of the treatments (Fig. 4B) confirms that dimedone reproducibly and fully blocks arsenite-induced signals.
Fig. 4.

Western blot (A) and quantitative analysis (B) of multiple experiments showing that dimedone treatment of samples from hearts treated with 100μM H2O2 for 5 min abolishes the arsenite-induced sulfenic acid signal. This occurs because dimedone reacts with the sulfenic acid to form a stable product that cannot be reduced by arsenite, as highlighted in Fig. 1. This observation helps confirm the specificity of arsenite for the reduction of protein sulfenic acids, and validates the methodology we have used.
Fig. 5A is a Western blot showing that the low molecular weight, membrane permeable thiol NAC can significantly attenuate protein sulfenic acid formation, when perfused through the heart for 5 min immediately before perfusion with H2O2. A quantitative analysis of data collected from three different experiments is shown in Fig. 5B.
Fig. 5.

Western blot (A) and quantitative analysis (B) of multiple experiments showing that the membrane permeable antioxidant compound, NAC, when loaded into the heart before administration of H2O2, was effective in significantly attenuating protein sulfenic acid formation.
Protein sulfenic acids were purified by affinity chromatography using a 1-ml column of streptavidin–agarose, resolved by SDS/PAGE and Coomassie stained (Fig. 6). Protein bands were excised and identified by using proteomic methods detailed above (see Table 1). We had hoped to map some of the actual cysteine residues within proteins where sulfenation occurred. However, this was not achieved, as we were unable to identify biotin-cysteine containing peptides in the MS spectra. This absence may be explained by a combination of the low coverage offered typically by MALDI-TOF MS and possible suppression of biotin peptide ionization. This does not prevent us unequivocally assigning the proteins as sulfenated, as these proteins were not found in control preparations and only appear in peroxide-treated tissue subject to arsenite reduction.
Fig. 6.
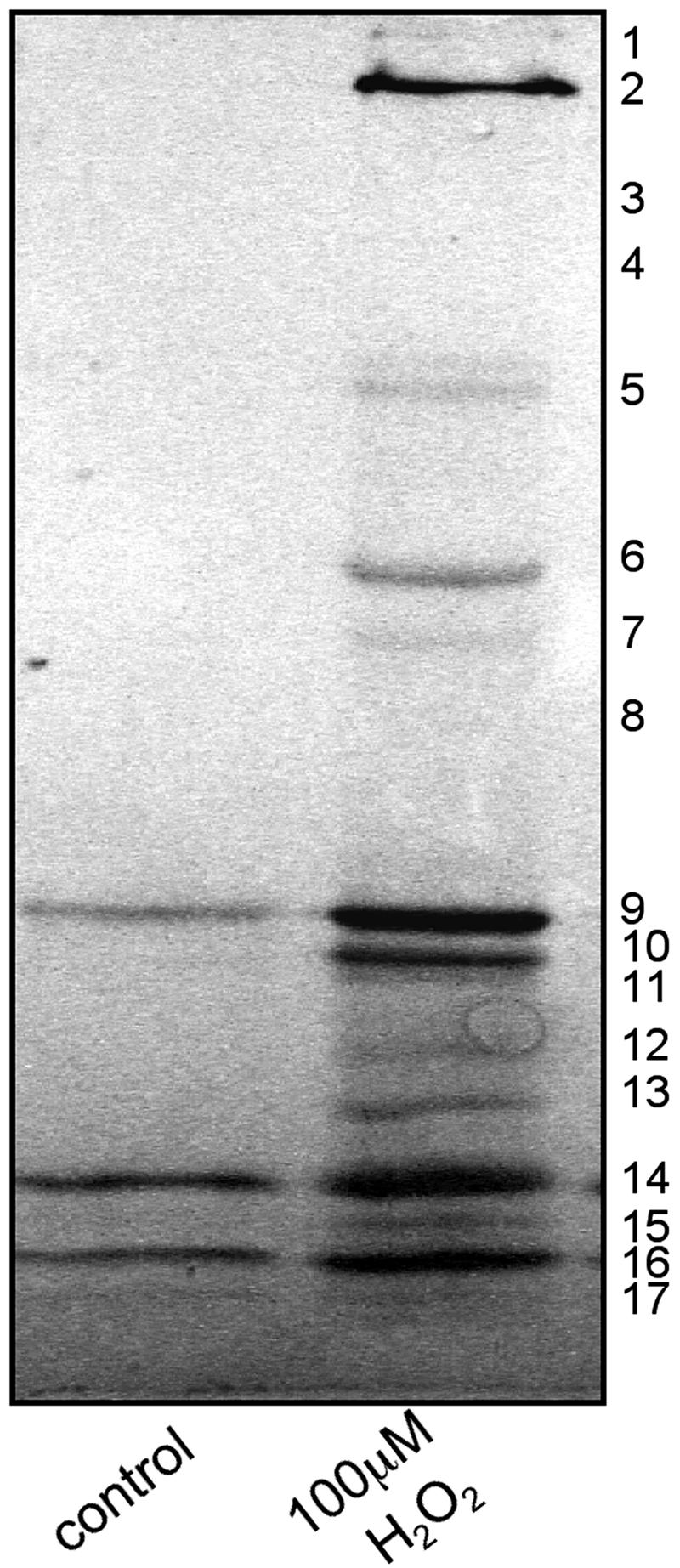
Coomassie-stained SDS/PAGE of protein sulfenic acids purified by using streptavidin–agarose affinity chromatography. Bands were excised, digested with trypsin, and analyzed by MALDI-TOF MS and database searching, leading to a number of identifications (see Table 1).
Table 1. Proteins that form sulfenic acids in isolated rat hearts administered H2O2.
| Band number | Score (>61) | Entry number | Molecular mass, Da | Description |
|---|---|---|---|---|
| 1 | 131 | P02563 | 223,507 | Myosin heavy chain α isoform |
| 2 | 241 | P02563 | 223,507 | Myosin heavy chain α isoform |
| 150 | P02564 | 223,082 | Myosin heavy chain β isoform | |
| 3 | 82 | Q9ER34 | 85,744 | Aconitase |
| 4 | 61 | P45953 | 70,749 | Acyl-CoA dehydrogenase |
| 5 | 118 | P15999 | 58,904 | ATP synthase α chain |
| 127 | P10719 | 56,318 | ATP synthase β chain | |
| 6 | 62 | P13221 | 46,197 | Aspartate aminotransferase |
| 7 | 67 | P04270 | 42,019 | Actin |
| 78 | P15650 | 47,873 | Acyl-CoA dehydrogenase | |
| 8 | 106 | P04270 | 42,019 | Actin, α cardiac |
| 9 | 113 | P04692 | 32,694 | Tropomyosin 1 α chain |
| 71 | P50753 | 35,578 | Troponin T | |
| 10 | 55 | Q05962 | 32,989 | ADP, ATP carrier protein T1 |
| 11 | 68 | P04636 | 35,655 | Malate dehydrogenase |
| 12 | 94 | P42123 | 36,481 | l-lactate dehydrogenase β chain |
| 13 | 109 | P04270 | 42,019 | Actin |
| 14 | 74 | Q05962 | 32,989 | ADP, ATP carrier protein T1 |
| 61 | P16409 | 22,025 | Myosin light polypeptide 3 | |
| 15 | 92 | P10888 | 19,514 | Cytochrome C oxidase subunit IV |
| 16 | 95 | P16409 | 22,025 | Myosin light polypeptide 3 |
| 17 | 82 | Q9QZ76 | 17,157 | Myoglobin |
Using immunoblotting with antibodies to some of the proteins we have identified as forming sulfenic acids, we calculated the proportion of the total pool of these proteins that form this oxidized derivative. As described in Materials and Methods, a comparison of the signals generated on blots from samples of total heart homogenate with those from affinity-purified protein sulfenic acids preparations allows the proportion of the protein that formed sulfenic acid to be calculated. Fig. 7 shows Western blots probed with antibodies to proteins identified as forming sulfenic acids. Quantitative analysis allowed the percentage conversion to sulfenic acids to be calculated: lactate dehydrogenase β chain, 28 ± 3%; ATP synthase α-subunit, 11 ± 3%; ADP, ATP carrier protein, 68 ± 5%; malate dehydrogenase, 67 ± 15%; actin, <1%; tropomyosin, <1%; myosin heavy chain, <1%; and aspartate aminotransferase, 13 ± 3%.
Fig. 7.

Quantitation of the proportion of specific proteins that formed sulfenic acid. Protein sulfenic acids were purified from 1 ml of a 20% (wt/vol) cardiac homogenate. After affinity purification, the purified proteins were resuspended in a final volume of 1 ml of SDS sample buffer. Ten microliters of the affinity-purified protein sample was resolved by SDS/PAGE adjacent to 20-, 10-, 5-, 2-, and 1-μl samples of the starting cardiac homogenate reconstituted in an equal volume of sample buffer. After blotting to poly(vinylidene difluoride), the samples were probed with antibodies to the respective proteins that form sulfenic acids. If 100% of a given protein formed sulfenic acid, the immunoblot signal present in the 10-μl sample of affinity-purified proteins would exactly match the amount present in a 20-μl sample of the starting sample (allowing for the 2-fold dilution in sample buffer). The varying amount of unfractionated material loaded serves as a calibrant, and allows the proportion of a protein (percentage of total pool) that formed sulfenic acid to be determined (see Results).
Discussion
Protein Thiols as Targets of H2O2. In terms of abundance, one of the most important redox couples in cells involves the equilibrium between reduced and oxidized forms of thiols (23). Thiols are primarily found on the amino acid cysteine, which is present in high abundance in glutathione and cellular proteins. The thiol redox status of cells varies during their lifetime, with set points depending on whether tissue is quiescent, proliferating, confluent, differentiating, apoptotic, or necrotic (23). Protein thiol oxidation is a widespread marker of many diseases (24), and H2O2 is utilized widely in model systems of disease and will itself directly oxidize protein thiols. Changes in cellular redox potential to a more oxidizing state can be triggered by a number of processes in healthy tissue, such as metabolism and signaling events (11).
Protein sulfenic acids, which are principal products formed by protein thiols on contact with H2O2, have generally been thought to occur as transient intermediates in redox reactions. This idea is supported by the propensity of these species to self-stabilize by sulfenyl-amide formation, dimerize to the thiosulfinate, react with oxygen to sulfinic or sulfonic acid, form disulfide bonds, or be reduced by vicinal or low-molecular-weight thiols. Despite their reactivity, it is now clear that some sulfenic acids are more stable, and these can be identified in proteins under physiological conditions (21, 25–28).
Protein Sulfenic Acid as a Posttranslational Regulatory Mechanism. There is a growing awareness that reversible cysteine-targeted modifications of proteins, particularly S-thiolation and S-nitrosylation, provide an additional mechanism by which proteins can be posttranslationally regulated (14). There is already some evidence that protein sulfenic acid formation is also important in regulation of protein function (13, 15), but the full extent of these regulatory mechanisms is yet to be determined. The number of proteins that are targets for regulatory cysteine oxidation could number those regulated by phosphorylation (29). In addition, because a variety of protein cysteine oxidation states can be formed, this offers the potential for oxidant-specific functional effects in which particular oxidants can differentially regulate the activity of proteins on the basis that they form varied structural motifs. Protein sulfenic acid formation has all of the essential elements of a regulatory system, including sensitivity, specificity, and reversibility. Protein sulfenation is also directly modulated by the cellular H2O2 concentration, which itself can be carefully controlled by classical signal transduction molecules, as discussed above. When a protein forms a sulfenic acid, the function of the protein may be altered, such that a function can be switched on and off or otherwise modified. Of course, most proteins contain cysteine residues. Not all protein cysteines are important in sulfenic acid regulatory control. Indeed, most cysteines are not functionally important and do not react with physiological concentrations of H2O2. Proteins that are regulated by H2O2 have characteristic cysteines with a particularly low pKa, rendering the thiol more reactive at physiological pHs.
Widespread Protein Sulfenic Acid Formation in Mammalian Tissues. Poole et al. (13) have recently exquisitely reviewed the current state of sulfenic acid research, particularly in the context of redox signaling. Protein sulfenic acid formation is clearly important in the redox regulation of transcription factors (OxyR, OhrR, Yap1), tyrosine phosphatases, members of the NAD(P)H/disulphide reductase family, cysteine based peroxidases (including Orp1), peroxiredoxins, and methionine sulfoxide reductases (12, 13, 30). However, this list is small, with the vast majority of proteins identified from bacterial systems, although some mammalian substrates have been characterized (26, 28, 31).
In this study, we report widespread protein sulfenic acid formation within mammalian tissues when H2O2 is elevated. We used sodium arsenite to convert protein sulfenic acids to the reduced form, because there is significant evidence that this reagent has specificity for reducing this from of oxidized cysteine and has been widely used for this application (18–22). To confirm the arsenite-specific reduction of sulfenic acids, we also carried out studies with dimedone. The nucleophile dimedone reacts preferentially with sulfenic to form a stable product, which when combined with spectraphotometry and MS has allowed detection of protein sulfenic acids (22). We found that dimedone was able to fully block arsenite-induced thiol labeling with biotin–maleimide, providing additional corroborative evidence as to the specify of these methods for protein sulfenic acids.
Although we have detected a large number of proteins that form protein sulfenic acid when H2O2 is present, it is possible that there are a number of proteins that are also modified in this way, which we did not detect. These may be particularly unstable sulfenic acids, which do not survive the labeling protocol. However, it is clear that a number of protein sulfenates are indeed stable enough to allow detection and purification. These more stable protein sulfenic acids may be important in posttranslational regulation, with the prospect that more transient, labile species not detected by these methods are important in enzyme catalysis. Our ability to detect some sulfenic acids at least may reflect the use of a high concentration of maleimide, which rapidly alkylates low molecular weight and protein thiols that might otherwise rapidly reduce thse species.
Influence of Concentration and Duration of H2O2 Administration on Sulfenic Acid Formation. Although we have observed sulfenic acid formation when H2O2 is perfused through isolated hearts, we have also found that, at higher concentrations, there was a gradual loss of this modification. The most likely explanation for this biphasic response is the further oxidation of the sulfenic acid (–SOH) to the sulfinic (–SO2H) or sulfonic (–SO3H) oxidation state. These more oxidized forms of cysteine have been regarded as irreversible, although recent evidence shows that sulfinic acids are, at least in the case of the peroxiredoxins, reversible by ATP-dependent enzymes and are important in redox regulation of function, with the potential of other proteins being similarly regulated (32, 33). We also found that sulfenic acid formation was substantially attenuated by preloading the intact heart with NAC directly before the administration of H2O2. NAC and other small thiol compounds are extensively utilized in redox cell biology studies, and have the general effect of blocking or blunting oxidant-induced changes. NAC may manifest the reduction in protein sulfenic acid formation by a direct scavenging reaction with H2O2 or by directly reducing them once formed.
Protein sulfenic acids were rapidly detected after H2O2 treatment, but were gradually lost during sustained perfusion. The biphasic response in sulfenic acid formation for both H2O2 concentration and duration of perfusion is consistent with the tissue having a discrete capacity for the maintenance of adequate reducing conditions to prevent thiol oxidation to sulfinic or sulfonic states. Thus, the heart may have a maximal H2O2 load it can tolerate, which once surpassed results in thiol oxidation past the sulfenic state. Progression through the oxidation states may correlate with transition from reversible to irreversible tissue injury.
Proteins Identified That Form Sulfenic Acids. The proteins we found to form sulfenic acids originate from diverse classes, and include cytosolic and mitochondrial metabolic enzymes, as well as cytoskeletal and myofilament proteins (see Table 1). In general, the proteins we have identified are highly expressed in cardiac tissue, illustrating the methodology preferentially detects and identifies the most abundant proteins modified. It is likely that there are substantial numbers of other proteins not detected or identified, because they are in low abundance in the tissue studied. It is also possible the methods we have used may not trap all forms of protein sulfenic acids with equal efficiency. Many of the proteins identified clearly possess cysteines susceptible to oxidation, because we and others have already found them to be susceptible to other forms of cysteine targeted oxidation, such as S-thiolation (17, 34, 35) or S-nitrosylation (36). Immunoblot analysis showed the extent to which sulfenic acid derivatives are formed varies considerably between proteins. It is difficult to reconcile the low percentage oxidation of actin, tropomyosin, or myosin heavy chain with a possible functional effect. It is a complex issue to broadly define the proportion of a protein that has to be modified for it to be functionally relevant, assuming there would in fact be a functional correlate of oxidation. Even small percentage changes could have a large functional effect if the oxidative event acts as an “on switch,” enabling an enzymatic activity not previously present. The low, but significant degree of oxidative modification observed for ATP synthase α-subunit (11%) and aspartate aminotransferase (13%) could conceivably have a functional impact, but further studies are required. A substantial proportion of both ADP/ATP carrier protein (adenine nucleotide translocator) and malate dehydrogenase formed sulfenic acid, observations that warrant further investigation. It is highly likely that there would be a functional consequence of sulfenic acid formation, because both of these proteins are known to have redox active thiols that can regulate their activity (35, 37).
However, in two cases (ATP synthase β-subunit and troponin T), it is difficult to explain their presence, because they do not contain cysteine residues. We previously found the β-subunit of the ATP synthase present after affinity purification of S-thiolated proteins (34), and reasoned that this likely occurred because it tightly binds the redox active α-subunit of the ATP synthase. The same explanation could hold true in these studies, as the ATP synthase α-subunit was also detected amongst the purified protein sulfenic acids. Similarly, troponin T is likely present following copurification with other myofilament components with a redox active cysteine. Potential and likely candidates for such proteins include tropomyosin, which directly binds troponin T, and actin, which itself binds troponin T. Both tropomyosin and actin are already known to have redox active cysteines (38, 39), and were identified as forming sulfenic acids in this study.
In summary, these studies have shown the utility of arsenite-induced reduction and subsequent labeling with biotin–maleimide in the detection, purification, and identification of proteins that interact directly with H2O2. We have used this approach to show that increased cellular H2O2 causes widespread protein sulfenic acid formation, which is likely to play a crucial role in redox signaling in health and disease.
Acknowledgments
This research was supported by grants from The Wellcome Trust, the U.K. Biotechnology and Biological Sciences Research Council, and Aventis Pharma.
Author contributions: A.T.S. and P.E. designed research; A.T.S., H.N., and J.P.B. performed research; A.T.S., H.N., and P.E. analyzed data; and A.T.S. and P.E. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: NAC, N-acetylcysteine.
References
- 1.Nishida, M., Maruyama, Y., Tanaka, R., Kontani, K., Nagao, T. & Kurose, H. (2000) Nature 408, 492–408. [DOI] [PubMed] [Google Scholar]
- 2.Minamino, T., Yujiri, T., Papst, P. J., Chan, E. D., Johnson, G. L. & Terada, N. (1999) Proc. Natl. Acad. Sci. USA 96, 15127–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka, H., Sakurai, K., Takahashi, K. & Fujimoto, Y. (2003) J. Cell Biochem. 89, 944–955. [DOI] [PubMed] [Google Scholar]
- 4.Gustafsson, Å., Tsai, J. G., Logue, S. E., Crow, M. T. & Gottlieb, R. A. (2004) J. Biol. Chem. 279, 21233–21238. [DOI] [PubMed] [Google Scholar]
- 5.Yang, H. Roberts, L. J., Shi, M. J., Zhou, L. C., Ballard, B. R., Richardson, A. & Guo, Z. M. (2004) Circ. Res. 95, 1075–1081. [DOI] [PubMed] [Google Scholar]
- 6.Suh, Y.-A., Arnold, R. S., Lassegue, B., Shi, J., Xu, X., Sorescu, D., Chung, A. B., Griendling, K. K. & Lambeth, J. D. (1999) Nature 401, 79–83. [DOI] [PubMed] [Google Scholar]
- 7.Arnold, R. S., Shi, J., Murad, E., Whalen, A. M., Sun, C. Q., Polavarapu, R., Parthasarathy, S., Petros, J. A. & Lambeth, J. D. (2001) Proc. Natl. Acad. Sci. USA 98, 5550–5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okuda, S., Nishiyama, N., Saito, H. & Katsuki, H. (1996) Proc. Natl. Acad. Sci. USA 93, 12553–12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krtolica, A., Parrinello, S., Lockett, S., Desprez, P.-Y. & Campisi, J. (2001) Proc. Natl. Acad. Sci. USA 98, 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahsan, H., Ali, A. & Ali, R. (2002) Clin. Exp. Immunol. 131, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhee, S. G., Bae, Y.-S., Lee, S.-R. & Kwon, J. (2000) Sci. STKE, www.stke.org/cgi/content/full/OC_sigtrans;2000/53/pe1. [DOI] [PubMed]
- 12.Claiborne, A., Yeh, J. I., Mallett, T. C., Luba, J., Crane, E. J., Charrier, V. & Parsonage, D. (1999) Biochemistry 38, 15407–15416. [DOI] [PubMed] [Google Scholar]
- 13.Poole, L. B., Karplus, P. A. & Claiborne, A. (2004) Annu. Rev. Pharmacol. Toxicol. 44, 325–347. [DOI] [PubMed] [Google Scholar]
- 14.Di Simplicio, P., Franconi, F., Frosali, S. & Di, G. (2003) Amino Acids 25, 323–339. [DOI] [PubMed] [Google Scholar]
- 15.Claiborne, A., Yeh, J. I., Mallett, T. C., Luba, J., Crane, E. J., Charrier, V. & Parsonage, D. (1999) Biochemistry 38, 15408–15416. [DOI] [PubMed] [Google Scholar]
- 16.Salmeen, A., Andersen, J. N., Myers, M. P., Meng, T.-C., Hinks, J. A., Tonks, N. K. & Barford, D. (2003) Nature 423, 769–773. [DOI] [PubMed] [Google Scholar]
- 17.Eaton, P., Byers, H. L., Leeds, N., Ward, M. A. & Shattock, M. J. (2002) J. Biol. Chem. 277, 9806–9811. [DOI] [PubMed] [Google Scholar]
- 18.Radi, R., Beckman, J. S., Bush, K. M. & Freeman, B. A. (1991) J. Biol. Chem. 266, 4244–4250. [PubMed] [Google Scholar]
- 19.Kuhn, D. M. & Geddes, T. J. (1999) J. Biol. Chem. 274, 29726–29732. [DOI] [PubMed] [Google Scholar]
- 20.Torchinsky, Y. M. (1981) in Sulfur in Proteins (Pergamon, Oxford), pp. 53.
- 21.Kim, S. O., Merchant, K., Nudelman, R., Beyer, J. F., Jr., Keng, T., DeAngelo, J., Hausladen, A. & Stamler, J. S. (2002) Cell 109, 383–396. [DOI] [PubMed] [Google Scholar]
- 22.Ellis, H. R. & Poole, L. B. (1997) Biochemistry 36, 15013–15018. [DOI] [PubMed] [Google Scholar]
- 23.Schafer, F. Q. & Buettner, G. R. (2001) Free Radical Biol. Med. 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- 24.Dalle-Donne, I., Scaloni, A., Giustarini, D., Cavarra, E., Tell, G., Lungarella, G., Colombo, R., Rossi, R. & Milzani, A. (2004) Mass Spectrom. Rev., in press. [DOI] [PubMed]
- 25.Denu, J. M. & Tanner, K. G. (1998) Biochemistry 37, 5633–5642. [DOI] [PubMed] [Google Scholar]
- 26.Allison, W. S. (1976) Acc. Chem. Res. 9, 293–299. [Google Scholar]
- 27.Claiborne, A., Miller, H., Parsinage, D. & Ross, R. P. (1999) FASEB J. 7, 1483–1490. [DOI] [PubMed] [Google Scholar]
- 28.Allison, W. & Benitez, L. V. (1972) Proc. Natl. Acad. Sci. USA 69, 3004–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang, F. L. & Huang, K.-P. (2002) Biochem. Pharmacol. 64, 1049–1056. [DOI] [PubMed] [Google Scholar]
- 30.Choi, H. J., Kim, S. J., Mukhopadhyay, P., Cho, S., Woo, J. R., Storz, G. & Ryu, S. E. (2001) Cell 105, 103–113. [DOI] [PubMed] [Google Scholar]
- 31.Stortz, G., Tartaglia, L. A. & Ames, B. N. (1990) Science 248, 189–194. [DOI] [PubMed] [Google Scholar]
- 32.Budanov, A. V., Sablina, A. A., Feinstein, E., Koonin, E. V. & Chumakov, P. M. (2004) Science 304, 596–600. [DOI] [PubMed] [Google Scholar]
- 33.Biteau, B., Labarre, J. & Toledano, M. B. (2003) Nature 425, 980–984. [DOI] [PubMed] [Google Scholar]
- 34.Eaton, P., Jones, M. E., McGregor, E., Dunn, M. J., Leeds, N., Byers, H. L., Leung, K. Y., Ward, M. A., Pratt, J. & Shattock, M. J. (2003) J. Am. Soc. Nephrol. 14, S290–S296. [DOI] [PubMed] [Google Scholar]
- 35.Eaton, P. & Shattock, M. J. (2002) Ann. N.Y. Acad. Sci. 973, 1–4. [DOI] [PubMed] [Google Scholar]
- 36.Kuncewicz, T., Sheta, E. A., Goldknopf, I. L. & Kone, B. C. (2003) Mol. Cell Proteom. 2, 156–163. [DOI] [PubMed] [Google Scholar]
- 37.McStay, G. P., Clarke, S. J. & Halestrap, A. P. (2002) Biochem. J. 367, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang, J., Boja, E., Tan, W. H., Tekle, E., Fales, H. M., English, S., Mieyal, J. J. & Chock, P. B. (2001) J. Biol. Chem. 276, 47763–47766. [DOI] [PubMed] [Google Scholar]
- 39.Fratelli, M., Demol, H., Puype, M., Casagrande, S., Eberini, I., Salmona, M., Bonetto, V., Mengozzi, M., Duffieux, F., Miclet, E., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 3505–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]