

Abstract
Rosuvastatin is a frequently used probe in transporter‐mediated drug‐drug interaction (DDI) studies. This report describes the development of a physiologically based pharmacokinetic (PBPK) model of rosuvastatin for prediction of pharmacokinetic (PK) DDIs. The rosuvastatin model predicted the observed single (i.v. and oral) and multiple dose PK profiles, as well as the impact of coadministration with transporter inhibitors. The predicted effects of rifampin and cyclosporine (6.58‐fold and 5.07‐fold increase in rosuvastatin area under the curve (AUC), respectively) were mediated primarily via inhibition of hepatic organic anion‐transporting polypeptide (OATP)1B1 (Inhibition constant (Ki) ∼1.1 and 0.014 µM, respectively) and OATP1B3 (Ki ∼0.3 and 0.007 µM, respectively), with cyclosporine also inhibiting intestinal breast cancer resistance protein (BCRP; Ki ∼0.07 µM). The predicted effects of gemfibrozil and its metabolite were moderate (1.88‐fold increase in rosuvastatin AUC) and mediated primarily via inhibition of hepatic OATP1B1 and renal organic cation transporter 3. This model of rosuvastatin will be useful in prospectively predicting transporter‐mediated DDIs with novel pharmaceutical agents in development.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Several attempts have been made previously to characterize and predict DDI outcomes between rosuvastatin and transporter inhibitors using both in vitro and in silico methods, some of which have qualitatively predicted interactions with rifampin and cyclosporine, but not the magnitude of the effect. These models were unsuccessful in predicting the effect of interaction with gemfibrozil, a weak OATP inhibitor.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This report describes the development of a PBPK model of rosuvastatin to incorporate the contributions of key transporters to its absorption, elimination, and distribution.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The updated rosuvastatin PBPK model incorporates additional transporters, including OST, OAT3, and MRP4, and increases the relative contribution of OATP1B1 compared with previous models, resulting in improved prediction of DDI with rifampin.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The new model provides an improvement over the previously published models of rosuvastatin and may be useful in prospective simulations to evaluate the potential for DDIs with novel pharmaceutical agents in development.
Rosuvastatin, a 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor, is a synthetic statin used to reduce low‐density lipoprotein cholesterol levels in the treatment of hyperlipidemia.1, 2 Metabolism, primarily by cytochrome P450 (CYP) 2C9, provides only a minor contribution to rosuvastatin clearance; rosuvastatin is predominantly eliminated through biliary and renal clearance pathways, mediated by influx and efflux drug transporters.3 Figure 1 shows an overview of the transporters involved in the absorption, distribution, and elimination of rosuvastatin.4, 5, 6, 7
Figure 1.
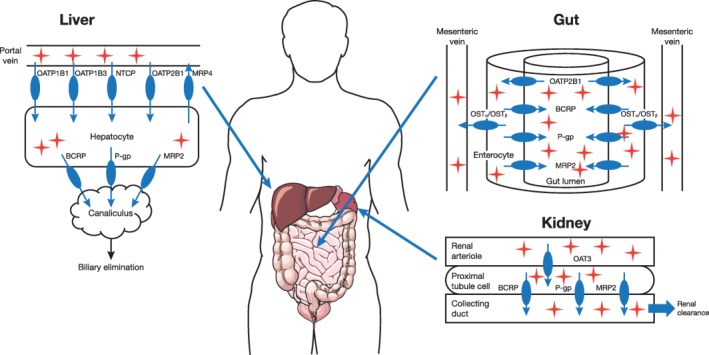
Transporters involved in the absorption, distribution, and elimination of rosuvastatin. BCRP, breast cancer resistance protein; MRP, multidrug resistance protein; NTCP, sodium‐taurocholate co‐transporting polypeptide; OATP, organic anion‐transporting polypeptide; OST, organic solute transporter; P‐gp, P‐glycoprotein.
Rosuvastatin is frequently used as a probe in transporter‐mediated drug‐drug interaction (DDI) studies, owing to its widespread usage and the wealth of available clinical data in various patient populations, its safety profile, and unique disposition properties.8 These factors also make it an attractive target for pharmacokinetic (PK) modeling, allowing researchers to verify and characterize the mechanisms involved. Several attempts have been made to predict DDI outcomes between rosuvastatin and transporter inhibitors using in vitro and in silico methods.9, 10, 11, 12 Jamei et al.10 developed physiologically based PK (PBPK) models to investigate the complex DDI between cyclosporine (which is known to inhibit transporters, including organic anion‐transporting polypeptide (OATP)1B1, OATP1B3, breast cancer resistance protein (BCRP), and P‐glycoprotein)13 and rosuvastatin using Simcyp Simulator. The rosuvastatin PBPK model developed included the hepatobiliary elimination pathway, and successfully captured the clinically observed plasma PK profiles of rosuvastatin. Although the outcome of cyclosporine and rosuvastatin DDI simulations published by Jamei et al.10 agreed qualitatively with the observed results, the magnitude of maximum concentration (Cmax) and area under the curve (AUC) changes were not accurately predicted. Another PBPK model for rosuvastatin was developed by Bosgra et al.14 using transporter kinetic data scaled from cell lines overexpressing the OATP1B1*1a, OATP1B3, OATP2B1, and OATP1B1*15 transporters, but was not verified with clinical DDI data.
This report describes the development of a PBPK model of rosuvastatin to include most of its known absorption and disposition properties, with a goal of improving the sensitivity to detect transporter‐mediated DDI. In addition to the simulation of interaction between cyclosporine and rosuvastatin, the current model was also used to simulate DDIs between rosuvastatin and rifampin (a strong OATP1B1 and OATP1B3 inhibitor) as well as gemfibrozil (a weak OATP1B1 inhibitor and OAT3 inhibitor).
METHODS
General physiologically based pharmacokinetic model assumptions and theories
All PBPK modeling and simulations were carried out using the Simcyp Simulator version 14r1 (Simcyp, Sheffield, UK). The principles for PBPK simulations in Simcyp were described previously.10 Briefly, a permeability‐limited liver model and mechanistic kidney model were used to construct the PBPK model of rosuvastatin, with the assumptions that only unbound drug species can passively permeate the plasma membrane; the concentrations in vascular space and extracellular space are in instantaneous equilibrium; transporters act only on unbound drug; drugs can be transported only across the canalicular membrane by active transporters but not passive permeation; the transporting processes are unidirectional, without limitation of cofactors or driving forces (such as pH or sodium gradient). Another key assumption made for rosuvastatin PBPK modeling by Jamei et al.10 that we included was that BCRP was the sole efflux transporter for rosuvastatin in the gut, canalicular membrane, and kidney proximal tubule cells. Other Simcyp modules used in the PBPK model included advanced dissolution, absorption and metabolism, and full PBPK distribution. The dissolution of rosuvastatin was assumed to be complete and fast, equivalent to solution dosing. DDI mediated by transporters was simulated by assuming that only unbound drug species at the transporter binding site can affect transporter function and that inhibition of each drug transporter is competitive. A summary of the optimized model parameters for rosuvastatin and their origins is shown in Supplementary Table S1.
Unique properties of the current rosuvastatin physiologically based pharmacokinetic model
The passive permeability of rosuvastatin across the intestinal wall is very low (∼0.25 × 10−6 cm/s)6; therefore, absorption of rosuvastatin in the model was mediated by multiple influx and efflux transporters on the basolateral and apical membranes of enterocytes. OATP2B1 mediates apical uptake of rosuvastatin from gut lumen into enterocytes, and is uniformly expressed along all segments of the gastrointestinal tract.15, 16 Conversely, BCRP on the apical membrane reduces the absorption of rosuvastatin from the gastrointestinal lumen. The distribution of BCRP in the gastrointestinal tract (higher in the duodenum and jejunum, lower in the ileum, and absent in the colon) may also delay absorption of rosuvastatin in humans.17 If the delayed absorption was solely due to BCRP activity, then inhibiting this transporter would shorten the time to maximum concentration (Tmax) significantly. However, in subjects with the c.421AA genotype, which is associated with reduced BCRP activity, Cmax and AUC of rosuvastatin are approximately doubled, whereas Tmax remained similar in individuals with the c.421CC (normal BCRP function) genotype.4 Therefore, BCRP is unlikely to be the sole cause of the delayed absorption of rosuvastatin.
Organic solute transporter (OST)α/OSTβ heteromer, a basolateral membrane facilitative transporter, was found to be involved in the transport of rosuvastatin from enterocytes to the blood circulation.6 OSTα/OSTβ is highly expressed in the middle to terminal parts of the ileum, moderately in the early part of the ileum, and expressed at lower levels in the jejunum, duodenum, and colon.18 This coincides with the absorption pattern of rosuvastatin, suggesting that basolateral transport may be an important factor in rosuvastatin absorption in humans. In Simcyp version 14r1, the transporter expression in the gastrointestinal tract is expressed as a value relative to the expression in the jejunum. Due to lack of quantitative expression data in the literature, the relative expression values of OSTα/OSTβ were determined by fitting to 10 mg, orally administered, PK for rosuvastatin as 1, 1, 1, 2, 5, 10, 9, and 0.01 for the duodenum, jejunum I, jejunum II, ileum I, ileum II, ileum III, ileum IV, and colon, respectively. OST is expressed in the ascending colon, but not the transverse or descending colon. However, the current gastrointestinal tract model used in Simcyp version 14r1 considers the colon as a single organ. The Michaelis–Menten constant (Km; 4 µM) of OST‐mediated basolateral transport implied saturation at gut intracellular concentrations of rosuvastatin greater than 4 µM. However, rosuvastatin exposure is dose proportional from 10 to 80 mg,19 which suggests that the absorption is not saturated, even at the 80‐mg dose. Therefore, an influx clearance was used in the PBPK model of rosuvastatin, instead of Km and maximal velocity values.
Rosuvastatin is primarily eliminated through transporter‐mediated biliary and renal routes, with only a minor contribution from metabolic clearance. Most of the rosuvastatin dose is eliminated unchanged in feces or urine,3 and renal clearance of rosuvastatin accounts for ∼28% of the total plasma clearance.20 In vitro study results indicate that OAT3 is a key basolateral membrane transporter for rosuvastatin,21 and that BCRP is a key efflux transporter on the brush‐border membrane of kidney cells.7 Transport across the basolateral membrane of kidney proximal tubule cells by OAT3 is the rate‐limiting step in renal clearance of rosuvastatin.7 Renal transport clearance of rosuvastatin in the model was estimated by fitting with a single 10‐mg dose in vivo data,19, 22, 23, 24 as in vitro‐in vivo extrapolation for transporter activity has not yet been established. As the rate‐limiting step was OAT3 transport, the transporter‐mediated clearance was assigned to be the same for OAT3 and BCRP (150 µL/min/million cells). The BCRP‐mediated efflux of rosuvastatin in the kidney was assigned to MRP4, because BCRP is not currently included in the renal model in Simcyp. As OAT3‐mediated uptake is the rate‐limiting step, this assignment has no impact on the simulation of rosuvastatin PK, nor the DDI simulation with cyclosporine or rifampin.
The relative contributions of OATP1B1, OATP1B3, and sodium‐taurocholate co‐transporting polypeptide (NTCP; and/or OATP2B1) to the overall hepatic uptake of rosuvastatin were estimated based on in vitro data.5, 14, 25 In the current PBPK model, the contributions from OATP1B1, OATP1B3, and NTCP were assigned as 70%, 20%, and 10%, respectively. In addition to basolateral hepatic uptake, there was a report in the literature investigating the contribution of multidrug resistance protein 4 (MRP4)‐mediated basal efflux of rosuvastatin,26 estimated to be about four times the biliary efflux. When the basal efflux process was included, it was necessary to increase the biliary clearance value of rosuvastatin to 3.8 µL/min/million cells based on model assessment, similar to the findings of Abe et al.27
As NTCP is not included in the model structure in Simcyp V14, the NTCP‐mediated uptake of rosuvastatin was assigned to organic cation transporter 1, assuming the same relative expression level. Likewise, in the DDI simulation of rosuvastatin and cyclosporine, the NTCP inhibition by cyclosporine was assigned to organic cation transporter 1, assuming the same Ki. Both approaches were used by Jamei et al.,10 implemented in Simcyp V12. To confirm that these approaches were appropriate, selected simulations (10‐mg single dose and rosuvastatin‐cyclosporine DDI) were repeated in Simcyp V15, in which absolute abundance of NTCP is included, with relatively similar results. The simulated mean values of rosuvastatin Cmax and AUC after a 10‐mg single oral dose from Simcyp V15 were 5.20 ng/mL and 24.5 ng/mL*h, respectively, compared with 4.74 ng/mL and 23.4 ng/mL*h (Table 1), respectively, from Simcyp V14. The simulated median ratios of rosuvastatin Cmax and AUC when coadministered with cyclosporine from Simcyp V15 were 7.96 and 6.07, respectively, compared to 7.14 and 5.07 (Table 2),28, 29, 30 respectively, from Simcyp V14.
Table 1.
Comparison of simulated and observed pharmacokinetic parameters for rosuvastatin after single doses in healthy volunteers
| Rosuvastatin dose | |||||
|---|---|---|---|---|---|
| 10 mg | 20 mg | 40 mg | 80 mg | ||
| Observed GMR (N = 15)a | Cmax (ng/mL) | 3.75 | 6.79 | 10.3 | 30.1 |
| AUC(0–72 h) (ng/mL*h) | 30.7 | 51.5 | 84.4 | 220 | |
|
Mean of simulated trials (range) (N = 15/trial)b |
Cmax (ng/mL) | 4.74 (3.72–6.59) | 9.60 (7.51–13.4) | 19.7 (15.0–27.6) | 41.3 (31.9–58.4) |
| AUC(0–72 h) (ng/mL*h) | 23.4 (19.4–30.2) | 42.7 (39.1–60.9) | 95.8 (79.2–124) | 197 (162–256) | |
AUC, area under the curve; Cmax, maximum concentration; GMR, geometric mean ratio.
There were 15 healthy male subjects in the single‐ascending dose study. bTen trials were simulated with 15 healthy male subjects per trial, aged 19–65 years. The mean of the simulated trials represents the arithmetic mean of the 10 geometric means from the simulated trials.
Table 2.
Ratios of rosuvastatin maximum concentration and area under the curve when coadministered with inhibitors vs. administered alone
| Rosuvastatin dose/frequency | Inhibitor, dose/frequency | Transporters inhibited | Cmax | AUC | |
|---|---|---|---|---|---|
| 10 mg q.d. | Cyclosporine, 200 mg p.o., b.i.d. | OATP1B1/3, BCRP, NTCP | Observed ratioa | 10.6 | 7.1 |
| Median ratios from simulated trials (range of mean ratios) | 7.14 (5.26–9.43) | 5.07 (3.92–6.96) | |||
| 5 mg single dose | Rifampin, 600 mg p.o., single dose | OATP1B1/3, BCRP | Observed GMR (90% CI)b | 9.93 (7.25–13.6) | 5.24 (3.66–7.49) |
| Median ratios from simulated trials (range of mean ratios) | 8.49 (6.35–11.1) | 6.58 (5.00–8.49) | |||
| 5 mg single dose | Rifampin, 600 mg i.v., single dose | OATP1B1/3, BCRP | Observed GMR (90% CI)b | 5.51 (4.38–6.93) | 3.30 (2.42–4.50) |
| Median ratios from simulated trials (range of mean ratios) | 4.05 (3.08–4.90) | 3.64 (2.86–4.37) | |||
| 80 mg single dose | Gemfibrozil, 600 mg p.o., b.i.d. | OATP1B1, OAT3 | Observed GMR (90% CI)c | 2.21 (1.81–2.69) | 1.88 (1.60–2.21) |
| Median ratios from simulated trials (range of mean ratios) | 2.04 (1.87–2.25) | 1.79 (1.69–1.97) |
AUC, area under the curve; BCRP, breast cancer resistance protein; CI, confidence interval; Cmax, maximum concentration; GMR, geometric mean ratio; NTCP, sodium‐taurocholate co‐transporting polypeptide; OAT3, organic anion‐transporting 3; OATP, organic anion‐transporting polypeptide.
Data from Simonson et al.28 2004: 10 mg rosuvastatin was administered to patients with heart failure, and the AUC and Cmax ratios were obtained by comparing with data from a study in healthy volunteers. bData from Prueksaritanont et al.29 2014: 5 mg rosuvastatin was administered to healthy volunteers in the presence or absence of rifampin. cData from Schneck et al.30 2004: 80 mg rosuvastatin was dosed to healthy volunteers in the presence or absence of gemfibrozil.
Model simulation and verification
The model was used to describe the PK and disposition of rosuvastatin in healthy subjects after a single i.v. or oral dose, or repeat once‐daily (q.d.) oral dosing. The performance of the model was assessed via visual inspection of the simulated population PK profiles and the observed PK profiles from single‐dose studies (oral and i.v.) and repeat‐dose studies (oral only) in healthy volunteers reported in the literature, along with the comparison of simulated and observed PK exposure parameters. The relevant reported studies and the population characteristics for each comparison are shown in Figure 2.20, 31 In each case, the simulations were conducted with similar characteristics as the subjects in the studies, including sex ratio and age.
Figure 2.
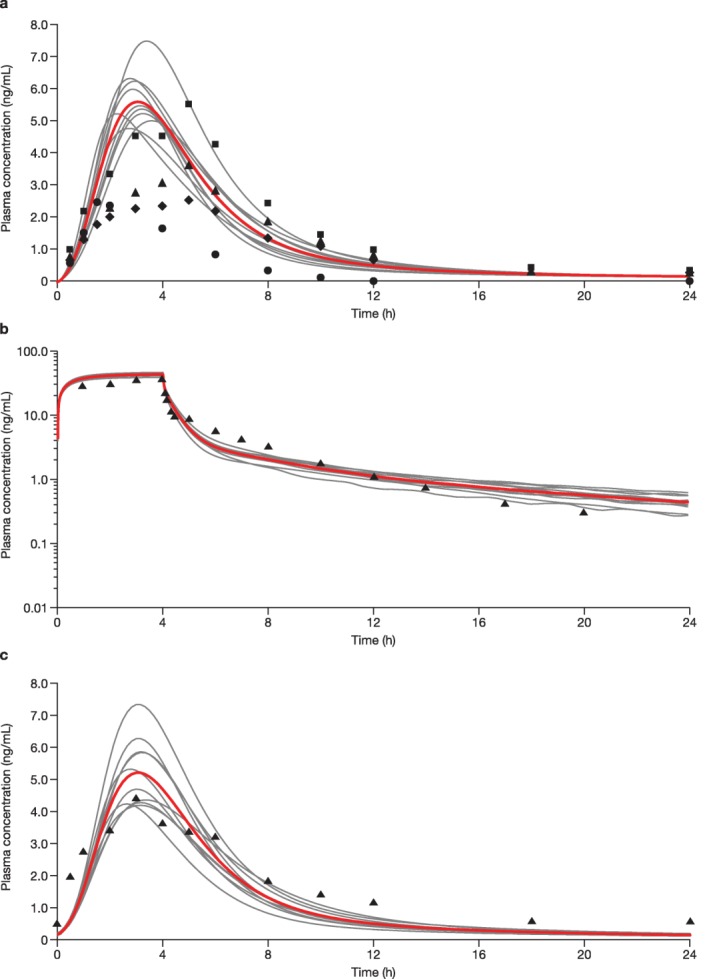
Simulated and observed rosuvastatin plasma concentration‐time profiles of rosuvastatin in healthy subjects after oral administration of (a) a single 10‐mg dose of rosuvastatin, (b) a single 8‐mg i.v. infusion, and (c) a 10‐mg once‐daily dosing for 14 days. (a) Ten trials were simulated with 15 healthy volunteers/trial (20% women; age, 19–65 years). Curves represent the simulated mean rosuvastatin profiles for the individual simulated trials with the overall mean shown in red; circles, diamonds, triangles, and squares represent the observed means from four studies. (b) Ten trials were simulated with 10 healthy volunteers/trial (20% women; age, 19–55 years). Curves represent the simulated mean rosuvastatin profiles for the individual simulated trials with the overall mean shown in red; triangles represent the observed mean from the absolute bioavailability study by Martin et al.20 2003. (c) Ten trials were simulated with 24 healthy volunteers/trial (10% women; age, 19–60 years). Curves represent the simulated mean rosuvastatin profiles for the individual simulated trials with the overall mean shown in red; triangles represent the observed mean from the pharmacokinetic/pharmacodynamic study by Martin et al.31 2002.
After the construction of the PBPK model of rosuvastatin, simulations were conducted to describe the single‐dose and multiple‐dose PK of rosuvastatin and interactions with perpetrator drugs. Model parameters for cyclosporine and rifampin were initially developed by Simcyp and the absorption and inhibitory effects were further modified according to Jamei et al.10 (Supplementary Table S2). Model parameters for gemfibrozil were also developed by Simcyp, with only minor modifications regarding its inhibitory effect on OATP1B1 and OAT3 (Supplementary Table S3). These inhibition parameters for OATP1B1, OATP1B3, and OAT3 were obtained from the literature and a similar procedure was used to scale OATP1B1 and OATP1B3 inhibition parameters, as in Jamei et al.10 The model parameters used for each DDI simulation are shown in Supplementary Tables S4–S6. Results from clinical DDI studies with cyclosporine, rifampin, and gemfibrozil were used to verify the model via visual inspection of an overlay of the simulated and observed PK profiles, and by comparison of simulated and observed PK parameters.
RESULTS
Simulation of single‐dose pharmacokinetic of rosuvastatin
The simulated rosuvastatin PK profiles after administration of a single 10‐mg oral dose are shown in Figure 2 a.19, 22, 23, 24 Simulated profiles after administration of 20, 40, and 80‐mg single doses of rosuvastatin are shown in Supplementary Figure S1. The ranges of simulated geometric mean values of the Cmax and AUC time curve from time zero to the time with the last quantifiable concentration (AUC(0–T)) after a 10, 20, 40, or 80‐mg single dose were compared with the observed data (Table 1).19 This single‐ascending dose study was conducted in healthy male volunteers (N = 15) aged 19–65 years; the simulation was conducted with the same study design.19 The predicted Tmax of rosuvastatin was also similar to the observed Tmax (2.6–3.7 hours vs. 3–4 hours, respectively).19 Mean plasma concentrations of rosuvastatin as simulated for an 8‐mg i.v. infusion agreed well with the mean observed data (Figure 2 b).20
Simulation of multiple‐dose pharmacokinetic of rosuvastatin
The plasma concentration‐time profiles comparing observed31 vs. simulated results on day 14 for the repeated 10‐mg rosuvastatin q.d. dose are shown in Figure 2 c. The simulations were conducted with a similar design as reported31 (i.e., 24 healthy volunteers in each trial for 10 simulated trials and 10% women in the population). The accumulation of rosuvastatin after multiple doses is minimal,32 and steady‐state plasma time profiles were similar to those after a single dose. The observed geometric mean values of the Cmax and AUC time curve to end of dosing interval (AUC(TAU)) on day 1431 were within the ranges of simulated Cmax and AUC(TAU) (results not shown).
Simulation of rosuvastatin and cyclosporine drug‐drug interaction
Simulated steady‐state plasma concentration‐time profiles for rosuvastatin 10 mg q.d. with and without cyclosporine 200 mg b.i.d. in healthy subjects are shown in Figure 3.28 The geometric mean ratios (GMRs) for rosuvastatin AUC and Cmax reported by Simonson et al.28 were slightly above the range of the simulated mean ratios (Table 2).28, 29, 30 However, this was not a dedicated DDI study, but a comparison of the historical mean rosuvastatin PK parameters from healthy volunteers with observed rosuvastatin PK parameters from patients who undergo heart transplantation.28 Therefore, the interaction reported may also reflect combined effects of the DDI, interindividual variability, and pathophysiological differences between healthy volunteers and patients who undergo heart transplantation.
Figure 3.
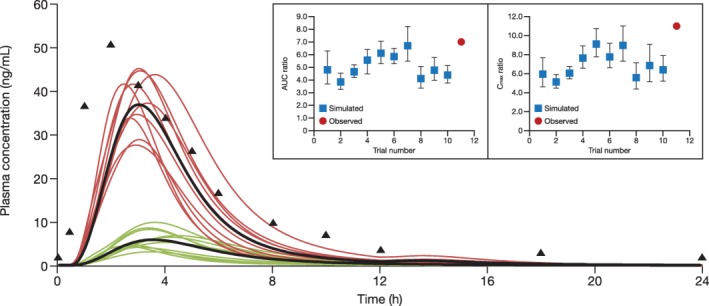
Simulated rosuvastatin plasma concentration‐time profiles in healthy subjects after administration of rosuvastatin 10 mg once daily in the presence and absence of cyclosporine 200 mg twice daily. Ten trials were simulated with 6 healthy subjects/trial (50% women; age, 19–55 years). Red curves represent the simulated mean rosuvastatin profiles for the individual simulated trials in the presence of cyclosporine. Green curves represent the individual simulated trials in the absence of cyclosporine. Curves in black represent the simulated mean profiles of all simulation trials. Triangles represent the observed mean data from Simonson et al.28 in 2004. Inset panels show simulated vs. observed area under the curve (AUC) and maximum concentration (Cmax) geometric mean ratios of rosuvastatin in the presence or absence of cyclosporine. Error bars in the inset represent 90% confidence intervals.
Simulation of rosuvastatin and rifampin drug‐drug interaction
The simulated plasma profiles of rosuvastatin 5 mg coadministered with a single dose of 600 mg rifampin administered i.v. or orally are shown in Figure 4 a,b,29 respectively, and compared with the observed rosuvastatin PK profiles from a healthy volunteer crossover study in the presence or absence of rifampin (i.v. or orally). The reported GMRs of rosuvastatin AUC and Cmax in the presence and absence of 600 mg oral rifampin were 5.24 and 9.93,29 respectively, which are within the range of simulated mean ratios of 5.00–8.49 for AUC and 6.35–11.1 for Cmax (Table 2).28, 29, 30 Similarly, the reported GMR of rosuvastatin AUC (3.30) coadministered with 600 mg i.v. rifampin was also within the range of simulated mean ratios of 2.86–4.37 (Table 2); however, the reported GMR of rosuvastatin Cmax (5.51) was slightly above the range of simulated mean ratios (3.08–4.90).
Figure 4.
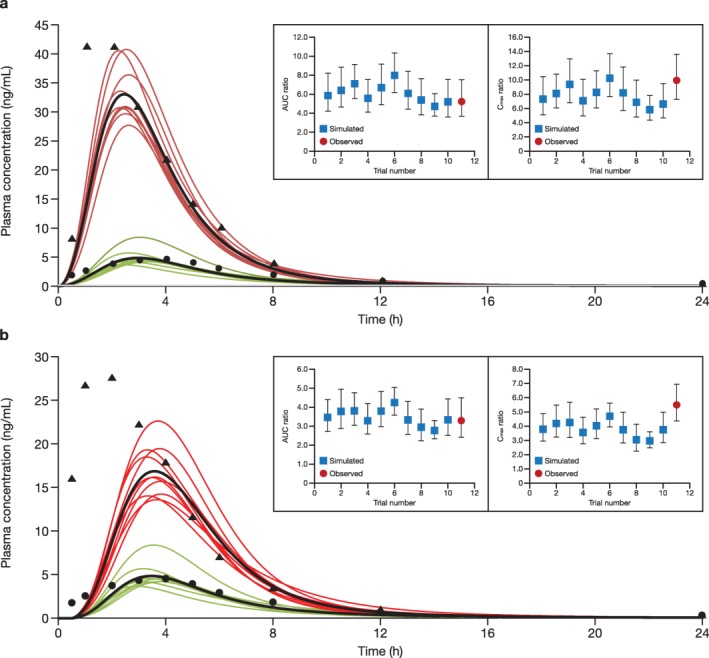
Simulated and observed plasma concentration‐time profile of 5 mg rosuvastatin when administered in the presence or absence of a single dose of (a) oral and (b) i.v. rifampin 600 mg. Ten trials were simulated with 10 healthy subjects/trial (50% women; age, 19–55 years). Red curves indicate the presence of rifampin and green curves indicate the absence of rifampin. Curves in black represent the simulated mean profiles of all simulation trials. Circles and triangles represent observed data in the absence and presence of rifampin from Prueksaritanont et al.29 in 2014. Inset panels show simulated vs. observed area under the curve (AUC) and maximum concentration (Cmax) ratios of rosuvastatin in the presence of rifampin. Error bars in the insets represent 90% confidence intervals.
Simulation of gemfibrozil and rosuvastatin drug‐drug interaction
The plasma concentration‐time profiles for a single 80‐mg dose of rosuvastatin in the presence and absence of gemfibrozil 600 mg b.i.d. are shown in Figure 5.30 The GMRs of rosuvastatin AUC and Cmax from a healthy subject study by Schneck et al.30 (N = 20), with 15% female subjects were reported as 1.88 and 2.21, respectively, which are within the ranges of simulated mean ratios of 1.69–1.97 for AUC and 1.87–2.25 for Cmax (Table 2).
Figure 5.
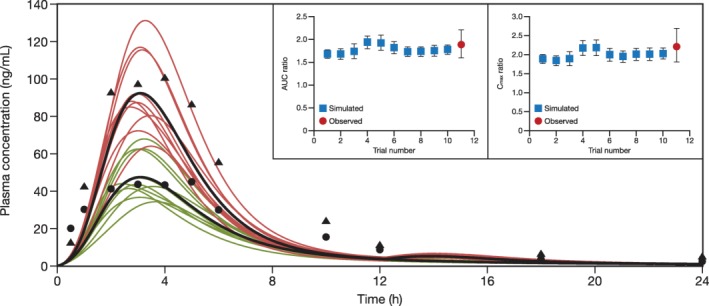
Simulated and observed plasma concentration‐time profile of a single 80‐mg dose of rosuvastatin when administered in the presence or absence of gemfibrozil 600 mg twice daily. Ten trials were simulated with 20 healthy subjects/trial (15% women; age, 19–55 years). Curves in black represent the mean rosuvastatin profiles for the simulation trials. Red curves indicate the presence of gemfibrozil. Green curves denote the absence of gemfibrozil. Black triangles represent observed concentrations of rosuvastatin in the presence of gemfibrozil. Black circles represent observed concentrations of rosuvastatin in the absence of gemfibrozil from Schneck et al.30 in 2004. Inset panels show simulated vs. observed area under the curve (AUC) and maximum concentration (Cmax) ratios of rosuvastatin in the presence of gemfibrozil. Error bars in the inset represent 90% confidence intervals.
DISCUSSION
This was a PBPK modeling study of DDIs between rosuvastatin and cyclosporine, rifampin, and gemfibrozil using the Simcyp simulator. Using the developed PBPK model, simulated PK estimates for rosuvastatin were in agreement with observed values for single oral or i.v. dosing of rosuvastatin, and for rosuvastatin administered orally q.d. Overall, there was slight overprediction of the Cmax, which might be due to the solution formulation used in the current model. However, the model was able to predict the Cmax after repeat dosing and when rosuvastatin was administered alone in the drug interaction studies.
The shorter Tmax of rosuvastatin after coadministering with cyclosporine was also observed with coadministration of i.v. and oral rifampin. This could be a consequence of a reduced volume of distribution due to inhibition of the hepatic uptake transporters. In the current PBPK model, Tmax was also affected by the gastrointestinal basal transporters OSTα/OSTβ, which are distributed unevenly along the gastrointestinal tract. Although we could not identify any data in the literature to suggest that cyclosporine affects OSTα/OSTβ activity in vivo, and, thus, did not include it in the PBPK model, the possibility cannot be ruled out entirely. The distribution of rosuvastatin in various organs was predicted using the Rodgers and Rowland method,33 with liver and kidney distribution determined independently by the permeability limited liver and kidney models in Simcyp, as both uptake and efflux transporters are involved in distribution of rosuvastatin in the liver and kidney. As the predictions by the Rodgers and Rowland method33 do not account for transporter‐mediated distribution, the predicted volume of distribution at steady state of rosuvatatin (0.227 L/kg) does not reflect its “true” volume of distribution at steady state. The volume of distribution at steady state was calculated independently by noncompartmental analysis from the simulated plasma concentrations and found to be about 2.0 L/kg, which was close to that observed in the absolute bioavailability study (134 L for an individual with average body weight of 77.6 kg).20 This included the majority of the rosuvastatin dose residing in the liver, consistent with previous reports that rosuvastatin was specifically distributed into liver tissues.34, 35
This model included OSTα/OSTβ for absorption, MRP4 for hepatic basal efflux, and OAT3 and BCRP for renal clearance. Although additional efflux transporters, such as P‐glycoprotein, MRP2, and MRP4, may also contribute to rosuvastatin renal efflux,7 we found that inhibition of a single efflux transporter had little effect on rosuvastatin plasma PK, owing to the redundancy of renal efflux transporters. Therefore, these transporters were not included in the final model. Furthermore, we found that, although inclusion of the MRP4‐mediated basolateral efflux helped to capture the distribution phase of the PK profile of rosuvastatin administered i.v., it was not critical to the PBPK model of rosuvastatin, having little impact on the DDI prediction with a hypothetical MRP4 transporter inhibitor based on the sensitivity analysis, and thus may be omitted from the rosuvastatin PBPK model.
Estimates of the hepatic uptake clearance of rosuvastatin showed some variation10, 14 and clearance varied among different ethnic populations.36 Further, recent publications on OATP1B1 in human hepatocytes have indicated a large interindividual variability in protein expression25, 37, 38 and in rosuvastatin hepatic uptake.39 In one recent study, the contribution of OATP1B1 to total rosuvastatin hepatic uptake was estimated as ∼70% using a protein expression scaling factor, but ∼88% if estimated using a relative activity scaling factor.25 This compared with the estimate of ∼50% used by Jamei et al.10 OATP1B3 and NTCP (and/or OATP2B1) contributed to the hepatic uptake of rosuvastatin to varying degrees. Using expression‐based scaling factors, the contribution of OATP1B3 to rosuvastatin uptake was estimated as ∼10%25 or 16–34%.5 The NTCP‐mediated uptake was estimated to be 10–35%,25, 40 but OATP2B1 expression was low in liver tissue and was considered to be an insignificant uptake transporter for rosuvastatin.5 The contribution of OATP1B3 and NTCP to rosuvastatin hepatic uptake could be further refined when in vivo DDI data with specific inhibitors are available. However, the highly variable expression of OATP1B1 and OATP1B3 suggested that a large sample pool would be needed to find the population mean expression values for both transporters.
Drug‐drug interaction models
Cyclosporine
The PBPK modeling by Jamei et al.10 qualitatively captured the DDI between cyclosporine and rosuvastatin, but was not quantitatively close to the observed data.28 The simulated rosuvastatin AUC and Cmax ratios by Jamei et al.10 ranged from 1.45–1.68 and 2.73–3.62, respectively, compared to the observed 7.1 and 10.6, respectively. It should be noted that the observed ratios were based on a comparison between patients with heart failure and healthy volunteers from different clinical trials.28 To our knowledge, no crossover study in the same population has investigated the interaction between cyclosporine and rosuvastatin. The current PBPK model has improved the absorption model of rosuvastatin, reassessed the contributions of the hepatic influx transporters OATP1B1, OATP1B3, NTCP, and OATP2B1, and modified the renal clearance of rosuvastatin. The effect of cyclosporine on intestinal BCRP resulted in an increased fraction of rosuvastatin absorbed in most gastrointestinal segments, excepting the colon (data not shown). The decreased fraction of rosuvastatin absorbed from the colon can be attributed to the fact that, in the presence of cyclosporine, most of the rosuvastatin dose was absorbed from the gastrointestinal segments before the colon, which was not the case in the absence of cyclosporine. The overall hepatic uptake clearance of rosuvastatin was decreased by about 70% and overall canalicular efflux was decreased by ∼20% in the presence of cyclosporine (data not shown). The simulated rosuvastatin AUC and Cmax ratios of 5.07 and 7.14, respectively, when coadministered with cyclosporine using the current model were relatively similar to the observed data.28
Rifampin
Based on in vitro data, rifampin inhibits hepatic uptake transporters OATP1B1 and OATP1B3 with half‐maximal inhibitory concentration (IC50) values of 1.1 and 0.3 µM, and is also a BCRP inhibitor with an IC50 value of 14 µM.29 These values were lowered by 10‐fold to derive the Ki in the simulations according to Jamei et al. 10 Intestinal BCRP is inhibited when rifampin is administered orally, resulting in decreased intestinal efflux of rosuvastatin with a concomitant increase in intestinal absorption. By contrast, absorption of rosuvastatin was unaffected by i.v. rifampin. Hepatic uptake clearance of rosuvastatin decreased with increasing plasma concentrations of rifampin owing to hepatic OATP1B1 and OATP1B3 inhibition, irrespective of the route of rifampin administration, falling to only 10–15% of the hepatic uptake clearance in the absence of rifampin (data not shown). The simulated DDI outcome between oral or i.v. rifampin and rosuvastatin in this model was similar to clinically observed data in the literature.29
Gemfibrozil
Based on in vitro data, gemfibrozil is an inhibitor of OATP1B1,12, 30, 41, 42 OATP2B1,43 and OAT3,43 but not of OATP1B3.44 The metabolite of gemfibrozil, gemfibrozil‐1‐O‐glucuronide, has also been shown to inhibit OATP1B1 and OAT3.42, 43 Gemfibrozil is not an inhibitor of BCRP, so it does not inhibit the intestinal absorption of rosuvastatin. However, gemfibrozil did decrease the OATP1B1‐mediated hepatic clearance uptake and OAT3‐mediated renal clearance of rosuvastatin in the simulation.
The inhibition constant (Ki) of gemfibrozil for OATP1B1 was 1.9 µM, calculated from OATP1B1‐overexpressing cells using rosuvastatin as substrate.12 Consistent with this, the Ki calculated from the experiment conducted with oocytes expressing OATP1B1 was shown to be around 2 µM.30 The Ki of gemfibrozil‐1‐O‐glucuronide for OATP1B1 was 0.385‐fold that of gemfibrozil.41 The Ki for OAT3 (1.47 µM) was taken directly from Watanabe et al.43 In the simulation, gemfibrozil decreased hepatic clearance by inhibiting hepatic uptake of rosuvastatin by up to 50%, and resulted in a decrease in the renal clearance of rosuvastatin of ∼35% via inhibition of OAT3. Using the in vitro‐observed Ki values, the simulated results agreed well with the observed data.30 If the renal transporter interaction was not included, the simulated GMR of AUC and Cmax would be smaller, and a much lower adjusted Ki of gemfibrozil on OATP1B1 would need to be used to account for the observed GMRs of rosuvastatin AUC and Cmax. The simulation outcome suggests that two major mechanisms are involved in the interaction between gemfibrozil and rosuvastatin: inhibition of OATP1B1‐mediated and OATP2B1‐mediated hepatic uptake of rosuvastatin, and inhibition of OAT3‐mediated renal uptake. Without either mechanism, the simulation was not able to fully capture the observed data.
The current rosuvastatin model incorporated additional transporters involved in the absorption and disposition of rosuvastatin, such as intestinal influx transporter OST, renal efflux transporter OAT3, and hepatic basal efflux transporter MRP4. Although including these transporters does not necessarily improve the DDI prediction with cyclosporine or rifampin, as neither drug inhibits these transporters, it is important from the mechanistic perspective of the model development. The improvement of the DDI prediction with cyclosporine and rifampin is more likely from the reassignment of the relative contribution of OATP1B1 to the hepatic uptake of rosuvastatin, from 50% by Jamei et al.10 to 70% in the current model. Recently, Li et al.45 used a top‐down modeling approach, integrating all available rosuvastatin PK data in subjects with wild‐type or polymorphic OATP1B1, and concluded that OATP1B1 contributed ∼50% to the total hepatic uptake of rosuvastatin, in agreement with the initial publication by Jamei et al.,10 but in contrast to the 70% proposed by our model. Additional simulations using our model for rosuvastatin after a single oral dose of 10 mg in subjects with wild‐type OATP1B1 and a low activity polymorphism showed Cmax and AUC ratios of 1.52 and 1.53, respectively, vs. 1.79 and 1.65, respectively, as observed by Li et al.45
When a DDI simulation with the same trial design, as described in Figure 3, was run using cyclosporine as a hypothetical inhibitor with only BCRP inhibition turned on, the predicted AUC and Cmax ratios of rosuvastatin were 2.22 and 2.96, respectively. In addition, rosuvastatin AUC and Cmax ratios from the simulations in which the intestinal BCRP was turned off compared with the current model were 2.99 and 3.47, respectively. These ratios are higher than the maximum predicted AUC ratio of two by Elsby et al.46 when the intestinal BCRP was assumed to be completedly inhibited. This difference was due to the inclusion of the intestinal influx transporter OST in the current model, thus the higher efflux by BCRP, as compared with the assumption by others that BCRP was the only transporter involved in rosuvastatin absorption. However, as the proposed rosuvastatin PBPK model has not been verified using all available relevant clinical datasets, it is difficult to judge whether it could be used in certain situations, including for prospective DDI simulations with potent BCRP inhibitors (in the absence of OATP inhibition), in all ethnic groups (e.g., in Asian subjects, who show twofold higher exposure), or to simulate PK or DDI scenarios in subjects with low activity polymorphisms of BCRP.
In conclusion, a PBPK model of rosuvastatin was developed to incorporate the contributions of key transporters to the absorption, elimination, and distribution of rosuvastatin, including OST, OAT3, and MRP4, and updating the contribution of OATP1B1. The new model was verified using data from DDI studies with cyclosporine, rifampin, and gemfibrozil. The new model provided an improvement over the previously published PBPK models of rosuvastatin and may be useful in prospective simulations to evaluate the potential for DDIs with novel pharmaceutical agents in development.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
This study was sponsored by Bristol‐Myers Squibb. Professional medical writing and editorial assistance was provided by Andy Shepherd at Caudex and was funded by Bristol‐Myers Squibb.
Conflict of Interest
All authors are employees of Bristol‐Myers Squibb at the time of writing this manuscript.
Author Contributions
Q.W., M.Z., and T.L. wrote the manuscript. Q.W. designed the research. Q.W. performed the research. Q.W., M.Z., and T.L. analyzed the data.
References
- 1. Blasetto, J.W. , Stein, E.A. , Brown, W.V. , Chitra, R. & Raza, A. Efficacy of rosuvastatin compared with other statins at selected starting doses in hypercholesterolemic patients and in special population groups. Am . J. Cardiol. 91, 3C–10C; discussion 10C (2003). [DOI] [PubMed] [Google Scholar]
- 2. Olsson, A.G. , McTaggart, F. & Raza, A. Rosuvastatin: a highly effective new HMG‐CoA reductase inhibitor. Cardiovasc. Drug Rev. 20, 303–328 (2002). [DOI] [PubMed] [Google Scholar]
- 3. Martin, P.D. et al Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin. Ther. 25, 2822–2835 (2003). [DOI] [PubMed] [Google Scholar]
- 4. Keskitalo, J.E. , Zolk, O. , Fromm, M.F. , Kurkinen, K.J. , Neuvonen, P.J. & Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 86, 197–203 (2009). [DOI] [PubMed] [Google Scholar]
- 5. Kitamura, S. , Maeda, K. , Wang, Y. & Sugiyama, Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 36, 2014–2023 (2008). [DOI] [PubMed] [Google Scholar]
- 6. Li, J. , Wang, Y. , Zhang, W. , Huang, Y. , Hein, K. & Hidalgo, I.J. The role of a basolateral transporter in rosuvastatin transport and its interplay with apical breast cancer resistance protein in polarized cell monolayer systems. Drug Metab. Dispos. 40, 2102–2108 (2012). [DOI] [PubMed] [Google Scholar]
- 7. Verhulst, A. , Sayer, R. , De Broe, M.E. , D'Haese, P.C. & Brown, C.D. Human proximal tubular epithelium actively secretes but does not retain rosuvastatin. Mol. Pharmacol. 74, 1084–1091 (2008). [DOI] [PubMed] [Google Scholar]
- 8. Food and Drug Administration . Guidance for industry. Drug interaction studies – study design, data analysis and implications for dosing and labeling: draft guidance. <http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf> (2012).
- 9. Izumi, S. et al Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug‐drug interactions. Drug Metab. Dispos. 43, 235–247 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Jamei, M. et al A mechanistic framework for in vitro‐in vivo extrapolation of liver membrane transporters: prediction of drug‐drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 53, 73–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karlgren, M. , Ahlin, G. , Bergström, C.A. , Svensson, R. , Palm, J. & Artursson, P. In vitro and in silico strategies to identify OATP1B1 inhibitors and predict clinical drug‐drug interactions. Pharm. Res. 29, 411–426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharma, P. , Butters, C.J. , Smith, V. , Elsby, R. & Surry, D. Prediction of the in vivo OATP1B1‐mediated drug‐drug interaction potential of an investigational drug against a range of statins. Eur. J. Pharm. Sci. 47, 244–255 (2012). [DOI] [PubMed] [Google Scholar]
- 13. Gertz, M. et al Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug‐drug interaction potential. Pharm. Res. 30, 761–780 (2013). [DOI] [PubMed] [Google Scholar]
- 14. Bosgra, S. et al Predicting carrier‐mediated hepatic disposition of rosuvastatin in man by scaling from individual transfected cell‐lines in vitro using absolute transporter protein quantification and PBPK modeling. Eur. J. Pharm. Sci. 65, 156–166 (2014). [DOI] [PubMed] [Google Scholar]
- 15. Gröer, C. et al LC‐MS/MS‐based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J. Pharm. Biomed. Anal. 85, 253–261 (2013). [DOI] [PubMed] [Google Scholar]
- 16. Meier, Y. et al Regional distribution of solute carrier mRNA expression along the human intestinal tract. Drug Metab. Dispos. 35, 590–594 (2007). [DOI] [PubMed] [Google Scholar]
- 17. Haller, S. et al Expression profiles of metabolic enzymes and drug transporters in the liver and along the intestine of beagle dogs. Drug Metab. Dispos. 40, 1603–1610 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Dawson, P.A. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb. Exp. Pharmacol. 201, 169–203 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin, P.D. , Warwick, M.J. , Dane, A.L. & Cantarini, M.V. A double‐blind, randomized, incomplete crossover trial to assess the dose proportionality of rosuvastatin in healthy volunteers. Clin. Ther. 25, 2215–2224 (2003). [DOI] [PubMed] [Google Scholar]
- 20. Martin, P.D. , Warwick, M.J. , Dane, A.L. , Brindley, C. & Short, T. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin. Ther. 25, 2553–2563 (2003). [DOI] [PubMed] [Google Scholar]
- 21. Windass, A.S. , Lowes, S. , Wang, Y. & Brown, C.D. The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. J. Pharmacol. Exp. Ther. 322, 1221–1227 (2007). [DOI] [PubMed] [Google Scholar]
- 22. Busti, A.J. et al Effects of atazanavir/ritonavir or fosamprenavir/ritonavir on the pharmacokinetics of rosuvastatin. J. Cardiovasc. Pharmacol. 51, 605–610 (2008). [DOI] [PubMed] [Google Scholar]
- 23. Cooper, K.J. , Martin, P.D. , Dane, A.L. , Warwick, M.J. , Schneck, D.W. & Cantarini, M.V. Effect of itraconazole on the pharmacokinetics of rosuvastatin. Clin. Pharmacol. Ther. 73, 322–329 (2003). [DOI] [PubMed] [Google Scholar]
- 24. Pham, P.A. et al Differential effects of tipranavir plus ritonavir on atorvastatin or rosuvastatin pharmacokinetics in healthy volunteers. Antimicrob. Agents Chemother. 53, 4385–4392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kunze, A. , Huwyler, J. , Camenisch, G. & Poller, B. Prediction of organic anion‐transporting polypeptide 1B1‐ and 1B3‐mediated hepatic uptake of statins based on transporter protein expression and activity data. Drug Metab. Dispos. 42, 1514–1521 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Pfeifer, N.D. , Yang, K. & Brouwer, K.L. Hepatic basolateral efflux contributes significantly to rosuvastatin disposition I: characterization of basolateral versus biliary clearance using a novel protocol in sandwich‐cultured hepatocytes. J. Pharmacol. Exp. Ther. 347, 727–736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abe, K. , Bridges, A.S. & Brouwer, K.L. Use of sandwich‐cultured human hepatocytes to predict biliary clearance of angiotensin II receptor blockers and HMG‐CoA reductase inhibitors. Drug Metab. Dispos. 37, 447–452 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simonson, S.G. et al Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 76, 167–177 (2004). [DOI] [PubMed] [Google Scholar]
- 29. Prueksaritanont, T. et al Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneck, D.W. et al The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin. Pharmacol. Ther. 75, 455–463 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Martin, P.D. , Mitchell, P.D. & Schneck, D.W. Pharmacodynamic effects and pharmacokinetics of a new HMG‐CoA reductase inhibitor, rosuvastatin, after morning or evening administration in healthy volunteers. Br. J. Clin. Pharmacol. 54, 472–477 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Warwick, M.J. , Dane, A.L. , Raza, A. & Schneck, D.W. Single‐ and multiple‐dose pharmacokinetics and safety of the new HMG‐CoA reductase inhibitor ZD4522. Atherosclerosis 151, 39 (2000). [Google Scholar]
- 33. Rodgers, T. , Leahy, D. & Rowland, M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J. Pharm. Sci. 94, 1259–1276 (2005). [DOI] [PubMed] [Google Scholar]
- 34. McTaggart, F. et al Preclinical and clinical pharmacology of rosuvastatin, a new 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor. Am. J. Cardiol. 87, 28B–32B (2001). [DOI] [PubMed] [Google Scholar]
- 35. Nezasa, K. et al Liver‐specific distribution of rosuvastatin in rats: comparison with pravastatin and simvastatin. Drug Metab. Dispos. 30, 1158–1163 (2002). [DOI] [PubMed] [Google Scholar]
- 36. Tomita, Y. , Maeda, K. & Sugiyama, Y. Ethnic variability in the plasma exposures of OATP1B1 substrates such as HMG‐CoA reductase inhibitors: a kinetic consideration of its mechanism. Clin. Pharmacol. Ther. 94, 37–51 (2013). [DOI] [PubMed] [Google Scholar]
- 37. Kimoto, E. et al Characterization of organic anion transporting polypeptide (OATP) expression and its functional contribution to the uptake of substrates in human hepatocytes. Mol. Pharm. 9, 3535–3542 (2012). [DOI] [PubMed] [Google Scholar]
- 38. Prasad, B. et al Interindividual variability in hepatic organic anion‐transporting polypeptides and P‐glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab. Dispos. 42, 78–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Menochet, K. , Kenworthy, K.E. , Houston, J.B. & Galetin, A. Use of mechanistic modeling to assess interindividual variability and interspecies differences in active uptake in human and rat hepatocytes. Drug Metab. Dispos. 40, 1744–1756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bi, Y.A. et al Quantitative assessment of the contribution of sodium‐dependent taurocholate co‐transporting polypeptide (NTCP) to the hepatic uptake of rosuvastatin, pitavastatin and fluvastatin. Biopharm. Drug Dispos. 34, 452–461 (2013). [DOI] [PubMed] [Google Scholar]
- 41. Kudo, T. , Hisaka, A. , Sugiyama, Y. & Ito, K. Analysis of the repaglinide concentration increase produced by gemfibrozil and itraconazole based on the inhibition of the hepatic uptake transporter and metabolic enzymes. Drug Metab. Dispos. 41, 362–371 (2013). [DOI] [PubMed] [Google Scholar]
- 42. Nakagomi‐Hagihara, R. , Nakai, D. , Tokui, T. , Abe, T. & Ikeda, T. Gemfibrozil and its glucuronide inhibit the hepatic uptake of pravastatin mediated by OATP1B1. Xenobiotica 37, 474–486 (2007). [DOI] [PubMed] [Google Scholar]
- 43. Watanabe, T. et al Prediction of the overall renal tubular secretion and hepatic clearance of anionic drugs and a renal drug‐drug interaction involving organic anion transporter 3 in humans by in vitro uptake experiments. Drug Metab. Dispos. 39, 1031–1038 (2011). [DOI] [PubMed] [Google Scholar]
- 44. Ho, R.H. et al Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology 130, 1793–1806 (2006). [DOI] [PubMed] [Google Scholar]
- 45. Li, R. , Barton, H.A. & Maurer, T.S. Toward prospective prediction of pharmacokinetics in OATP1B1 genetic variant populations. CPT Pharmacometrics. Syst. Pharmacol. 3, e151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elsby, R. , Martin, P. , Surry, D. , Sharma, P. & Fenner, K. Solitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug‐drug interaction with rosuvastatin by causing up to a 2‐fold increase in statin exposure. Drug Metab. Dispos. 44, 398–408 (2016). [DOI] [PubMed] [Google Scholar]
- 47. Huang, L. , Wang, Y. & Grimm, S. ATP‐dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab. Dispos. 34, 738–742 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information