

Abstract
BACKGROUND AND OBJECTIVE: Aurora A, as a member of serine/threonine kinase family and a common characteristic of epithelial cancers, plays a critical role in cell mitosis. However, the clinical significance of Aurora A in non–small cell lung cancer (NSCLC) remains undetermined. METHODS: The expression of Aurora A in NSCLC and paired normal adjacent lung tissues was determined by immunohistochemistry, Western blot, and reverse transcriptase polymerase chain reaction. Receiver operating characteristic (ROC) curve analysis was employed to determine a cutoff score for Aurora A expression in a training set (n = 135). For validation, the ROC-derived cutoff score was subjected to analysis of the association of Aurora A expression with patient outcome and clinicopathological characteristics in a testing set (n = 128) and overall patients (n = 263). The correlation of Aurora A with cisplatin resistance and epithelial-mesenchymal transition (EMT) was examined in vitro in NSCLC cells by overexpression or knockdown of Aurora A. RESULTS: Aurora A expression was significantly upregulated in tumor tissues compared with paired normal tissues (P < .01). The expression of Aurora A was closely associated with clinical stage, lymph node metastasis, and recurrence and was an independent prognostic parameter in multivariate analysis. High level of Aurora A expression predicted poorer overall survival and disease-free survival in NSCLC patients treated with cisplatin-based adjuvant chemotherapy. In vitro data showed that overexpression or knockdown of Aurora A resulted in increased or decreased cellular resistance to cisplatin. Furthermore, inhibition of Aurora A reversed the EMT process. CONCLUSIONS: Aurora A was identified as an inferior prognostic and cisplatin-resistant biomarker in NSCLC patients, which provided potential evidences for therapeutic target and reversing drug resistance.
Introduction
Lung cancer is the leading cause of cancer-related deaths [1], [2]. Non–small cell lung cancer (NSCLC) accounts for 80% to 85% of lung cancers [3]. Most of the patients are diagnosed with advanced-stage disease, and the average 5-year survival rate remains dismal [4]. Multiple genetic abnormalities, including tumor suppressor genes, oncogenes, cell adhesion molecules, cell-cycle regulators, and growth factors, are responsible for the development and progression of NSCLC [5]. Molecular epidemiological studies have provided evidence that multiple genetic alterations make risk assessment of lung cancer patients more accurate. Aberrant proliferation of NSCLC is frequently associated with mutational activation of receptor tyrosine kinases signaling including genes encoding transmembrane receptor tyrosine kinases (ALK [6], EGFR [6], [7], ROS1 [8]) or intracellular signaling proteins such as KRAS [9] or its effectors A- B- or C-RAF [10] or PIK3CA [11]. BRAF is estimated to be mutated in ~2% of NSCLCs, 25% of which express the BRAFT1799A oncogene encoding the BRAFV600E oncoprotein kinase [12], [13]. However, like many cancers, mutational activation of proto-oncogenes such as ERBB1, KRAS, or BRAF is generally accompanied by silencing of tumor suppressor genes such as TP53, CDKN2A, or PTEN that cooperatively serve to promote the stepwise malignant transformation of normal lung epithelial cells to malignant lung cancer cells [13]. Although much is known about the causal factors, clinical features, and pathogenesis of NSCLC, the molecular marker that has major clinical prognostic predictive value remains substantially limited. Thus, it is of great clinical value to further identify more valuable prognostic biomarkers.
Mitosis is a hallmark of epithelial cancers, raising the possibility that regulators of relative kinases have a role in tumorigenesis. The Aurora kinases play a key role in mitosis [14]; in particular, Aurora kinase A (Aurora A) is involved in various mitotic events, such as centrosome function and maturation, spindle assembly, chromosome alignment, and mitotic entry [14], [15]. In cells, Aurora A expression and kinase activity are increased during late G2 to M phase, and its subcellular localization dynamically changes during the cell cycle [16]. Overexpression or amplification of Aurora A has been noted across a range of different tumor types, such as colon, breast, bladder, ovarian, and pancreatic cancers, and is linked with tumor progression and poor prognosis [17], [18], [19]. Furthermore, previous studies showed that overexpression of Aurora A increases migration and leads to resistance to chemotherapeutics [20], [21]. Inhibition of Aurora A resulted in abnormal spindle formation, mitotic defects, and cell death, which serves as a promising target in cancer therapy, and several small-molecule inhibitors for Aurora A kinase are currently being investigated within clinical trials [22], [23], [24]. Thus, inhibition of Aurora kinase A is a rational target for anticancer treatment.
The aim of the present study was to investigate the expression of Aurora A in NSCLC specimens and determine its correlation with clinical characteristics. The current results showed that Aurora A, as detected by immunohistochemistry, was significantly higher in NSCLC tissues compared with the adjacent normal tissues and closely associated with tumor recurrence rate. High expression of Aurora A predicted an inferior overall survival (OS) and disease-free survival (DFS) in NSCLC patients treated with cisplatin-based adjuvant chemotherapy. Moreover, multivariate analysis revealed that Aurora A was an independent prognostic factor for NSCLC.
Methods
Patients
A total of 283 primary NSCLC patients were initially recruited in our study. All patients underwent initial surgical resection from March 2003 to January 2013. We further screened patients using a strict eligibility criteria protocol as follows: microscopically pathologically confirmed NSCLC, without any distant metastatic diseases, no prior chemotherapy or radiation therapy history, and having over 5-year follow-up period. Smoking history was based on records at patients' first clinic visit, and having smoked more than 100 cigarettes in a lifetime was used to define smokers. Performance status was evaluated using the Eastern Cooperative Oncology Group criteria. Ultimately, 20 patients with loss of follow-up were not included in the present study, leading to 263 NSCLC patients subjected to further clinical and survival analysis.
Of the 263 NSCLC patients (median age, 48.0 years; range, 20-72 years), those with positive lymph node metastasis were treated with four to six cycles of cisplatin + vinorelbine adjuvant chemotherapies after surgical resection, whereas patients with negative lymph node metastasis did not receive adjuvant chemotherapies. No adjuvant radiotherapy was administered to any of the patients after surgery. The cohort of 263 NSCLC patients was then randomly divided into training set (n = 135) and testing set (n = 128) by computer (SPSS 17.0 software). The histological and stage types were determined according to the classification of NSCLC by the World Health Organization and International Union Against Cancer Tumor-Node-Metastasis staging system. This study was approved by the ethics committee and Institutional Review Board of Beijing Cancer Hospital.
Tissue Microarray Construction
The tissue microarrays (TMAs) were constructed according to a method described previously [25]. Briefly, two representative core tissue biopsies (0.6 mm in diameter) were punched from represented NSCLC tissues and one cylinder with the same diameter from adjacent normal lung tissues. Multiple sections (5 μm thick) were cut from the TMA blocks and mounted on the microscope slides.
Immunohistochemical Analysis
In brief, after deparaffinization, rehydration, antigen retrieval, and blocking, the TMA slides were incubated overnight at 4°C with a polyclonal antibody against human Aurora A (1:500; Cell Signaling Technology, No. 12100) in a moist chamber. Then slides were incubated in corresponding secondary antibody at room temperature for 30 minutes. Specimens were visualized through staining with 3,3-diaminobenzidine. Finally, the sections were counterstained with hematoxylin, dehydrated, and mounted. Negative controls were obtained by replacing the Aurora A antibody with a normal murine IgG. Known immunostaining-positive slides were used as positive controls.
Staining Evaluation
The brown granules in both nucleus and cytoplasm of Aurora A were considered as positive staining. The staining intensity was graded as follows: negative (score 0), bordering (score 1), weak (score 2), moderate (score 3), and strong (score 4). In addition, the staining extent was also graded into five levels according to the percentage of cells with elevated Aurora A staining, including negative (score 0), 0% to 25% (score 1), 26% to 50% (score 2), 51% to 75% (score 3), and 76% to 100% (score 4). Aurora A staining was assessed by two pathologists who were unaware of any clinical details related to the patients. The assessment was congruent in 87% (229 identical scores in 263 cases) of the cases, suggesting a highly reproducible scoring system. Discrepant cases were reviewed for a second time. The value was selected until both pathologists agreed with the result.
Selection of a Cutoff Score for Aurora A Expression
The receiver operating characteristic (ROC) curve analysis was subjected to the selection of Aurora A cutoff score for analysis of OS and DFS in the training set, as described previously [26]. ROC curve analysis has been shown to be reproducible to evaluate immunohistochemistry protein expression and to select biologically or clinically a relevant cutoff score with maximum sensitivity and specificity. ROC curve analysis was facilitated by dichotomizing the features of patients' outcome into survival [death versus others (censored, alive, or death from other causes)] and progression (local failure or distant metastasis).
Follow-Up
All patients had follow-up records of over 5 years. After the completion of therapy, patients were observed at 3-month intervals during the first 3 years and at 6-month intervals thereafter. OS was defined as the time from the tumor resection to the date of death or when censored at the latest date if patients were still alive; DFS was defined as the time from the tumor resection to the date of disease relapse/progression or the date of death or when censored at the latest date.
Cell Lines and Cell Culture
The human lung adenocarcinoma cell lines H1299, A549, SK-MES, A427, H2170, H358, and H23 were obtained from Peking University Cancer Hospital. Cell lines were cultured in respective culture supplemented with 10% fetal bovine serum at 37°C in humidified 5% CO2 incubator.
Transient Transfection
A549 cells were transfected with an empty vector or FLAG-Aurora A. The negative control siRNA and siRNA against Aurora A (5′-AUGCCCUGUCUUACUGUCA-3′) were synthesized by GenePharma Company (Shanghai) for H23 cells to reduce the expression of Aurora A.
Western Blot and Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) Analysis
Cells or tissues were ground and lysed with the RIPA buffer on ice before being subjected to protein gel blot analysis. The protein concentration was detected by the Bradford method. Equal amounts of cell and tissue extract were subjected to electrophoresis in sodium dodecyl sulfate polyacrylamide gel and transferred to nitrocellulose membrane for antibody blotting. The membrane was then blocked and incubated with mouse anti–β-actin antibody, rabbit anti–Aurora A antibody (1:1000, Cell Signaling Technology, No. 12100), and EMT Antibody Sampler Kit (1:1000, Cell Signaling Technology, No. 9782T). RT-PCR by a real-time PCR system (Applied Biosystems, Foster City, CA) was described previously [27].
ATPlite Cell Proliferation Assay
The stable cells were pooled and seeded into 96-well plates with 2000 cells per well in triplicate, along with the vector control cells. After 24-hour incubation, cells were treated with DMSO control or cisplatin (2.5, 5, 10 μM) for 48 hours and then subjected to ATPlite cell proliferation assay (Perkin-Elmer).
Statistical Analysis
ROC analysis was used to get an optimal cutoff score of Aurora A expression for survival analysis in the training set (n = 135). For validation, the relationship between Aurora A expression and OS and DFS was evaluated in the testing set (n = 128) and the overall patient cohort (n = 263). Relationship between Aurora A expression and clinicopathological variables was analyzed by the χ2 test or Fisher's exact test. Kaplan-Meier analysis was employed to evaluate the relationship between Aurora A expression and OS and DFS. Differences in survival probabilities between patient subsets were assessed by the log-rank tests. The multivariate Cox proportional-hazards model was utilized to estimate the hazard ratios and 95% confidence intervals (CIs) for patient outcome. All P values quoted were two sided, and P < .05 was considered statistically significant. Statistical analysis was performed using SPSS v. 17.0 (SPSS, Inc., Chicago, IL).
Results
Patient Characteristics
A total of 263 patients with NSCLC were enrolled in this study from March 2003 to January 2013 at Beijing Cancer Hospital, and their clinicopathologic characteristics are listed in Table 1. The patients, 181 males (68.8%) and 82 females (31.2% %), ranged in age from 20 to 72 years (mean 48 years). Histological examination was performed on formalin-fixed tissues in all cases, and tumors were diagnosed and classified according to the American Joint Committee on Cancer classification. Briefly, in the training set, there are 92 males and 43 females, with 48 cases of stage I, 39 cases of stage II, and 48 cases of stage III. Meanwhile, there are 89 males and 39 females in the testing set, with 46 cases of stage I, 30 cases of stage II, and 52 cases of stage III.
Table 1.
Patient Characteristics
| Characteristics | No. of Patients (n = 263) | Percentage (%) |
|---|---|---|
| Age (years) | ||
| Median | 48 | |
| Range | 20-72 | |
| Gender | ||
| Male | 181 | 68.8% |
| Female | 82 | 31.2% |
| Smoking history | ||
| Never smoker | 152 | 57.8% |
| Smoker | 111 | 42.2% |
| Histology | ||
| Adenocarcinoma | 120 | 45.6% |
| Squamous cell carcinoma | 122 | 46.4% |
| Adenosquamous carcinoma | 21 | 8.0% |
| Stage | ||
| I | 94 | 35.7% |
| II | 69 | 26.3% |
| III | 100 | 38.0% |
Aurora A Expression in NSCLC and Adjacent Normal Tissues
Immunohistochemistry was employed to examine the protein expression of Aurora A in primary NSCLC specimens and adjacent normal tissues. Aurora A staining was located in both nucleus and cytoplasm, predominantly in nucleus. Moreover, Aurora A was overexpressed in the NSCLC (Figure 1, A and A’), whereas normal paired adjacent tissues showed nearly negative expression (Figure 1, B and B’).
Figure 1.

Aurora A expression in human NSCLC and adjacent normal tissue. (A) Aurora A was detected and overexpressed in the NSCLC tissue (100 ×). (B) Adjacent normal tissue showed nearly negative expression of Aurora A (100 ×). (A’ and B′) The higher magnification (200×) from the area of the box in A and B, respectively.
Consistent with immunohistochemistry results, Western blot analysis revealed a similar finding in NSCLC and normal lung tissues (Figure 2A). For the RT-PCR results, we also observed that Aurora A overexpressed in NSCLC tissues, whereas it expressed negatively in normal lung tissues (Figure 2B).
Figure 2.
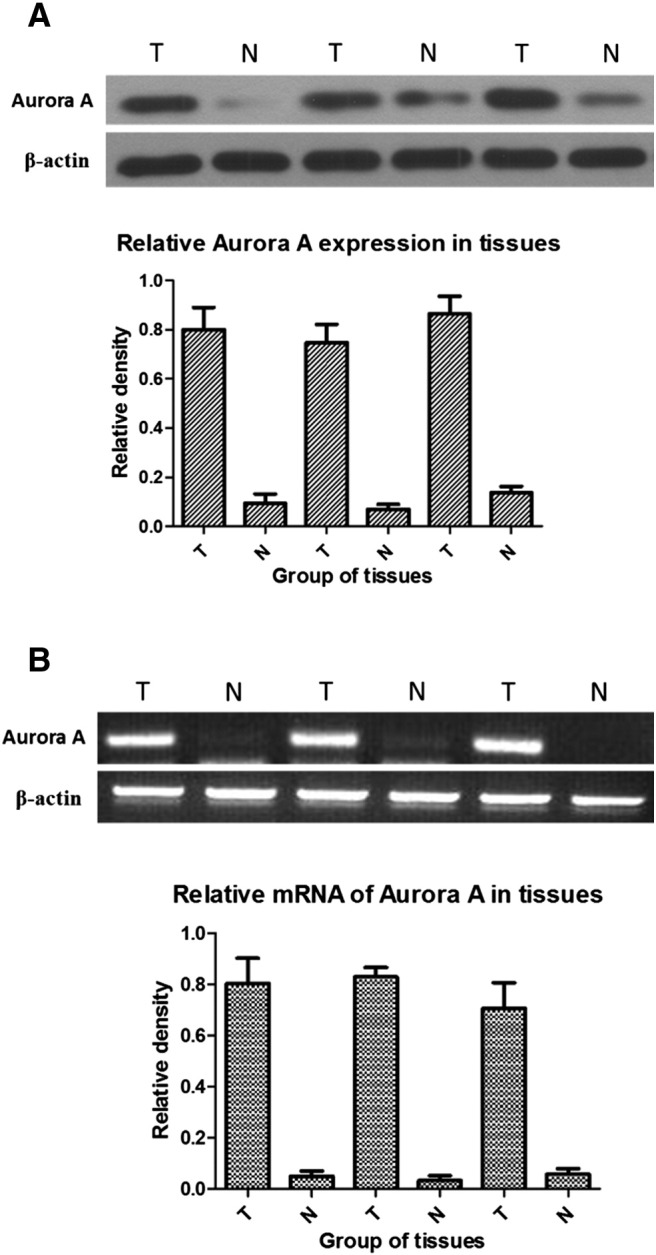
Aurora A expression in human NSCLC and adjacent normal tissue. (A) Western blot analysis of Aurora A expression in representative primary NSCLC tissue (T) and adjacent normal tissue (N). (B) The mRNA results were consistent with protein results.
Cutoff Score for Aurora A “Positive” Expression
To further assess survival analysis and avoid the problems of multiple cutoff point selection, ROC curve analysis was employed to determine a cutoff score for Aurora A expression in the training set. As shown in Figure 3, A and B, the Aurora A cutoff score for OS and DFS in the training set (n = 135) was 3.8 (P < .001) and 3.5 (P < .001), respectively. Thus, a score of 4 (>4 vs ≤4) for Aurora A expression was selected as the uniform cutoff point to distinguish NSCLC patients as having high or low expression.
Figure 3.

ROC curves analysis of Aurora A cutoff score in the training set. (A) Aurora A cutoff point for OS in the training set. (B) Aurora A cutoff point for DFS in the training set. At each immunohistochemical score, the sensitivity and specificity for the outcome being studied were plotted, thus generating an ROC curve. Aurora A cutoff score for OS and DFS was 3.8 and 3.5, respectively.
Aurora A Expression and Clinical Features
The clinical features including age, gender, smoking history, CEA level, initial clinical stage, histology, differentiation, tumor stage, lymph node metastasis, recurrence, and Aurora A expression were summarized in Table 2. The ROC-derived Aurora A cutoff score of 4 derived from the training set successfully segregated the testing set into two subgroups. In brief, patients with a cutoff score >4 are regarded as having Aurora A high expression (57/128, 44.5%), and those with a score ≤4 are viewed as having Aurora A low expression (71/128, 55.5%). High expression of Aurora A was mainly found in high CEA concentration (95/145, 65.5% in >5.0 ng/ml vs 33/118, 28.0% in ≤5.0 ng/ml, P = .004). High expression of Aurora A was mainly detected in patients with late clinical stage (71/100, 71% in patients with stage III vs 27/94, 28.7% in patients with stage I, P < .001). In addition, Aurora A expression also positively correlated with the initial tumor stage (P < .001). High expression of Aurora A was associated with metastatic spread to the lymph nodes (P < .001). Aurora A level also correlated with differentiation (P = .012) in the testing set. Furthermore, correlation analysis demonstrated that Aurora A expression was positively correlated with tumor recurrence in both sets (P < .001 in the training set and P < .001 in the testing set). We could not show any correlation between Aurora A expression and other patient characteristics including age, gender, smoking history, etc.
Table 2.
Association of Aurora A Expression with Clinicopathologic Characteristics in NSCLC Patients
| Variable | All Cases | Training Set (n = 135) |
Testing Set (n = 128) |
||||
|---|---|---|---|---|---|---|---|
| High | Low | P | High | Low | P | ||
| Age (years) | |||||||
| ≥62.0 | 129 | 34 | 30 | 29 | 36 | ||
| <62.0 | 134 | 37 | 34 | .724 | 28 | 35 | .689 |
| Gender | |||||||
| Male | 181 | 50 | 42 | 38 | 51 | ||
| Female | 82 | 21 | 22 | .583 | 19 | 20 | .325 |
| Smoking history | |||||||
| Yes | 152 | 45 | 35 | 30 | 42 | ||
| No | 111 | 26 | 29 | .528 | 27 | 29 | .315 |
| CEA (ng/ml) | |||||||
| >5.0 | 145 | 50 | 22 | 45 | 28 | ||
| ≤5.0 | 118 | 21 | 42 | .044 | 12 | 43 | .025 |
| Initial clinical stage | |||||||
| I | 94 | 16 | 32 | 11 | 35 | ||
| II | 69 | 17 | 22 | 13 | 17 | ||
| III | 100 | 38 | 10 | <.001 | 33 | 19 | .003 |
| Histology | |||||||
| Squamous cell carcinoma | 120 | 37 | 34 | 21 | 28 | ||
| Adenocarcinoma | 122 | 34 | 30 | .452 | 27 | 31 | .821 |
| Adenosquamous cell carcinomas | 21 | 0 | 0 | 9 | 12 | ||
| Differentiation | |||||||
| Highly | 62 | 17 | 12 | 6 | 27 | ||
| Moderately | 94 | 28 | 20 | 18 | 28 | ||
| Poorly | 107 | 26 | 32 | .165 | 33 | 16 | .012 |
| Tumor stage | |||||||
| T1 + T2 | 128 | 20 | 37 | 14 | 57 | ||
| T3 + T4 | 135 | 51 | 27 | .031 | 43 | 14 | .042 |
| Lymph node metastasis | |||||||
| Negative | 137 | 26 | 48 | 17 | 46 | ||
| Positive | 126 | 45 | 16 | .011 | 40 | 25 | .012 |
| Recurrence | |||||||
| Positive | 132 | 51 | 15 | 40 | 26 | ||
| Negative | 131 | 20 | 49 | <.001 | 17 | 45 | <.001 |
Aurora A Expression and Survival Analysis: Univariate Survival Analysis
As shown in Figure 4, Kaplan-Meier analysis showed that high expression of Aurora A strongly predicted an inferior OS and DFS in overall patients (P < .001 for OS and P = .002 for DFS, Figure 4, A and B). In the overall patient cohort, the median duration of overall survival for patients with high and low expression of Aurora A was 24.6 versus 78.8 months, respectively. Also, elevated expression of Aurora A predicted an inferior DFS with the median duration of DFS of 13.4 versus 56.8 months, respectively. Further analysis was performed between Aurora A expression and subsets of NSCLC patients within each clinical stage. High expression of Aurora A also served as a poor prognostic factor in each stage of NSCLC patients in the testing set: stage I (median OS: 102 vs 48.5 months, median DFS: 68.4 vs 42.8 months, P = .033 for OS and P = .024 for DFS; Figure 5, A and B), stage II (median OS: 82.6 vs 40.0 months, median DFS: 58.0 months vs 22.5 months, P = .026 for OS and P = .050 for DFS; Figure 5, C and D), and stage III (median OS: 36.3 vs 11. 2 months, median DFS: 9.8 vs 5.2 months, P = .012 for OS and P = .047 for DFS; Figure 5, E and F).
Figure 4.
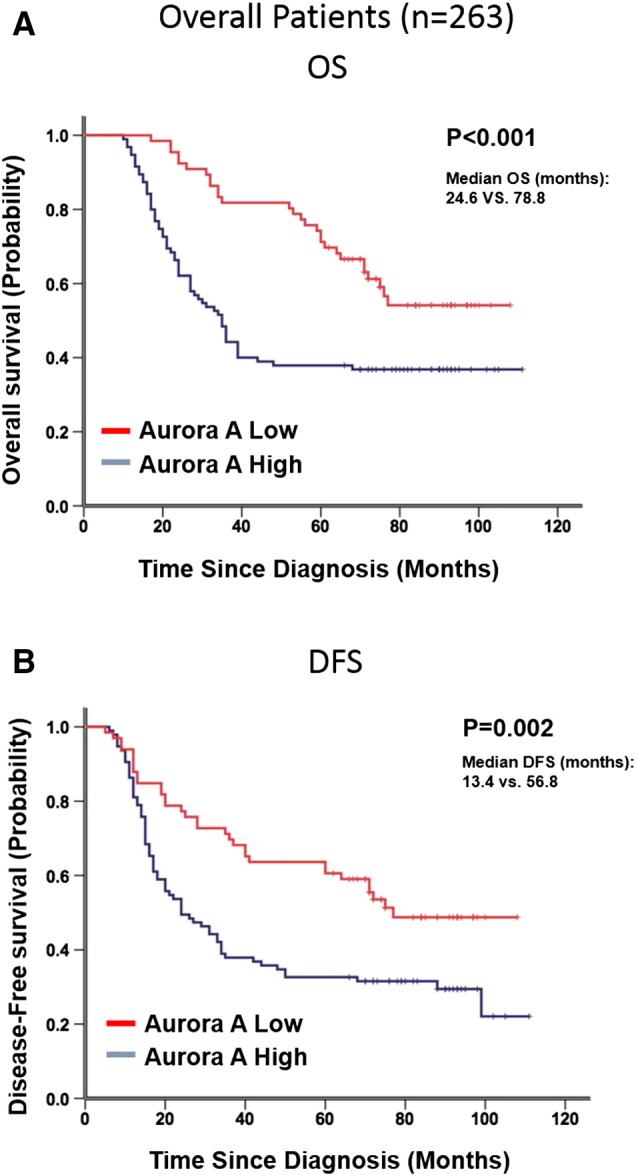
Kaplan-Meier survival analysis of Aurora A expression in overall patients. Patients with higher Aurora A expression acquired an inferior (A) OS and (B) DFS in overall patients. The median duration of OS and DFS for patients with high and low expression of Aurora A was 24.6 versus 78.8 months (P < .001) and 13.4 versus 56.8 months (P = .02), respectively.
Figure 5.

Kaplan-Meier survival analysis of Aurora A expression in subsets of NSCLC patients with stage I, II, and III disease (log-rank test). Probability of (A) OS and (B) DFS of patients with stage I NSCLC in the overall patients: low expression, n = 67; high expression, n = 27. Probability of (C) OS and (D) DFS of stage II patients with NSCLC in the overall patients: low expression, n = 39; high expression, n = 30. Probability of (E) OS and (F) DFS of patients with stage III NSCLC in the overall patients: low expression, n = 29; high expression, n = 71.
Multivariate Cox Regression Analysis
To avoid the influence caused by univariate analysis, the expression of Aurora A as well as other parameters was examined in multivariate Cox analysis (Tables 3 and 4). In the testing set, Aurora A was found to be a significant independent prognostic factor for poor OS (hazard ratio, 4.221; 95% CI, 2.493-6.920; P < .001; Table 3) and DFS (hazard ratio, 3.892; 95% CI, 1.027-5.237; P < .001; Table 3). Similar results were also observed in overall patients (hazard ratio, 4.271; 95% CI, 2.541-5.684; P < .001 for OS and hazard ratio, 3.789; 95% CI, 2.007-4.487; P < .001 for DFS; Table 4). Of other parameters, CEA and smoking history were found to be an independent prognostic factor for patient survival in the testing set and in overall patients.
Table 3.
Results of Multivariate Cox Proportional-Hazards Analysis in Testing Set (n = 128).
| Variable | For OS |
For DFS |
||||
|---|---|---|---|---|---|---|
| Hazard Ratio | 95% CI | P | Hazard Ratio | 95% CI | P | |
| Age (years) ≥60.00 (vs <60.0) | 1.134 | (0.892-2.142) | .472 | 1.241 | (0.784-2.024) | .812 |
| Gender male (vs female) | 2.532 | (1.432-5.241) | .221 | 2.532 | (1.893-4.342) | .032 |
| Smoking history yes (vs no) | 3.982 | (1.993-5.326) | .003 | 2.143 | (1.423-3.253) | .012 |
| CEA (ng/ml) >5 (vs ≤5) | 2.755 | (1.435-3.253) | .001 | 2.532 | (1.245-3.914) | .003 |
| Initial clinical stage | ||||||
| I | 1.053 | (0.298-3.143) | .462 | 1.336 | (0.624-3.573) | .927 |
| II | 0.932 | (0.422-2.986) | .765 | (0.356-2.650) | .772 | |
| III | 1 | 1 | 1 | 1 | ||
| Histology | ||||||
| Squamous cell carcinoma | 1.381 | (0.386-5.334) | .622 | 1.142 | (0.705-3.098) | .723 |
| Adenocarcinoma | 1.588 | (0.672-5.924) | .325 | 1.724 | (0.223-3.981) | .471 |
| Adenosquamous carcinoma | 1 | 1 | 1 | 1 | ||
| Differentiation | ||||||
| Well | 0.837 | (0.472-1.987) | .342 | 0.434 | (0.341-1.243) | .083 |
| Moderately | 1.232 | (0.583-2.538) | .602 | 0.736 | (0.315-1.430) | .277 |
| Poorly | 1 | 1 | 1 | 1 | ||
| Tumor stage T4 + T3 (vs T2 + T1) | 1.098 | (1.002-3.940) | .561 | 1.303 | (1.002-2.874) | .026 |
| Lymph node metastasis positive (vs negative) | 2.906 | (2.093-6.232) | <.001 | 3.092 | (1.409-6.243) | .033 |
| Aurora A low (vs high) | 4.221 | (2.493-6.920) | <.001 | 3.892 | (1.027-5.237) | .001 |
Table 4.
Results of Multivariate Cox Proportional-Hazards Analysis in Overall Patients
| For OS |
For DFS |
|||||
|---|---|---|---|---|---|---|
| Variable | Hazard Ratio | 95% CI | P | Hazard Ratio | 95% CI | P |
| Age (years) ≥60.00 (vs <60.0) | 1.376 | (0.953-1.999) | .092 | 1.041 | (0.709-1.544) | .842 |
| Gender male (vs female) | 1.453 | (0.832-2.563) | .257 | 2.532 | (0.745-2.239) | .379 |
| Smoking history yes (vs no) | 1.496 | (1.023-2.384) | .041 | 1.107 | (0.648-1.927) | .421 |
| CEA (ng/ml) >5 (vs ≤5) | 2.188 | (1.489-3.210) | <.001 | 2.046 | (1.386-2.994) | <.001 |
| Initial clinical stage | ||||||
| I | 0.768 | (0.466-1.686) | .583 | 0.962 | (0.345-2.625) | .915 |
| II | 0.846 | (0.468-1.585) | .609 | 0.982 | (0.474-1.427) | .761 |
| III | 1 | 1 | 1 | 1 | ||
| Histology | ||||||
| Squamous cell carcinoma | 1.293 | (0.389-4.072) | .726 | 0.978 | (0.345-2.782) | .723 |
| Adenocarcinoma | 1.072 | (0.308-3.867) | .926 | 0.632 | (0.198-1.098) | .742 |
| Adenosquamous carcinoma | 1 | 1 | 1 | 1 | ||
| Differentiation | ||||||
| Well | 0.756 | (0.343-1.734) | .353 | 0.642 | (0.389-1.129) | .132 |
| Moderately | 0.595 | (0.319-1.584) | .085 | 0.913 | (0.604-1.476) | .547 |
| Poorly | 1 | 1 | 1 | 1 | ||
| Tumor stage T4 + T3 (vs T2 + T1) | 1.221 | (0.704-1.762) | .613 | 1.159 | (0.687-1.984) | .482 |
| Lymph node metastasis positive (vs negative) | 2.765 | (1.726-3.856) | <.001 | 1.682 | (1.268-2.748) | .007 |
| Aurora A low (vs high) | 4.271 | (2.541-5.684) | <.001 | 3.789 | (2.007-4.487) | <.001 |
Overexpression of Aurora A Increases Resistance and Epithelial-Mesenchymal Transition (EMT)
We sought to explore the association between Aurora A expression and cisplatin resistance in vitro. Aurora A expression was different in multiple human lung cancer cell lines, with low expression in A549 cell line and high expression in H23 cells (Figure 6A). Thus, FLAG-Aurora A markedly increased the Aurora A expression in A549 cells, and siRNA knockdown of Aurora A caused the decreased expression in H23 (Figure 6B). The ectopic expression of Aurora A induced A549 cells resistant to cisplatin. However, knockdown of Aurora A could significantly sensitize H23 cells to cisplatin-induced growth suppression (Figure 6B). On the other hand, Aurora A was found to mediate EMT by detecting some related proteins, as evidenced by decreased levels of E-cadherin (E-cad) and increased levels of Snail and Slug (Figure 6C). Taken together, these results indicated that Aurora A induced cisplatin resistance and EMT process.
Figure 6.

(A) Protein levels of Aurora A in multiple lung cancer cell lines. Cell lysates were prepared from indicated cell lines, followed by Western blotting with equal amount protein loaded, using indicated antibodies. (B) Overexpression of Aurora A induced cisplatin resistance in lung cancers, whereas Aurora A knockdown sensitizes lung cancer cells to cisplatin-induced apoptosis. (C) Overexpression of Aurora A contributed to increased Snail and Slug with decreasing E-cad, which indicated EMT process. However, knockdown of Aurora A upregulated E-cad but downregulated Snail and Slug, which inhibited EMT process.
Discussion
Although treatment for NSCLC has improved in recent years with the development of targeted drugs for patients with amenable mutations, only a small proportion of patients have these mutations, and most tumors become resistant to targeted treatment [28], [29], [30]. Thus, more novel molecular markers that can identify tumor progression and predict the prognosis individually are still urgently needed. In the present study, we evaluated Aurora A expression in NSCLC, showing that, at both transcript and protein levels, Aurora A expression was significantly upregulated in NSCLC tumor samples compared with matched lung normal tissue. Furthermore, Aurora A overexpression was associated with poor survival in NSCLC patients. Aurora A overexpression decreased the sensitivity of lung cancer cells to cisplatin and induced EMT in vitro.
Genetic amplification and mRNA and protein overexpression of Aurora kinase A are implicated in the genesis of various neoplasms, which are significantly associated with aneuploidy, high tumor grade, increased invasiveness, and poor prognosis [18], [31], [32]. Activation of the Aurora A kinase regulates cellular biological processes like centrosome maturation, entry into mitosis, formation and function of the bipolar spindle, and cytokinesis [16]. There are overwhelming evidences which report overexpression and gene amplification of Aurora A in several human cancers and suggest that Aurora A could be a bona fide oncogene involved in tumorigenesis. For instance, aberrant Aurora A mRNA and protein are common in various malignant tumors including breast [33], colon [34], ovarian [31], and pancreatic [18] cancers, indicating that it is important for tumor formation or progression. Also, Aurora A increases migration and leads to resistance to chemotherapeutic drugs [20], [35]. Furthermore, numerous substrates of Aurora A kinase have been identified, which are predominantly related to cell cycle progression, whereas some of them are transcription factors. Aurora A–mediated phosphorylation can either directly or indirectly regulate the function of its substrates [36], [37], [38]. These properties have led Aurora A to be considered a high-value target for development of cancer therapeutics, with multiple agents currently in early-phase clinical trials [22], [39]. Alisertib is an investigational, oral, selective inhibitor of Aurora kinase A. In xenograft models, this agent showed potent inhibition of Aurora kinase A and high antitumor activity across a range of tumor types [40]. Also, it is currently undergoing evaluation in a phase I/II trial in various nonhematological and hematological malignant diseases [23], [41], [42], [43].
In this study, to develop an objective Aurora A cutoff score for survival analysis, we used the ROC curve analysis to generate a cutoff score in the training set. Aurora A expression, which was classified as high and low level by the ROC-derived cutoff score, was mainly found to be higher in more advanced tumor stages, indicating that Aurora A might be involved in NSCLC progression. Correlation analysis further demonstrated that high Aurora A expression was associated with clinical stage and tumor invasion in NSCLC. Furthermore, in the testing set and overall patients, high Aurora A expression predicted a significant OS and DFS disadvantage over the low–Aurora A expression subgroup. Importantly, worse prognostic impact of increased Aurora A expression was demonstrated in patients with stage III and IV tumors, indicating that Aurora A might be a novel factor for risk definition in NSCLC. In addition, multivariate analyses in the testing set and overall patients revealed that Aurora A expression was an independent prognostic parameter. Taken together, our findings in this study provided evidence that overexpression of Aurora A in NSCLC might facilitate an increased malignant and worse prognostic phenotype of this tumor, which was consistent with the previous studies.
As for the underlying mechanism of Aurora A involved in chemoresistance, recurrence, and metastasis, it remains complicated and varies in various types of human cancers. Inhibition of Aurora A in cells expressing mutant JAK2 abolishes the resistance to cisplatin [44] and enhances cisplatin-induced cell death in esophageal carcinoma cells [39], [45]. It has been reported that Aurora A induces cisplatin chemoresistance by inhibition of p53, leading to downregulation of PTEN and activation of Akt in human ovarian cancer cells [46]. Some studies show that Aurora A promotes chemotherapeutic drug resistance via a NF-κB signaling pathway in p53 knockdown lung cancer cells [47]. EMT is a malignant cancer phenotype characterized by aggressive invasion and metastasis, and resistance. Metastatic tumors are invariably more resistant to the chemotherapy compared with primary tumors as evidenced by the marked decrease of chemotherapy response rate [48], [49]. Furthermore, recent studies have shown a relation between cisplatin resistance and the EMT phenotype [50]. Resistance to cisplatin was observed in cell lines undergoing EMT [50]. In our study, we found that Aurora A–related cisplatin resistance may be conferred by increasing of EMT. Thus, therapeutic combinations using EMT signaling inhibitors may reverse the resistance of some types of cancer to chemotherapy.
In summary, our results provide a basis for the concept that increased expression of Aurora A in human NSCLC may be important in the tumor progression and serves as an independent biomarker for poor survival. Thus, overexpression of Aurora A identifies patients at high risk and is a novel therapeutic molecular target for NSCLC. Also, our data suggest that increased expression of Aurora A is related to cisplatin resistance. Consequently, inhibition of Aurora A kinase may be a promising regimen to overcome recurrence, metastasis, and resistance in NSCLC patients.
Acknowledgements
No conflicts of interests are present.
The authors are grateful to all the patients and investigators for their participation in this study.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura H, Saji H. Worldwide trend of increasing primary adenocarcinoma of the lung. Surg Today. 2014;44(6):1004–1012. doi: 10.1007/s00595-013-0636-z. [DOI] [PubMed] [Google Scholar]
- 4.Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. doi: 10.1007/978-3-319-24223-1_1. [DOI] [PubMed] [Google Scholar]
- 5.Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet. 2013;382(9893):720–731. doi: 10.1016/S0140-6736(13)61715-8. [DOI] [PubMed] [Google Scholar]
- 6.Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8(7):823–859. doi: 10.1097/JTO.0b013e318290868f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 8.Dhanasekaran SM, Balbin OA, Chen G, Nadal E, Kalyana-Sundaram S, Pan J, Veeneman B, Cao X, Malik R, Vats P. Transcriptome meta-analysis of lung cancer reveals recurrent aberrations in NRG1 and Hippo pathway genes. Nat Commun. 2014;5:5893. doi: 10.1038/ncomms6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grabner B, Schramek D, Mueller KM, Moll HP, Svinka J, Hoffmann T, Bauer E, Blaas L, Hruschka N, Zboray K. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat Commun. 2015;6:6285. doi: 10.1038/ncomms7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imielinski M, Greulich H, Kaplan B, Araujo L, Amann J, Horn L, Schiller J, Villalona-Calero MA, Meyerson M, Carbone DP. Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J Clin Invest. 2014;124(4):1582–1586. doi: 10.1172/JCI72763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heist RS, Engelman JA. SnapShot: non–small cell lung cancer. Cancer Cell. 2012;21(3):448.e442. doi: 10.1016/j.ccr.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research N Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolanos-Garcia VM. Aurora kinases. Int J Biochem Cell Biol. 2005;37(8):1572–1577. doi: 10.1016/j.biocel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 15.Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL, Jr., Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70(4):661–687. doi: 10.1007/s00018-012-1073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marumoto T, Zhang D, Saya H. Aurora-A — a guardian of poles. Nat Rev Cancer. 2005;5(1):42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 17.Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, Washington K, Castells A, Pera M, El-Rifai W. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008;112(8):1688–1698. doi: 10.1002/cncr.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoque A, Carter J, Xia W, Hung MC, Sahin AA, Sen S, Lippman SM. Loss of aurora A/STK15/BTAK overexpression correlates with transition of in situ to invasive ductal carcinoma of the breast. Cancer Epidemiol Biomarkers Prev. 2003;12(12):1518–1522. [PubMed] [Google Scholar]
- 19.Mazumdar A, Henderson YC, El-Naggar AK, Sen S, Clayman GL. Aurora kinase A inhibition and paclitaxel as targeted combination therapy for head and neck squamous cell carcinoma. Head Neck. 2009;31(5):625–634. doi: 10.1002/hed.21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3(1):51–62. doi: 10.1016/s1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- 21.Guan Z, Wang XR, Zhu XF, Huang XF, Xu J, Wang LH, Wan XB, Long ZJ, Liu JN, Feng GK. Aurora-A, a negative prognostic marker, increases migration and decreases radiosensitivity in cancer cells. Cancer Res. 2007;67(21):10436–10444. doi: 10.1158/0008-5472.CAN-07-1379. [DOI] [PubMed] [Google Scholar]
- 22.Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, Obermannova R, DeMichele A, Zatloukal P, Zhang B. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non–small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. 2015;16(4):395–405. doi: 10.1016/S1470-2045(15)70051-3. [DOI] [PubMed] [Google Scholar]
- 23.Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA, Ray-Coquard I, Kutarska E, Liu H, Fingert H, Zhou X. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol. 2012;127(1):63–69. doi: 10.1016/j.ygyno.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 24.Dickson MA, Mahoney MR, Tap WD, D'Angelo SP, Keohan ML, Van Tine BA, Agulnik M, Horvath LE, Nair JS, Schwartz GK. Phase II study of MLN8237 (alisertib) in advanced/metastatic sarcoma. Ann Oncol. 2016;27(10):1855–1860. doi: 10.1093/annonc/mdw281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie D, Sham JS, Zeng WF, Lin HL, Che LH, Wu HX, Wen JM, Fang Y, Hu L, Guan XY. Heterogeneous expression and association of beta-catenin, p16 and c-myc in multistage colorectal tumorigenesis and progression detected by tissue microarray. Int J Cancer. 2003;107(6):896–902. doi: 10.1002/ijc.11514. [DOI] [PubMed] [Google Scholar]
- 26.Zlobec I, Steele R, Terracciano L, Jass JR, Lugli A. Selecting immunohistochemical cut-off scores for novel biomarkers of progression and survival in colorectal cancer. J Clin Pathol. 2007;60(10):1112–1116. doi: 10.1136/jcp.2006.044537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X, Gao L, Wang S, Lee CK, Ordentlich P, Liu B. HDAC inhibitor SNDX-275 induces apoptosis in erbB2-overexpressing breast cancer cells via down-regulation of erbB3 expression. Cancer Res. 2009;69(21):8403–8411. doi: 10.1158/0008-5472.CAN-09-2146. [DOI] [PubMed] [Google Scholar]
- 28.Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Noguchi M, Hirsch FR. Hepatocyte growth factor expression in EGFR-mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Clin Oncol. 2011;29(15_Suppl):7531. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 29.Hirsch FR, Franklin WA, Witta SE, Helfrich B, Lapadat R, Coldren CD, Sugita M, Bunn PA., Jr. Defining gefitinib sensitivity/resistance in non–small cell lung cancer (NSCLC) cell lines by Affymetrix gene arrays. J Clin Oncol. 2004;22(14_Suppl):7027. [Google Scholar]
- 30.Taniguchi H, Takeuchi S, Fukuda K, Nakagawa T, Arai S, Nanjo S, Yamada T, Yamaguchi H, Mukae H, Yano S. Amphiregulin triggered epidermal growth factor receptor activation confers in vivo crizotinib-resistance of EML4-ALK lung cancer and circumvention by epidermal growth factor receptor inhibitors. Cancer Sci. 2016 doi: 10.1111/cas.13111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9(4):1420–1426. [PubMed] [Google Scholar]
- 32.Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors—rising stars in cancer therapeutics? Mol Cancer Ther. 2010;9(2):268–278. doi: 10.1158/1535-7163.MCT-09-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka T, Kimura M, Matsunaga K, Fukada D, Mori H, Okano Y. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 1999;59(9):2041–2044. [PubMed] [Google Scholar]
- 34.Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17(11):3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka E, Hashimoto Y, Ito T, Okumura T, Kan T, Watanabe G, Imamura M, Inazawa J, Shimada Y. The clinical significance of Aurora-A/STK15/BTAK expression in human esophageal squamous cell carcinoma. Clin Cancer Res. 2005;11(5):1827–1834. doi: 10.1158/1078-0432.CCR-04-1627. [DOI] [PubMed] [Google Scholar]
- 36.Katayama H, Sasai K, Czerniak BA, Carter JL, Sen S. Aurora-A kinase phosphorylation of Aurora-A kinase interacting protein (AIP) and stabilization of the enzyme-substrate complex. J Cell Biochem. 2007;102(5):1318–1331. doi: 10.1002/jcb.21421. [DOI] [PubMed] [Google Scholar]
- 37.Morrison C, Henzing AJ, Jensen ON, Osheroff N, Dodson H, Kandels-Lewis SE, Adams RR, Earnshaw WC. Proteomic analysis of human metaphase chromosomes reveals topoisomerase II alpha as an Aurora B substrate. Nucleic Acids Res. 2002;30(23):5318–5327. doi: 10.1093/nar/gkf665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lioutas A, Vernos I. Aurora A kinase and its substrate TACC3 are required for central spindle assembly. EMBO Rep. 2013;14(9):829–836. doi: 10.1038/embor.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sehdev V, Peng D, Soutto M, Washington MK, Revetta F, Ecsedy J, Zaika A, Rau TT, Schneider-Stock R, Belkhiri A. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Mol Cancer Ther. 2012;11(3):763–774. doi: 10.1158/1535-7163.MCT-11-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ. Characterization of alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17(24):7614–7624. doi: 10.1158/1078-0432.CCR-11-1536. [DOI] [PubMed] [Google Scholar]
- 41.Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, Rosello S, Andreu J, Jung J, Sanchis-Garcia JM. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(17):4764–4774. doi: 10.1158/1078-0432.CCR-12-0571. [DOI] [PubMed] [Google Scholar]
- 42.Dennis M, Davies M, Oliver S, D'Souza R, Pike L, Stockman P. Phase I study of the Aurora B kinase inhibitor barasertib (AZD1152) to assess the pharmacokinetics, metabolism and excretion in patients with acute myeloid leukemia. Cancer Chemother Pharmacol. 2012;70(3):461–469. doi: 10.1007/s00280-012-1939-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dees EC, Cohen RB, von Mehren M, Stinchcombe TE, Liu H, Venkatakrishnan K, Manfredi M, Fingert H, Burris HA, III, Infante JR. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res. 2012;18(17):4775–4784. doi: 10.1158/1078-0432.CCR-12-0589. [DOI] [PubMed] [Google Scholar]
- 44.Sumi K, Tago K, Kasahara T, Funakoshi-Tago M. Aurora kinase A critically contributes to the resistance to anti-cancer drug cisplatin in JAK2 V617F mutant-induced transformed cells. FEBS Lett. 2011;585(12):1884–1890. doi: 10.1016/j.febslet.2011.04.068. [DOI] [PubMed] [Google Scholar]
- 45.Wang XX, Liu R, Jin SQ, Fan FY, Zhan QM. Overexpression of Aurora-A kinase promotes tumor cell proliferation and inhibits apoptosis in esophageal squamous cell carcinoma cell line. Cell Res. 2006;16(4):356–366. doi: 10.1038/sj.cr.7310046. [DOI] [PubMed] [Google Scholar]
- 46.Yang H, He L, Kruk P, Nicosia SV, Cheng JQ. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer. 2006;119(10):2304–2312. doi: 10.1002/ijc.22154. [DOI] [PubMed] [Google Scholar]
- 47.Sun C, Chan F, Briassouli P, Linardopoulos S. Aurora kinase inhibition downregulates NF-kappaB and sensitises tumour cells to chemotherapeutic agents. Biochem Biophys Res Commun. 2007;352(1):220–225. doi: 10.1016/j.bbrc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150(1):165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei Y, Hu G, Kang Y. Metadherin as a link between metastasis and chemoresistance. Cell Cycle. 2009;8(14):2132–2133. [PubMed] [Google Scholar]
- 50.Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69(6):2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]