

Abstract
The purpose of this study was to create an open access repository of validated liquid chromatography tandem mass spectrometry (LC‐MS/MS) multiple reaction monitoring (MRM) methods for quantifying 284 important proteins associated with drug absorption, distribution, metabolism, and excretion (ADME). Various in silico and experimental approaches were used to select surrogate peptides and optimize instrument parameters for LC‐MS/MS quantification of the selected proteins. The final methods were uploaded to an online public database (QPrOmics; www.qpromics.uw.edu/qpromics/assay/), which provides essential information for facile method development in triple quadrupole mass spectrometry (MS) instruments. To validate the utility of the methods, the differential tissue expression of 107 key ADME proteins was characterized in the tryptic digests of the pooled subcellular fractions of human liver, kidneys, intestines, and lungs. These methods and the data are critical for development of physiologically based pharmacokinetic (PBPK) models to predict xenobiotic disposition.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Quantitative methods to measure the expression of ADME proteins in various human tissues are not available. There has been limited emphasis on compiling validated quantitative methods outside of a small number of ADME genes.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We have developed quantitative proteomics methods for 284 important human DMEs, transporters, and nuclear receptors. Further, we have validated and applied many of these methods in multiple human tissues.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This study compiled an open access database of surrogate peptides, optimized LC‐MS/MS parameters, and tissue expression data of DMEs and transporters in human liver, kidneys, intestines, and lungs.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The methods provided are useful for generating inter‐tissue and interindividual variability data on the expression of drug disposition‐related proteins, which are crucial for developing accurate PBPK models applicable in drug development.
Physiologically based pharmacokinetic (PBPK) modeling is an emerging approach for predicting the potential in vivo PKs of drugs.1, 2, 3 By characterizing the differential tissue expression and interindividual variability of proteins involved in drug disposition (i.e., drug metabolizing enzymes (DMEs) and drug transporters), the accuracy of PBPK predictions can be significantly improved. Both phase I and phase II DMEs regulate drug concentration through chemical biotransformation of the drugs. Phase I enzymes modify a compound through oxidation, reduction, or hydrolysis. Cytochrome P450 (CYP) enzymes are the most commonly studied enzymes with regard to drug metabolism, as they are widely expressed and broadly specific. CYP enzymes, along with flavin‐containing monooxygenases (FMOs), alcohol dehydrogenases (ADHs), aldehyde dehydrogenases (ALDHs, respectively), and several other families, oxidize xenobiotics to break them down into more readily eliminated compounds. Esterases, such as carboxylesterases (CESs) and paraoxonases (PONs), are hydrolytic enzymes. In contrast to phase I enzymes, phase II enzymes conjugate compounds in order to make them more polar, and, thus more readily eliminated. UDP‐glucuronosyltransferases are the most widely studied phase II enzymes. Glutathione s‐transferases, sulfotransferases, and N‐acetyltransferases are also grouped in phase II enzymes. In addition to DMEs, there are drug transporters that control uptake (phase 0) or efflux (phase III) of drugs in tissues, such as the liver, intestines, kidneys, and brain. Therefore, DMEs and transporters often collectively constitute rate‐determining processes with regard to drug efficacy (on‐target effects) and toxicity (off‐target effects).4, 5, 6, 7, 8
Human primary cell‐based in vitro models, such as human hepatocytes or proximal tubular cells, can be used to predict drug metabolism and transport in vivo.3, 9 However, in vitro protein expression can change depending on the culture conditions.10, 11 Therefore, primary cells often lead to poor prediction of in vivo clearance, especially when changes in protein expression are not quantified.12 Moreover, primary cells are not available for several organs that are important to drug disposition, such as the intestines and lungs. An emerging approach relies on estimating in vitro clearance of DMEs or transporters using recombinant enzymes or cells.10, 11 These in vitro clearance data can be extrapolated to in vivo by incorporating tissue protein expression in the PBPK models, as discussed elsewhere.13, 14, 15 Therefore, protein expression data in human tissues are important for in vitro‐in vivo extrapolation and PBPK modeling from either primary cells or recombinant systems. In addition, interindividual variability in drug disposition can also be predicted using expression data generated using the banked donor tissues. By characterizing tissue cohorts for the protein expression, the effect of covariates, such as age, sex, genotype, and disease conditions can be integrated into PBPK models. Further, quantification of DMEs and transporters is important for the characterization of in vitro models, such as cell‐lines, primary cells, or subcellular fractions.
To quantify proteins, multiple reaction monitoring (MRM) quantitative proteomics has become increasingly popular in the last few years.16, 17 MRM analysis of proteins utilizes a triple quadrupole mass spectrometry (MS) instrument to achieve greater selectivity (i.e., quantifying a specific parent mass and selected daughter ions from the same). When coupled to liquid chromatography (LC), the specificity of the MS can allow for the rapid, multiplexed quantification of absorption, distribution, metabolism, and excretion (ADME) proteins with excellent selectivity and reproducibility.16, 17 Further, MRM methods are not limited by the availability of antibodies as with immunoblotting (e.g., Western blotting) and multiple proteins can be quantified simultaneously by liquid chromatography tandem mass spectrometry (LC‐MS/MS).17 Despite the aforementioned advantages over the conventional methods, method development in MRM proteomics is an iterative exercise involving a significant initial outlay of time and money. MRM method development becomes more challenging if the “best performing” surrogate peptides are not readily identifiable. Although comprehensive databases already exist, such as SRMAtlas (www.srmatlas.org/), they do not provide experimentally optimized instrument conditions and data demonstrating the usefulness of surrogate peptides. Therefore, we present here the development of optimized quantification methods for 284 human ADME proteins (Table 1 , Figure 1), which are compiled in a publicly accessible database (QPrOmics; www.qpromics.uw.edu/qpromics/assay/). The key information includes unique surrogate peptides, parent and fragment m/z values, and predicted or observed collision energy and cone voltage or declustering potential for both light and heavy peptides. In addition, the database provides information on unique surrogate peptides to distinguish selected nonsynonymous single nucleotide polymorphisms (SNPs), annotations of potential post‐translation modifications (PTMs), subcellular localization of each protein, and whether the peptide is conserved in different species. These methods have extensive applications in the field of systems pharmacology, in which quantitative understanding of DMEs and transporters is critical. Also presented here is the application of these MRM methods to determine inter‐tissue differences in protein expression of DMEs and transporters in human tissue samples.
Table 1.
List of major absorption, distribution, metabolism, and excretion proteins for which surrogate peptides were selected.
| Phase I Enzymes |
| ADH1A, ADH1B*, ADH1C*, ADH4, ADH5, ADH6, ADH7, ADHFE1, ALDH1A1*, ALDH1A2*, ALDH1A3*, ALDH1B1, ALDH2, ALDH3A1, ALDH3A2, ALDH3B1, ALDH3B2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDH8A1, ALDH9A1, AOX1*, CBR1, CBR3, CES1*, CES2, CYB5R3, CYP1A1*, CYP1A2*, CYP1B1, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP19A1, CYP20A1, CYP20A1, CYP21A2, CYP24A1, CYP26A1, CYP26C1, CYP27A1, CYP27B1, CYP2A13, CYP2A6*, CYP2A7, CYP2B6, CYP2C8*, CYP2C9*, CYP2C18*, CYP2C19, CYP2D6*, CYP2D7P1, CYP2E1*, CYP2F1, CYP2J2*, CYP2R1, CYP2S1, CYP3A4*, CYP3A5*, CYP3A7, CYP3A43*, CYP4A11, CYP4B1, CYP4F11, CYP4F12, CYP4F2, CYP4F3, CYP4F8, CYP4Z1, CYP7A1, CYP7B1, CYP8B1, CYP39A1, CYP46A1, CYP51A1, DDO, DHRS1, DHRS2, DHRS3, DHRS4, DHRS4L1, DHRS4L2, DHRS7, DHRS7B, DHRS7C, DHRS9, DHRS12, DHRS13, DHRSX, DPEP1, DPYD*, EPHX1*, EPHX2*, FMO1*, FMO2*, FMO3*, FMO4, FMO5, FMO6P, GPX1, GPX2, GPX3, GPX4, GPX5, GPX6, GPX7, GSR, GSS, HAGH, HSD11B1, HSD17B11, HSD17B14, LOC728667, LOC731356, LOC731931, METAP1, NOS1, NOS2A, NOS3, PDE3A, PDE3B, PLGLB1, PON1, PON2, PON3, SULF1, XDH |
| Phase II Enzymes |
| CHST1, CHST2, CHST3, CHST4, CHST5, CHST6, CHST7, CHST8, CHST9, CHST10, CHST11, CHST12, CHST13, GSTA1*, GSTA2, GSTA3*, GSTA4, GSTA5*, GSTCD, GSTK1, GSTM1*, GSTM2*, GSTM3*, GSTM4*, GSTM5, GSTO1, GSTO2, GSTP1*, GSTT1*, GSTT2, GSTZ1, HNMT, MGST1, MGST2, MGST3, NAT1*, NAT2*, NNMT, PNMT, SULT1A1*, SULT1A2*, SULT1A3, SULT1B1, SULT1C1/C2*, SULT1E1, SULT2A1*, SULT2B1*, SULT4A1, TPMT, UGT1A1*, UGT1A3*, UGT1A4*, UGT1A5*, UGT1A6, UGT1A7, UGT1A8*, UGT1A9*, UGT1A10*, UGT2A1*, UGT2B4*, UGT2B7*, UGT2B10, UGT2B11, UGT2B15*, UGT2B17, UGT2B28, UGT8* |
| Transporters |
| ABCA1 (CERP), ABCA4, ABCB1 (P‐gp, MDR1)*, ABCB2 (TAP1), ABCB3 (TAP2), ABCB4 (MDR3), ABCB5, ABCB6 (PRP, UMAT), ABCB7, ABCB8 (MABC1), ABCB11 (BSEP), ABCC1 (MRP1), ABCC2 (MRP2)*, ABCC3 (MRP3), ABCC4 (MRP4)*, ABCC5 (MRP5), ABCC6 (MRP6), ABCC8 (SUR1), ABCC9 (SUR2), ABCC10 (MRP7), ABCC11 (MRP8), ABCC12 (MRP9), ABCC13, ABCG1 (WHT1), ABCG2 (BCRP)*, SLC2A4 (GLUT4), SLC2A5 (GLUT5), SLC5A6 (SMVT), SLC6A6, SLC7A5 (LAT1), SLC7A7, SLC7A8 (LAT2), SLC10A1 (NTCP), SLC10A2 (NTCP2), SLC13A1 (NAS1), SLC13A2 (NADC1), SLC13A3 (NADC3), SLC15A1 (PEPT1), SLC15A2 (PEPT2), SLC16A1 (MCT1), SLC19A1 (FLOT1), SLC22A1 (OCT1)*, SLC22A2 (OCT2)*, SLC22A3 (OCT3)*, SLC22A4 (OCTN1)*, SLC22A5 (OCTN2)*, SLC22A6 (OAT1)*, SLC22A7 (OAT2), SLC22A8 (OAT3)*, SLC22A9 (OAT7)*,SLC22A10 (OAT5), SLC22A11 (OAT4), SLC22A12 (URAT1)*, SLC22A13 (OAT10), SLC22A14 (OCTL2), SLC22A15 (FLIPT1), SLC22A16 (OCT6), SLC22A17 (BOIT), SLC22A18 (BWR1A), SLC22A18AS (BWR1B), SLC27A1 (FATP1), SLC28A1 (CNT1)*, SLC28A2 (CNT2)*, SLC28A3 (CNT3)*, SLC29A1 (ENT1), SLC29A2 (ENT2), SLC47A1 (MATE1), SLC47A2 (MATE2K), SLCO1A2 (OATP1A2)*, SLCO1B1 (OATP1B1)*, SLCO1B3 (OATP1B3)*, SLCO1C1 (OATP1C1), SLCO2A1 (OATP2A1), SLCO2B1 (OATP2B1), SLCO3A1 (OATP3A1), SLCO4A1 (OATP4A1), SLCO4C1 (OATP4C1), SLCO5A1 (OATP5A1), SLCO6A1 (OATP6A1) |
|
Nuclear Receptors
AHR, CAR, FXR, GCR, HNF4α, PGR, PXR |
|
Proteins shown in bold were detected in different human tissues and the asterisk (*) indicates availability of surrogate peptide with change in amino acid due to single nucleotide polymorphism. UniProt IDs provided in Supplementary Table S2. |
Figure 1.
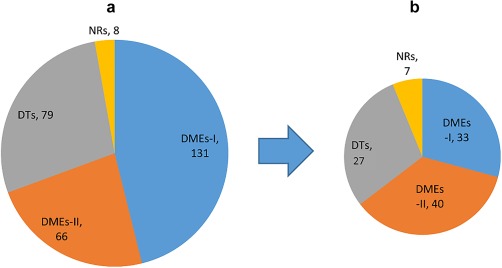
Distribution of drug metabolizing enzymes (DMEs), drug transporters (DTs), and nuclear receptors (NRs) for which quantifiable targeted peptides are identified (a) or detected in human tissue subcellular fractions (b). Values indicate the number of proteins. DME‐I and DME‐II represent phase I and phase II DMEs, respectively.
MATERIALS AND METHODS
The total protein quantification bicinchoninic acid assay kit, iodoacetamide, dithiothreitol, and ammonium bicarbonate (ABC) were purchased from Pierce Biotechnology (Rockford, IL). The ProteoExtract native membrane protein extraction kit was procured from Calbiochem (Temecula, CA). Synthetic signature peptides and heavy labeled surrogate peptides were obtained from Thermo Fisher Scientific (Rockford, IL). Formic acid was purchased from Sigma‐Aldrich (St. Louis, MO). Chloroform, MS‐grade acetonitrile, methanol, and formic acid were purchased from Fischer Scientific (Fair Lawn, NJ). Sodium deoxycholate (98% purity) was obtained from MP Biomedicals (Santa Ana, CA).
Surrogate peptide selection
A simple outline of the peptide selection strategy is depicted in Figure 2. Initially, target proteins were selected based on mRNA databases of in vivo signal in multiple body tissues.18 Two hundred eighty‐four proteins were chosen as the most significant with regard to drug disposition. There proteins include 131 phase I DMEs, 66 phase II DMEs, 79 transporters, and 8 nuclear receptors (Figure 1). SRMAtlas (Institute for Systems Biology, Seattle, WA) was used to predict the 10–20 best peptides for each protein, and the 10 best fragments from each peptide. These predicted peptide sequences were subsequently imported into the Skyline program (University of Washington, Seattle, WA). The Skyline program analyzed the entered peptide sequences and produced initial MS settings (i.e., parent and product m/z, collision energy, and cone voltage) for both Waters Xevo TQ‐S MS instrument (Waters, Hertfordshire, UK) and AB Sciex 6500 Triple Quadrupole instrument (Framingham, MA).
Figure 2.
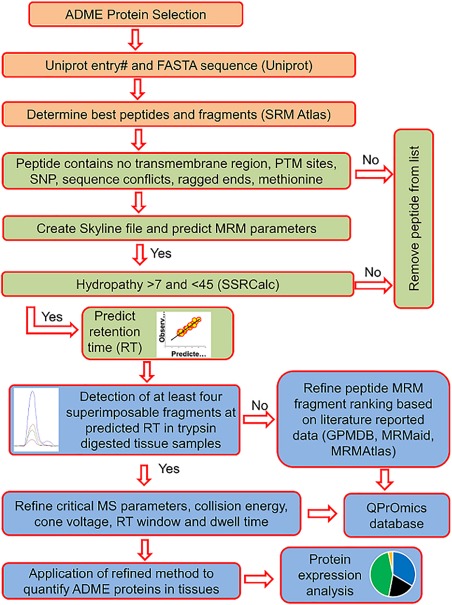
Quantitative proteomics workflow for surrogate peptide selection, method optimization, and validation for quantification of native peptides. For peptides relevant to post‐translation modifications (PTMs), and non‐synonymous single nucleotide polymorphisms (SNPs), the workflow can be modified to specifically target these sites. ADME, absorption, distribution, metabolism, and excretion; MRM, multiple reaction monitoring; MS, mass spectrometry.
The peptides predicted by SRMAtlas were further analyzed using MS Homology (National Center for Biotechnology Information) to determine if the peptide sequence is unique to a specific protein. This project focused on human data, but also compiled peptides that are conserved in common preclinical species (rat, mouse, dog, and monkey). If a peptide sequence was unique to a protein, it was further analyzed to determine if there were any potentially problematic areas, including PTM sites, SNPs, sequence conflicts, mutagenesis sites, and splice variants. Additionally, surrogate peptides for transporters were analyzed to ensure they were not located in the predicted transmembrane region of the protein. Although the aforementioned criteria were taken into consideration for peptide selection, some optimized peptides do include these areas as no alternative was available. Complete information on peptide features is contained in the database.
After this initial selection process, the Sequence Specific Retention Calculator (Manitoba Centre for Proteomics and Systems Biology, Winnipeg, Manitoba, Canada) was used to calculate the hydrophobicity index of each peptide. Peptides with a calculated hydrophobicity between 7 and 45 were given priority importance, as those that fall outside of this range are prone to poor LC characteristics and/or solubility problems. The number of peptides per protein was reduced to a maximum of three based on the outlined criteria, as well as an analysis of tandem mass spectrometry (MS/MS) fragmentation data reported by PeptideAtlas, MRMaid data, and SRMAtlas ranking.
Development of generic liquid chromatography methods and optimization of tandem mass spectrometry parameters for shortlisted peptides
From the original list of proteins, 107 clinically relevant DMEs were selected for further method validation and application to in vivo samples. DMEs were defined as “clinically relevant” if they were demonstrated to be relevant to the metabolism of a currently marketed drug. Methods were optimized using heavy peptides on two triple‐quadrupole LC‐MS instruments (Xevo TQ‐S and AB ScieX 6500 Triple Quadrupole MS instruments both coupled to ACQUITY UPLC; Waters, Hertfordshire, UK) in electrospray ionization‐positive ionization mode. From the calculated hydrophobicity (i.e., Sequence Specific Retention Calculator) for each peptide, in‐house developed calibration curves were used to generate a predicted retention time for two specific columns used (Supplementary Table S1; Supplementary Figure S1). The predicted retention times allowed the creation of scheduled MRM methods to maximize the number of peptides analyzed per run without compromising dwell time. LC methods were developed to improve peptide separation (Supplementary Table S1). Collision energy and cone voltage/declustering potential, depending on the platform, were optimized for each peptide in order to improve sensitivity in the two different MS/MS systems (Supplementary Table S2). Although the three best fragments for light peptides were retained, dwell time was improved by selecting only two fragments for the respective heavy peptides. The optimized methods are provided in Supplementary Table S2, and are also available for download from the QPrOmics database (www.qpromics.uw.edu/qpromics/assay/). Representative screenshots of QPrOmics database are provided in Supplementary Figures S4 and S5.
Procurement of human tissue subcellular fractions
Human liver and kidney samples were obtained from the tissue bank of School of Pharmacy University of Washington (Seattle, WA) and the University of Washington Kidney Research Institute, respectively. In addition to these samples, pooled subcellular fractions of human livers, kidneys, intestines, and lungs were obtained from Xenotech (Kansas City, KS). The Xenotech samples are considered as nonhuman subjects, precluding the need for approval. The number of samples in each pool is detailed in Supplementary Table S3.
Quantification of differential tissue expression of absorption, distribution, metabolism, and excretion proteins using liquid chromatography tandem mass spectrometry
All tissue samples were analyzed using Waters Xevo TQ‐S LC‐MS/MS. For peptide quantification, total membrane protein was isolated from six kidney and six liver samples, as per previously published protocol.19 Specifically, the tissue was prepared using the ProteoExtract native membrane protein extraction kit. The tissue was homogenized in 2 mL of Extraction Buffer I mixed with 10 uL of protease inhibitor cocktail, and then incubated for 10 minutes while gently rocking. The resulting homogenate was centrifuged at 16,000 xg for 15 minutes at 4°C, and the supernatant was discarded. The remaining pellet was resuspended in 1 mL of Extraction Buffer II mixed with 10 uL of protease inhibitor cocktail. This solution was then incubated while gently shaking at 4°C for 30 minutes, and subsequently centrifuged at 16,000 xg for 15 minutes at 4°C.
For tissues extracted in‐house, total protein was quantified using Pierce bicinchoninic acid protein assay kit. Commercially sourced fractions included company‐provided protein concentrations. The extracted tissue membrane and commercially sourced subcellular fractions were diluted to a concentration of 2 mg/mL of total protein. The protein sample (100 µL) was denatured, reduced, and alkylated, as per previously established protocol.20 The proteins were subsequently digested with trypsin in a 1:25 trypsin to protein ratio for 18 hours at 37°C. Digestion was then stopped by adding 30 µL of quenching solvent consisting of 80% acetonitrile and 0.1% formic acid. The quenching solvent contained a pool of all the heavy internal standard peptides listed in Supplementary Table S2. The final concentration of individual internal standards in the LC‐MS sample was ∼2–200 ng/mL. The samples were centrifuged for 5 minutes at 5,000 xg and 4°C, and the supernatant was analyzed for quantification of 107 shortlisted proteins using optimized LC‐MS/MS methods, as discussed above. The pooled tissue samples were processed and analyzed in triplicates.
Data analysis
The tissue expression data were processed using MassLynx software version 4.1 (Waters). Superimposability of multiple fragments and their co‐elution with heavy peptide peaks were taken into consideration to assign qualitative accuracy. Briefly, the heavy labeled standard peaks were used to define the elution time for the respective native peptides. Any peaks deviating more than 0.05 minutes from their heavy labeled standard were considered unreliable and discarded. MS response was calculated using the average of peak areas of at least three representative fragments for native peptides and two fragments for heavy peptides. Some peptides retain more than three fragments for the native form because there were multiple daughter ions that demonstrated similar MS response. The area ratios of these fragments (e.g., average three fragments of light peptide vs. average of two fragments of heavy peptides) were considered to address LC‐MS/MS related (i.e., technical) variability. By including the same known amount of heavy peptide to each sample, the inter‐run variability and matrix effects (ion suppression) were controlled. The average of the area ratios of multiple peptides were used to determine differential tissue expression of each protein. Limit of detection and lower limit of quantification were defined as the points where the peptide peak was more than twofold and fivefold higher than the surrounding background. Technical replicates of each subcellular fraction were run to validate measurements. Deviation more than 20% was used as the variability cutoff. If the replicates varied by more than 20%, the data was considered unreliable.
Once area ratios for each peptide were established, the relative expression level for the liver was set as “100,” and the values from other tissues for the same protein were compared. If no expression was seen in a tissue, it was determined to be “0.” If the liver had no expression, the kidney was considered as “100,” and other tissue expression was compared to the kidney. If neither liver nor kidney had expression, then the intestinal value was set to “100.” The protein expression across tissues was compared by Student's t‐test, with P values < 0.05 considered to be significant.
RESULTS
QPrOmics workflow for development of peptide quantification methods
A high throughput strategy was adopted to select and validate surrogate peptides that can be used to quantify in vivo expression levels of proteins. Using sequence specific in silico predicted hydropathy (SSRT number), peptide retention time was reasonably predicted in two LC columns (Supplementary Figure S1; Supplementary Table S1). This allowed a relatively narrow scan window, and increased the number of peptides that could be quantified with each method without affecting optimum dwell time and sensitivity. By multiplexing the methods using predicted retention time, energy and cone voltage, or declustering potential could be efficiently optimized, reducing the overall time and cost. A list of the optimized parameters for both MS instruments is given in Supplementary Table S2. The methods were reduced to a minimum of three light peptide fragments and two heavy peptide fragments, with the fragments giving the best response selected. Note that some peptides have more than three fragments selected; this was done because similar response was produced by multiple fragments, and it was determined that using these additional fragments would improve reproducibility without compromising the overall run time. By using multiple fragments per peptide, the identity of the peptide can be assured. Using this optimized workflow, in silico methods were generated to quantify surrogate peptides for a total of 284 important DMEs and transporters. From this initial list, heavy labeled peptides for 107 of these proteins were used to refine MS settings. Once the MS settings were optimized, heavy labeled peptide cocktails were spiked into tissue samples to serve as retention time calibrators, and native peptides were detected in the samples using the presented methods. All the detected proteins are validated by multiple peptides and multiple fragments of each peptide to ensure the accurateness of the information. The fully optimized peptides include surrogates for phase I and phase II DMEs, transporters, and nuclear receptors.
Further, a MySQL based (Cupertino, CA), open‐access database repository for optimized methods was developed. The key output of the database includes protein name, Uniprot ID, surrogate peptide sequence, parent and fragment m/z, column specific retention time, and MS‐specific parameters (i.e., cone voltage, collision energy). In addition, the database will also provide information on cellular localization of the particular protein, relevant information about surrogate peptide modifications (i.e., SNPs, PTMs, splice variants, and mutagenesis), transmembrane regions, and interspecies sequence homology. As an open access database, these parameters can be downloaded in Excel format (Microsoft, Redmond, WA). Currently, the database contains parameters for two different LC columns, two MS instruments (Xevo TQ‐S and AB Sciex 6500), and five species (human, dog, monkey, rat, and mouse). The user‐friendly database can be searched using a variety of criteria, including protein name, Uniprot ID, or peptide sequence, and the output can be refined by specifying MS instrument or species.
Differential subcellular expression of drug metabolizing enzymes in human liver
Although subcellular fractions were procured for multiple organs, only the liver had all three fractions (cytosolic, microsomal, and S9) available. Because S9 fraction is a crude homogenate containing proteins from multiple subcellular fractions, it was used primarily for method development and not for the inter‐tissue comparison. Both CES, ADH1A and ADH1B, were expressed at detectable levels in both cytosol and microsome (Supplementary Figure S2).
Differential tissue expression of drug metabolizing enzymes
The comparison of differential tissue expression for DMEs was done based on analysis of microsomal fractions. Seventy‐three DMEs were detected in human liver, kidney, intestine, and/or lung microsomal samples (Table 1 and Figures 3 and 4 ; Supplementary Figure S3). As shown in Figure 3, of 73 detected DMEs, the liver expressed 62, the intestine 53, the kidney expressed 49, and the lung only expressed 15. Thirty‐five DMEs were present in at least three of the major elimination organs that were analyzed. The relative inter‐tissue variability is shown in Figure 4 and Supplementary Figure S3. CYP1A2, CYP2B6, CYP3A7, and uridine 5′‐diphosphate glucuronosyltransferase (UGT)2B4 were primarily detectable in the liver, whereas ALDH1A2 and UGT1A10 were distinctly present in the kidneys and intestines, respectively. No proteins were found to be unique to the lungs, although FMO2 was found only in the lungs and kidneys.
Figure 3.
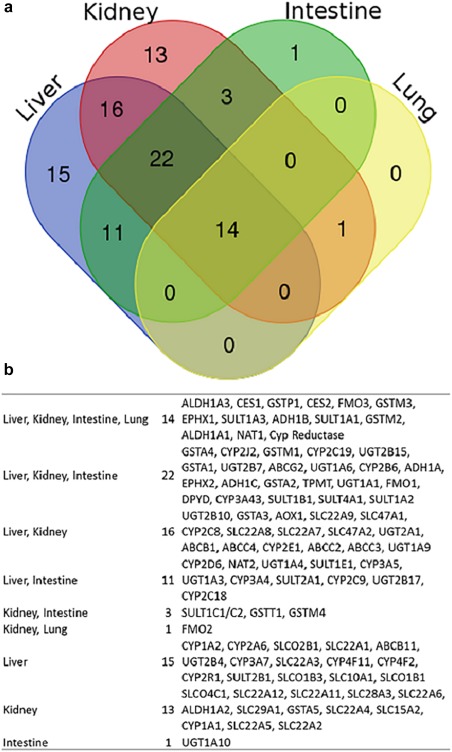
Qualitative tissue protein expression analysis of multiple absorption, distribution, metabolism, and excretion (ADME) proteins. (a) Diagram indicating distribution of number of identified proteins in various tissues. (b) List of ADME proteins detected in various tissues.
Figure 4.
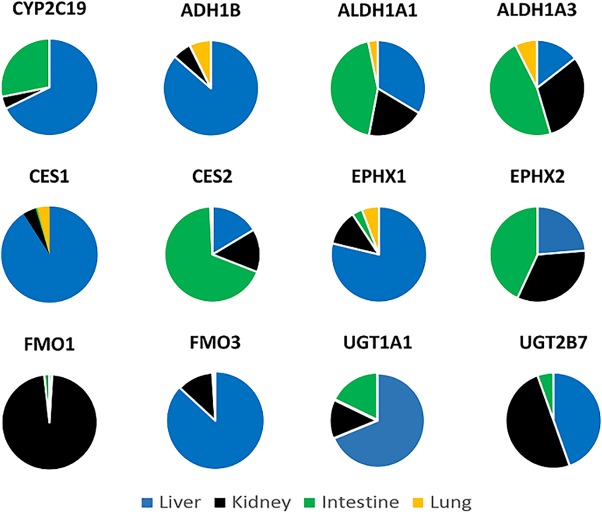
Differential tissue expression of selected absorption, distribution, metabolism, and excretion proteins in the human liver, kidneys, intestines, and lungs using the population mean protein values. The data only represent variability of individual proteins across tissues and not the relative abundance of multiple proteins. ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; CES, carboxylesterases; CYP, cytochrome P450; FMO, flavin‐containing monooxygenase; UGT, uridine 5′‐diphosphate glucuronosyltransferase.
Differential tissue expression of drug transporters
Drug transporter expression was quantified using membrane fractions isolated from pools of six livers and six kidneys (Supplementary Table S3). Of 27 detected transporters, 9 were present in both the liver and kidneys, whereas 8 were specific to the liver and 10 were specific to the kidneys. In our limited sample size, SLC22A4 and SLC47A2 had relatively higher abundance in the liver, whereas SLC22A8, SLC47A1, ABCB1, and ABCC2 were more highly expressed in the kidneys (Figure 5). ABCC4 and ABCG2 were present in both liver and kidneys, but the liver expression was below the lower limit of detection for ABCC4 and the kidney expression was below the lower limit of detection for ABCG2. For this study, the requirement for detectability was that the analyte peak was greater than twofold higher than the surrounding background.
Figure 5.

Relative expression (fold difference) of transporters detected in the liver and kidneys. Relative expression is defined as either the kidney expression divided by liver expression, or liver expression divided by kidney expression. The fold differences are statistically significant except for ABCC2 (Student's t‐test; P < 0.05). The table provides list of transporters detected in the liver and kidneys.
DISCUSSION
The quantification of DME and transporter expression in human tissues is important for predicting the in vivo disposition characteristics of xenobiotics.21, 22, 23, 24 Moreover, quantification of enzyme or transporter induction or suppression can be selectively performed using proteomics methods.25 Recently, such methods have been applied to quantify the interindividual or interspecies differences between human and preclinical models in order to better scale up experimental data.10, 19, 20, 26 Although immunoblotting techniques are available for the quantification of some ADME proteins, this method suffers distinct disadvantages over quantitative proteomics.17 These limitations include lack of selectivity, lack of reproducibility, low throughput, and cumbersome experimental procedures. These shortcomings can be avoided by replacing immunoblotting methods with LC‐MS/MS proteomics. However, developing methods for surrogate peptide quantification can be time and cost prohibitive.16 Additionally, conventional proteomics laboratories utilize high‐end instruments, such as NanoLC coupled to high resolution MS. Similarly, although mRNA expression is often used as a surrogate of protein expression, mRNA data do not always correlate well with protein expression and activity, particularly in human tissues due to the instability of mRNA and the time elapsed between tissue acquisition and storage.27 Therefore, quantification methods for important ADME proteins have been developed here using triple quadrupole MS instruments, which are commonly available in ADME laboratories both in industry and academia settings.28, 29, 30
These methods are available in an open access database and a detailed method can be downloaded and quickly personalized by any laboratory with a triple quadrupole MS instrument. The database is continuously growing with the uploading of multiple methods developed in‐house, covering several species and commonly used MS instruments. Additionally, the database will allow other laboratories to submit validated methods in order to increase its scope.
Currently, with regard to drug disposition, a few dozen ADME proteins dominate the field (e.g., CYPs, UGTs, and P‐glycoprotein). Because any ADME protein can be clinically relevant based on its individual contribution to the metabolism (fraction metabolized) or transport (fraction transported) of a particular drug, the availability of high throughput, broadly applicable targeted proteomics methods will allow rapid and precise characterization of the expression profile of multiple proteins in precious biological samples. In contrast, although global proteomics can characterize multiple proteins simultaneously, the technique currently lacks the sensitivity to quantify low abundance proteins.31 Moreover, global proteomics methods require expensive equipment and highly specific expertise in both the operation of the instruments and the data analysis.
A cursory evaluation of differential tissue expression data verifies that the liver is the primary elimination organ, followed closely by the intestines and kidneys, with the lungs having the least diversity of expressed DMEs. The DME profiles of the liver and intestines overlap significantly, with the majority of enzymes in the CYP and UGT families. This mirrors the importance of CYPs and UGTs in first pass metabolism of xenobiotics. Although the kidneys are typically viewed as organs of elimination through transport, we detected 53 major DMEs expressed in kidney tissue. The majority of the detected proteins in the kidneys are phase II enzymes, which serve to add polar groups that modify xenobiotics and improve their renal elimination characteristics. Beyond the traditional organs of elimination, the lungs were found to express 15 DMEs, including CES, EPHX1, NAT1, and multiple glutathione S‐transferase (GST)s. Although the abundance of DMEs is generally lower in the lungs, the huge surface area and high blood perfusion make the lungs potentially important organs for drug disposition and toxicity, including allergic response. The potential for the lungs to have metabolic impact is increased further by the fact that an i.v. dosed drug will pass through the lungs before reaching systemic circulation, resulting in a lung “first pass” effect. Although there have not yet been significant issues caused by lung metabolism, their role as organs of elimination cannot be discounted.
The ubiquity of key players like CYPs and UGTs is already well characterized, as is their significance in drug disposition, but the prevalence of multiple non‐CYP and non‐UGT DMEs in three or more major elimination organs warrants further analysis. For example, the presence of both major isoforms of CES in all four organs studied here is important for the prediction of activation, deactivation, and toxification of ester and amide containing moieties, including prodrugs and common environmental toxins, like flame retardants and pesticides. CES present unique opportunities and difficulties due to their broad specificity and ubiquity.32
When compared with historical data, these data align well with expression levels of FMOs, many CYPs and UGTs, most GSTs, and CES. This validates the utility of quantitative proteomics to accurately determine inter‐tissue variability. Moreover, some unique expression profiles for multiple under‐researched DMEs were found. Although mRNA assays have historically provided the majority of differential tissue expression data, they cannot be used as a direct surrogate of protein activity. On the other hand, direct activity measurement is not practical due to the requirement of having highly specific probe substrates and individual assays for each protein. Whereas CYP enzymes have a panel of available activity assays, non‐CYP enzymes are severely underserved. Furthermore, even for CYPs, activity assays cannot be taken as surrogates for in vivo expression. Therefore, proteomic data provides a cost‐effective and practical solution to the current limitations of selectivity and high‐throughput. In looking at historical data, FMO1 mRNA expression is reported to be highest in the kidneys, FMO2 in the kidneys and lungs and FMO3 primarily in the liver.33, 34 The data on CYP2B6, CYP2C19, CYP3A4, CYP2C9, GSTM2, and GSTM3 from this experiment also matches well with the protein data reported by Song et al.35
The methodology and data presented here have some limitations. For example, quantitative proteomics is targeted and as such cannot be used in quantification of proteins in a shotgun‐style approach. Additionally, the number of samples available served as a limitation, particularly regarding the lungs (n = 4), which can cause aberrations in the data and subsequent interpretation. Although quantification methods for nuclear receptors were validated using protein standards, these signaling proteins were undetected in in vivo samples. This lack of detection indicates low abundance of these proteins and requires further enrichment strategies, such as conventional or chromatin immunoprecipitation.36
Although there are techniques that produce global proteome results, such as sequential windowed acquisition of all theoretical fragment ion mass spectra, they require highly specialized equipment and experience and cannot provide the same sensitivity. Triple quadrupole MS are widely available and do not require the same specialization to use. In the validation of these methods, the inter‐tissue variability of a wide variety of proteins was characterized. These data are critical to the creation of broadly applicable PBPK models. From here, the next steps will involve profiling more body tissues and the integration of the protein data into a comprehensive PBPK model.
Supporting information
Supporting Information
Conflict of Interest
The authors declared no conflict of interest.
Source of Funding
This work was funded by NIH/National Institute of Child Health and Human Development grant R01HD081299.
References
- 1. Rowland, M. Physiologically‐based pharmacokinetic (PBPK) modeling and simulations principles, methods, and applications in the pharmaceutical industry. CPT Pharmacometrics Syst. Pharmacol. 2, e55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rowland, M. , Balant, L. & Peck, C. Physiologically based pharmacokinetics in drug development and regulatory science: a workshop report (Georgetown University, Washington, DC, May 29–30, 2002). AAPS PharmSci. 6, E6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Varma, M.V. , Lin, J. , Bi, Y.A. , Kimoto, E. & Rodrigues, A.D. Quantitative rationalization of gemfibrozil drug interactions: consideration of transporters‐enzyme interplay and the role of circulating metabolite gemfibrozil 1‐O‐β‐glucuronide. Drug Metab . Dispos. 43, 1108–1118 (2015). [DOI] [PubMed] [Google Scholar]
- 4. SEARCH Collaborative Group et al SLCO1B1 variants and statin‐induced myopathy–a genomewide study. N. Engl. J. Med. 359, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 5. He, J. et al PET imaging of Oatp‐mediated hepatobiliary transport of [(11)C] rosuvastatin in the rat. Mol. Pharm. 11, 2745–2754 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Prasad, B. & Unadkat, J.D. The concept of fraction of drug transported (ft) with special emphasis on BBB efflux of CNS and antiretroviral drugs. Clin. Pharmacol. Ther. 97, 320–323 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Shirasaka, Y. et al Interindividual variability of CYP2C19‐catalyzed drug metabolism due to differences in gene diplotypes and cytochrome P450 oxidoreductase content. Pharmacogenomics J. 16, 375–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Martino, M.T. et al Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan‐associated gastrointestinal toxicity in colorectal cancer patients: a DMET microarray profiling study. Cancer Biol. Ther. 12, 780–787 (2011). [DOI] [PubMed] [Google Scholar]
- 9. Fisel, P. , Renner, O. , Nies, A.T. , Schwab, M. & Schaeffeler, E. Solute carrier transporter and drug‐related nephrotoxicity: the impact of proximal tubule cell models for preclinical research. Expert Opin. Drug Metab. Toxicol. 10, 395–408 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Li, N. , Bi, Y.A. , Duignan, D.B. & Lai, Y. Quantitative expression profile of hepatobiliary transporters in sandwich cultured rat and human hepatocytes. Mol. Pharm. 6, 1180–1189 (2009). [DOI] [PubMed] [Google Scholar]
- 11. Li, N. , Singh, P. , Mandrell, K.M. & Lai, Y. Improved extrapolation of hepatobiliary clearance from in vitro sandwich cultured rat hepatocytes through absolute quantification of hepatobiliary transporters. Mol. Pharm. 7, 630–641 (2010). [DOI] [PubMed] [Google Scholar]
- 12. Qiu, X. , Bi, Y.A. , Balogh, L.M. & Lai, Y. Absolute measurement of species differences in sodium taurocholate cotransporting polypeptide (NTCP/Ntcp) and its modulation in cultured hepatocytes. J. Pharm. Sci. 102, 3252–3263 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Badée, J. , Achour, B. , Rostami‐Hodjegan, A. & Galetin, A. Meta‐analysis of expression of hepatic organic anion‐transporting polypeptide (OATP) transporters in cellular systems relative to human liver tissue. Drug Metab. Dispos. 43, 424–432 (2015). [DOI] [PubMed] [Google Scholar]
- 14. Chen, Y. , Liu, L. , Nguyen, K. & Fretland, A.J. Utility of intersystem extrapolation factors in early reaction phenotyping and the quantitative extrapolation of human liver microsomal intrinsic clearance using recombinant cytochromes P450. Drug Metab. Dispos. 39, 373–382 (2011). [DOI] [PubMed] [Google Scholar]
- 15. T'jollyn, H. et al Physiology‐based IVIVE predictions of tramadol from in vitro metabolism data . Pharm. Res. 32, 260–274 (2015). [DOI] [PubMed] [Google Scholar]
- 16. Prasad, B. & Unadkat, J.D. Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J. 16, 634–648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aebersold, R. , Burlingame, A.L. & Bradshaw, R.A. Western blots versus selected reaction monitoring assays: time to turn the tables? Mol. Cell. Proteomics 12, 2381–2382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schröder, A. et al Genomics of ADME gene expression: mapping expression quantitative trait loci relevant for absorption, distribution, metabolism and excretion of drugs in human liver. Pharmacogenomics J. 13, 12–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prasad, B. et al Interindividual variability in hepatic organic anion‐transporting polypeptides and P‐glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab. Dispos. 42, 78–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang, L. et al Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab. Dispos. 43, 367–374 (2015). [DOI] [PubMed] [Google Scholar]
- 21. Salem, F. , Johnson, T.N. , Barter, Z.E. , Leeder, J.S. & Rostami‐Hodjegan, A. Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug‐drug interactions. J. Clin. Pharmacol. 53, 857–865 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Harwood, M.D. et al In vitro‐in vivo extrapolation scaling factors for intestinal P‐glycoprotein and breast cancer resistance protein: part I: a cross‐laboratory comparison of transporter‐protein abundances and relative expression factors in human intestine and Caco‐2 cells. Drug Metab. Dispos. 44, 297–307 (2016). [DOI] [PubMed] [Google Scholar]
- 23. Al Feteisi, H. , Achour, B. , Rostami‐Hodjegan, A. & Barber, J. Translational value of liquid chromatography coupled with tandem mass spectrometry‐based quantitative proteomics for in vitro‐in vivo extrapolation of drug metabolism and transport and considerations in selecting appropriate techniques. Expert Opin. Drug Metab. Toxicol. 11, 1357–1369 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Margaillan, G. et al Multiplexed targeted quantitative proteomics predicts hepatic glucuronidation potential. Drug Metab. Dispos. 43, 1331–1335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Williamson, B.L. et al Quantitative protein determination for CYP induction via LC‐MS/MS. Proteomics 11, 33–41 (2011). [DOI] [PubMed] [Google Scholar]
- 26. Prasad, B. , Lai, Y. , Lin, Y. & Unadkat, J.D. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): effect of age, sex, and genotype. J. Pharm. Sci. 102, 787–793 (2013). [DOI] [PubMed] [Google Scholar]
- 27. Caixeiro, N.J. , Lai, K. & Lee, C.S. Quality assessment and preservation of RNA from biobank tissue specimens: a systematic review. J. Clin. Pathol. 69, 260–265 (2016). [DOI] [PubMed] [Google Scholar]
- 28. Zhang, J. et al Cassette incubation followed by bioanalysis using high‐resolution MS for in vitro ADME screening assays. Bioanalysis 4, 581–593 (2012). [DOI] [PubMed] [Google Scholar]
- 29. Ma, S. & Zhu, M. Recent advances in applications of liquid chromatography‐tandem mass spectrometry to the analysis of reactive drug metabolites. Chem. Biol. Interact. 179, 25–37 (2009). [DOI] [PubMed] [Google Scholar]
- 30. Mauriala, T. , Chauret, N. , Oballa, R. , Nicoll‐Griffith, D.A. & Bateman, K.P. A strategy for identification of drug metabolites from dried blood spots using triple‐quadrupole/linear ion trap hybrid mass spectrometry. Rapid Commun. Mass Spectrom. 19, 1984–1992 (2005). [DOI] [PubMed] [Google Scholar]
- 31. Boja, E.S. & Rodriguez, H. Mass spectrometry‐based targeted quantitative proteomics: achieving sensitive and reproducible detection of proteins. Proteomics 12, 1093–1110 (2012). [DOI] [PubMed] [Google Scholar]
- 32. Laizure, S.C. , Herring, V. , Hu, Z. , Witbrodt, K. & Parker, R.B. The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy 33, 210–222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yeung, C.K. , Lang, D.H. , Thummel, K.E. & Rettie, A.E. Immunoquantitation of FMO1 in human liver, kidney, and intestine. Drug Metab. Dispos. 28, 1107–1111 (2000). [PubMed] [Google Scholar]
- 34. Zhang, J. & Cashman, J.R. Quantitative analysis of FMO gene mRNA levels in human tissues. Drug Metab. Dispos. 34, 19–26 (2006). [DOI] [PubMed] [Google Scholar]
- 35. Song, W. , Yu, L. & Peng, Z. Targeted label‐free approach for quantification of epoxide hydrolase and glutathione transferases in microsomes. Anal. Biochem. 478, 8–13 (2015). [DOI] [PubMed] [Google Scholar]
- 36. Ranish, J.A. et al The study of macromolecular complexes by quantitative proteomics. Nat. Genet. 33, 349–355 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information