

Abstract
Natural rubber has unique physical properties that cannot be replaced by products from other latex-producing plants or petrochemically produced synthetic rubbers. Rubber from Hevea brasiliensis is the main commercial source for this natural rubber that has a cis-polyisoprene configuration. For sustainable production of enough rubber to meet demand elucidation of the molecular mechanisms involved in the production of latex is vital. To this end, we firstly constructed rubber full-length cDNA libraries of RRIM 600 cultivar and sequenced around 20,000 clones by the Sanger method and over 15,000 contigs by Illumina sequencer. With these data, we updated around 5,500 gene structures and newly annotated around 9,500 transcription start sites. Second, to elucidate the rubber biosynthetic pathways and their transcriptional regulation, we carried out tissue- and cultivar-specific RNA-Seq analysis. By using our recently published genome sequence, we confirmed the expression patterns of the rubber biosynthetic genes. Our data suggest that the cytoplasmic mevalonate (MVA) pathway is the main route for isoprenoid biosynthesis in latex production. In addition to the well-studied polymerization factors, we suggest that rubber elongation factor 8 (REF8) is a candidate factor in cis-polyisoprene biosynthesis. We have also identified 39 transcription factors that may be key regulators in latex production. Expression profile analysis using two additional cultivars, RRIM 901 and PB 350, via an RNA-Seq approach revealed possible expression differences between a high latex-yielding cultivar and a disease-resistant cultivar.
Keywords: RNA-Seq, rubber biosynthesis, latex, transcription factor, full-length cDNA
1. Introduction
Natural rubber is an indispensable biomass material produced from Hevea brasiliensis, or the para rubber tree, and its cis-1,4-polyisoprene polymer has superior characteristics that cannot be replaced by synthetic rubber produced from petroleum oils.1,2 The demand for it is increasing in accordance with advances in road and air transport, and medical devices.3
In Malaysia rubber has been bred for better productivity and quality. It has been studied for about 90 years since the first rubber tree was imported. Rubber breeders have been crossing a few varieties to achieve high productivity and quality. In 2013, the first draft genome sequence of H. brasiliensis (Willd.) Muell.-Arg. RRIM 600 was reported using 43-fold sequence coverage data.4 Recently, we reported a much more comprehensive RRIM 600 genome assembly based on ∼155-fold coverage5 and, in addition, an assembly of Reyan7-33-97, another rubber cultivar genome, has been released that has 94-fold coverage.6 The burgeoning research interest in the para rubber tree reflects its significance as an important commodity crop globally.
The rubber tree has very specialized cells in the bark around the vascular bundle called laticifer cells and latex is produced from these cells. It is composed of rubber serum and rubber particles. The particles are composed of lipid monolayers and it is suggested that there are several proteins localized on the surface of these. They include the rubber elongation factor (REF), small rubber particle protein (SRPP), and cis-prenyl transferase (CPT), and they are highly induced in latex compared with leaf and bark.7,8 REF and SRPP are the cause of rubber allergy.9,10 CPT functions as a polymerizing enzyme for the condensation reaction of isopentenyl diphosphate to form polyisoprene. Recently, the Nogo-B receptor (or CPTL) was reported as a component of the rubber particle11 but the complete picture of how natural rubber is produced is not clear.
To further improve knowledge on latex production, we performed transcriptome analyses of full-length cDNA (FL-cDNA) and RNA-Seq. The sequence information derived from FL-cDNA libraries provides exact transcribed regions, such as transcription units, splicing variants and transcription start sites (TSSs). We constructed rubber FL-cDNA libraries and collected 19,487 clones as bioresources. Using the Sanger and high throughput sequences from the rubber FL-cDNA libraries, we confirmed the transcripts of 7,704 loci and updated 5,466 with 22 novel loci. To increase the coverage of expressed genes, we further added public expressed sequence tags (ESTs),12–14 confirmed the expression of 23,790 loci and updated the annotation of 17,201 loci.
RNA-Seq reveals the presence and quantity of genome-wide gene expression in biological samples. To predict the candidate genes related to rubber biosynthesis, we performed transcriptome analysis on four tissues (latex, bark, petiole and leaf) and on three rubber cultivars (RRIM 600, PB 350 and RRIM 901). RRIM 600 is an average latex-yielding cultivar with a production of ∼1,350 kg of latex per hectare per year in estates over 20 years old. This cultivar is susceptible to disease caused by the pathogenic fungal Phytophthora spp. On the other hand, PB 350 is a latex-timber (∼1,862 kg/ha/year), disease-resistant cultivar and RRIM 901 produces a higher latex yield (∼1,971 kg/ha/year) when compared with RRIM 600.15 We also focus on the transcriptional regulatory factors of rubber biosynthesis and list transcription factors (TFs) that are highly expressed only in latex.
2. Materials and Methods
2.1. RNA isolation and construction of two FL-cDNA libraries
Samples from leaf, petiole, bark and/or latex of RRIM 600 were collected from 2-, 5-, and 15-year-old trees located at Liman Plantation, Kedah, Malaysia (Supplementary Table S1). They were collected in the morning during the rainy season (November 2012 to January 2013). Total RNA from each tissue was extracted using a protocol optimized for rubber tree.16 RNA samples were purified with Qiagen RNeasy plant mini kits (Qiagen Inc., Hilden, Germany) following the manufacturer's instructions in order to obtain high quality RNA. We mixed seven samples (Supplementary Table S1) to make two FL-cDNA libraries. Construction of the libraries from poly(A)+ RNA was performed using the biotinylated CAP-trapper method.17 The double-stranded FL-cDNAs were digested with BamHI and XhoI and inserted into the BamHI and SalI sites of a λ-FLC-III vector.18 The phage library was amplified on solid plates and phage particles were subsequently eluted with SM buffer. Plasmids were generated from the amplified phage library by in vitro excision. They were transformed into DH10B™ T1 phage-resistant Escherichiacoli and eluted with SOC medium with 13% glycerol. The titer of the two FL-cDNA plasmid libraries were (no. 1) 1.3 × 105 cfu/ml and (no 2) 7.6 × 105 cfu/ml (Fig. 1). When we checked the lengths of cDNAs in the two libraries with electrophoresis (data not shown), library no. 1 contained various lengths of cDNAs whereas library no. 2 had relatively short cDNAs. Since library no. 1 was contaminated with E. coli genomic DNA, we decided to use library no. 2 for Sanger sequencing of individual clones, and both libraries for Illumina sequencing (Fig. 1). For Sanger sequencing, we randomly selected 19,968 clones (52 plates of a 384 format) and cultured them in LB medium with 7% glycerol at 30 °C overnight.
Figure 1.
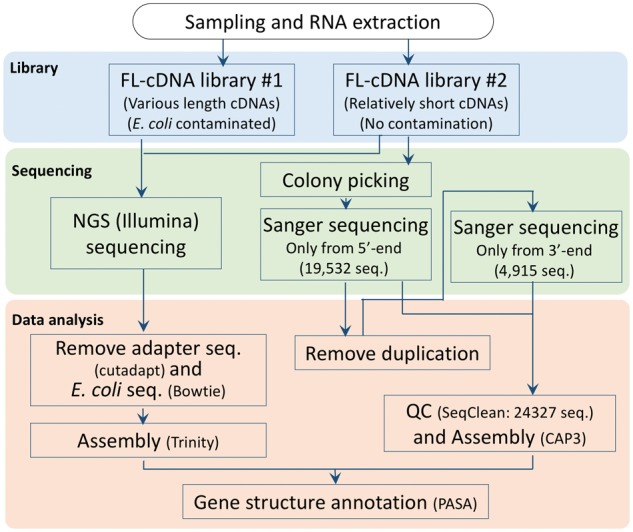
Flowchart of the steps in the construction of the FL-cDNA libraries, sequencing and data analysis.
2.2. FL-cDNA library no. 2: Sanger sequencing and quality control
End sequencing was performed by the Sanger method using ABI 3730xl capillary sequencers (Applied Biosystems, Foster City, CA, USA). The M13Fw (-20) primer (5′-GTAAAACGACGGCCAG-3′) and the M13Rvdt primer (5′-GCGGATAACAATTTCACACAGG-3′) were used for forward and reverse sequencing, respectively. The cDNA library was sequenced by the Sanger method from the 5′ end and a total of 19,532 sequences were generated that had a Phred quality of ≥20. To reduce the number of clones to sequence from 3′ end, we remove the duplicated sequences that show blast e-value ≤ 1e-100 and identity ≥95%. 5,395 clones were selected and Sanger sequenced from 3′ end. Of which, 4,915 sequences had a Phred quality of ≥20. We applied 19,532 5′ ESTs and 4,915 3′ ESTs to SeqClean (http://sourceforge.net/projects/seqclean/), which validates and trims DNA sequences. Then, 19,487 5′ ESTs and 4,480 3′ ESTs were assembled by CAP319 with the following parameters (MinDistance = 100 bp and MaxDistance = 15000 bp).
2.3. FL-cDNA library Nos. 1 and 2: Illumina sequencing and assembly
Total DNA from two FL-cDNA libraries was prepared using the Illumina TruSeq DNA LT Sample Prep Kit (Illumina, USA) following the manufacturer’s instructions. Sequencing was performed on the HiSeq 2000 (Illumina, USA) with 100 bp read length. Adapter sequences and poly-A were trimmed with cutadapt (v1.3),20 and FLC-III vector18 and E. coli genome21 sequences were removed using Bowtie 1.0.022 mapping software for no. 1 library . After these trimming and filtering processes, we assembled ESTs using Trinity (r2013-02-25) software23 with default parameters.
2.4. Genome annotation with the FL-cDNA sequences and public ESTs
We applied both the Sanger sequenced FL-cDNA ESTs and Illumina sequenced data to the PASA (Programme to Assemble Spliced Alignments) pipeline24 and updated the gene models. In this pipeline, we used two alignment software programmes, GMAP25 and BLAT,26 with default parameters and we accepted only transcripts with over 75% coverage (–MIN_PERCENT_ALIGNED = 75) and over 95% identity (–MIN_AVG_PER_ID = 95). To increase the gene coverage, we collected three public EST datasets12–14: (i) Chow et al.12 obtained 9,860 ESTs (NCBI accession: EC600050–EC609910) of RRIM 600 latex; (ii) Triwitayakorn et al.13 sequenced 2,311,497 reads from the vegetative shoot apical tissue of RRIM 600 (DDBJ: DRA000170) and (iii) Salgado et al.14 extracted RNAs from 33 organs and seedlings from open pollination of RRIM 600 and obtained 19,708 assembled sequences. We applied these three publicly accessible EST sources to the PASA pipeline with the same flow as our analysis.
2.5. TSS annotation with 5′ FL-cDNA sequences
We counted TSSs using 17,667 5′ FL-cDNA sequences that were successfully mapped to the genome with over 75% coverage and over 95% identity. As a first step, we eliminated redundancy and determined 9,486 TSSs. These were located at 5,482 loci. More specifically, 8,864 TSSs were located 1000 bp upstream or inside 5,042 genic regions, 54 were located on the reverse strand of 42 genic regions (candidates for antisense transcripts) and the other 618 TSSs were clustered in 398 intergenic regions that were defined using a simple distance-based approach with a maximum allowed distance of 1,000 bp between two neighboring TSSs.
2.6. RNA-Seq preparation and sequence analysis
Sampling, total RNA extraction and preparation of RNA-Seq libraries were performed using the same methods as for the FL-cDNA libraries (Supplementary Table S2). These libraries were high-throughput sequenced on the Illumina HiSeq 2000 platform using directed paired-end technology (2 × 100 bp).
To obtain more accurate results, we applied two mapping software programmes, TopHat v2.0.1327 and STAR version 020201.28 Cufflinks v2.2.129 assembled the mapped transcripts and calculated FPKM values for each sample.
For quality control, we drew a clustering dendrogram and a distance matrix to identify outlier replicates using an R module of CummeRbund29 (Supplementary Fig. S2). Except for RRIM 901 latex, each two replicates showed the closest Jensen-Shannon (JS) distance in Supplementary Figure S2. Since the JS distance of the two RRIM 901 replicates (JS distance = 0.16) was smaller than the JS distances in other tissue combinations (JS distance > 0.2), we decided to take both data for subsequent analysis (Supplementary Fig. S2b).
For gene set enrichment tests, we carried out a hypergeometric test to identify significantly enriched TF families or GO categories as follows:
, where N is the total number of all genes with TF family (or GO category) annotation, n is the number of differentially expressed genes (DEGs) in N, M is the number of genes in a given TF family (or GO category), and m is the number of DEGs in M. For this statistical calculation, we used SciPy (http://www.scipy.org/), which is a scientific library for Python. We then controlled the proportion of false positives by calculating the false discovery rate (FDR) corresponding to each P-value.29 The TF families (or GO categories) with a q-value of ≤ 0.05 were defined as significantly enriched. The TF family and GO annotation were carried out in our previous work.5
3. Results and Discussion
3.1. Cloning and data analysis of Sanger sequencing of rubber FL-cDNA library
FL-cDNA sequences are important for correct annotation and identification of authentic transcripts from plant tissues. We constructed two FL-cDNA libraries from H. brasiliensis RRIM 600 whose draft genome was determined recently.5 Since library no. 1 was contaminated with E. coli genomic DNA, we used library no. 2 for constructing FL-cDNA clones and applied both libraries to Illumina sequencing (Fig. 1).
After quality control, we obtained 19,487 FL-cDNA Sanger sequences from the 5′ ends (Table 1). We removed 5′ EST duplicates and selected 5,395 FL-cDNA clones. 4,840 clones were Sanger sequenced from their 3′ ends with Phred quality ≥ 20. A total of 24,327 FL-cDNA clones based on 5′/3′ ESTs were assembled into 4,590 contigs with CAP3, a DNA sequence assembly programme31 that also output 3,570 singletons. Of the 4,590 contigs, 3,011 (65.6%) were reached from both ends. We applied the PASA pipeline to annotate 8,160 unigenes (the sum of contigs and singletons) and they were mapped to 6,883 clustered positions. They were annotated as 3,877 known genes with 116 new isoforms and 258 structural gene modifications.
Table 1.
Summary of sequence resources of FL-cDNAs
| No. of sequences | Min. length (bp) | Max. length (bp) | Mean length (bp) | Mapping ratio (by PASA) | ||
|---|---|---|---|---|---|---|
| Sanger FL-cDNA | 5′ ESTs | 19,487 | 102 | 920 | 667.4 | |
| 3′ ESTs | 4,840 | 111 | 876 | 668.7 | ||
| Sanger FL-cDNA contigsa | 4,590 | 111 | 2,026 | 868.4 | ||
| Sanger FL-cDNA singletons | 3,570 | 103 | 884 | 655.7 | ||
| Assembled Illumina FL-cDNA contigs | 15,683 | 201 | 11,217 | 966.3 | ||
| Merged FL-cDNA (contigs) | 23,843 | 103 | 11,217 | 893.9 | 91.4% | |
aThe number of reads per contig was 4.5.
We also confirmed the functional bias of our FL-cDNA clones with GO slim31 (Supplementary Fig. S1). In most of the GO terms, the percentage of the cloned FL-cDNAs was around 10% of all GO annotation. The following 11 terms were enriched with a FDR adjusted q-value ≤ 0.05 in biological function: generation of precursor metabolites and energy (GO:0006091), biological_process (GO:0008150), photosynthesis (GO:0015979), catabolic process (GO:0009056), cellular homeostasis (GO:0019725), response to abiotic stimulus (GO:0009628), response to stress (GO:0006950), cellular component organization (GO:0016043), secondary metabolic process (GO:0019748). We infer that the enriched ‘response to abiotic stimulus’, ‘response to stress’, ‘response to external stimulus’ terms are related to our sampling of rubber tapping and latex. Also, ‘pollen-pistil interactions’ (GO:0009875) showed the lowest percentage and matched our sampling.
3.2. Data analysis of Illumina-sequenced rubber FL-cDNA library
Additionally to the Sanger-sequenced FL-cDNA clones, we sequenced our two FL-cDNA libraries with Illumina HiSeq 2000 and obtained 169,017,764 raw reads. After quality control, we used 81,624,108 trimmed and filtered read pairs and assembled them with Trinity.23 As a result, we obtained 15,683 assembled contigs. When we compared the Sanger- and Illumina-sequenced FL-cDNA contigs, it was found that 99% of the Sanger unigenes overlapped with the Illumina contigs.
We mapped all 23,843 FL-cDNA unigenes to the newly updated rubber genome,5 and 91.4% of them successfully mapped on the new draft genome while 80.4% mapped on the previous draft sequence (Supplementary Table S3). This high percentage suggests that the NGS sequenced data are efficient at collecting authentic transcripts. The results also suggest that the newest RRIM 600 draft genome has more comprehensive structural gene information than the previous one.
3.3. Structural gene modification with RIKEN FL-cDNA libraries and public EST data
We applied the PASA pipeline24 to update the current gene annotation. The PASA improved the 5,466 known genes and the 22 newly defined genes including four isoforms (Table 2). Details of the 5,466 modifications are as follows: 165 genes were fused into 78 genes and 3 genes were divided into 6 genes; 238 isoforms against 218 loci were newly annotated; 2,605 loci were improved in their 5′ and/or 3′ UTRs and a total of 2,901 annotated protein sequences were updated.
Table 2.
Statistics of updated structural gene annotation in H. brasiliensis.
| Categories | RIKEN data | + public ESTs |
|---|---|---|
| H. brasiliensis gene annotation | 84,440 | |
| No. of loci detected the expression | 7,704 | 23,790 |
| Total no. of loci updated with the PASA analysis | 5,466 | 17,201 |
| No. of fused genes | 78 | 236 |
| FL-cDNAs split single gene into multiple genes | 6 | 6 |
| No. of genes modified in UTRs | 2,605 | 7,525 |
| No. of genes modified in exon structure | 1,998 | 6,087 |
| No. of gene with newly defined isoforms | 218 | 1,967 |
| Proteins with CDS modification | 2,901 | 7,645 |
| Novel genes | 22 | 22 |
| Novel isoforms in the novel genes | 4 | 5 |
Since our FL-cDNA sequences cover only 9.1% (7,704 genes out of 84,440 predicted rubber genes) of all the structural genes in H. brasiliensis, we used publicly available ESTs from three publications7–9 and compared these with the new rubber genome. As a result, the coverage was raised to 28.2% (23,790 genes) and the annotation of 17,201 genes was improved (Table 2). We think that the excessive numbers of current gene predictions cause the limited gene coverage (see Section 3.5).
3.4. TSSs from FL-5′ ESTs
The PASA pipeline can use complete FL-cDNA sequences but not the 5′ TSS information from 5′ ESTs. For this reason, we separately analyzed rubber TSSs using 5′ ESTs. We mapped 19,487 5′ ESTs of FL-cDNA clones to the rubber genome. 17,667 5′ ESTs were successfully mapped and this corresponds to 9,486 unique TSSs. Only 55 of our TSSs exactly matched with the current TSS annotation. This very limited overlap was similar to the result obtained with Sorghum FL-cDNAs.32,33 The 9,486 TSSs were located at 5,482 loci, which contain 5,042 annotated genes, 42 antisense and 398 intergenic regions. Of these 398, 28 mapped to rubber draft scaffolds that have no annotated genes.
3.5. The gene coverage of multi-transcriptomic data
It is estimated that there are 84,440 genes in the new RRIM 600 draft genome.5 This number is much larger compared with the 27,416 genes in Arabidopsis thaliana,34 21,573 genes in Jatropha curcas35 and 31,344 genes in Ricinus communis (castor bean).36 Since there are more and more transcriptome data, false-positive gene predictions result in fewer problems than false-negative predictions. When we map RNA-Seq data to a genome, we can ignore the false-positives because they show 0 expression. On the other hand, problems still arise relating to quality and prediction of new genes using only RNA-Seq data. Therefore, the current number may be the result of reducing false-negative predictions. It will be greatly reduced after further annotation as was the case in the human genome (currently 26,000 genes).37,38 Transcriptomic data such as FL-cDNAs, ESTs and RNA-Seq are key information required to distinguish functional genes and pseudogenes.
We used six samples (leaf, petiole, bark and latex from RRIM 600 and latex from RRIM 901 and PB350) for RNA-Seq (Supplementary Table S2). In total, we obtained 118.8 Gb of RNA-Seq data (an average of 48 million reads) from the six samples with two replicates. These data were analyzed using two mapping software programmes, TopHat26 and STAR27. With our RNA-Seq data, STAR showed ∼10% better mapping ratio than TopHat (Supplementary Table S4). Engström et al.39 reported that TopHat showed lower tolerance for mismatches than STAR. As we consider that the current rubber draft genome still has many mismatches, we decided to use the mapping results from STAR for further analyses.
As a result of RNA-Seq, we confirmed that 36,544 genes were expressed in at least one of our samples with the criterion of FPKM > 0. Combining these with the 23,790 expressed genes obtained from the FL-cDNAs and ESTs, a total of 38,986 expressed genes (46.2% of the annotated genes) are validated in our analysis.
3.6. Tissue- and cultivar-specific gene expression based on RNA-Seq
To investigate the important genes involved in rubber biosynthesis, we compared RNA expression of leaves, petioles, bark and latex in 15-year-old RRIM 600 plants. We found 24,821 genes were commonly expressed in all tissues and 745 genes were uniquely expressed in latex (Fig. 2a). GO enrichment analysis of the 745 latex-specific genes showed that enzymatic activities (transferase activity (GO:0016740), kinase activity (GO:0016301), enzyme regulatory activity (GO:0030234), hydrolase activity (GO:0016787)) and binding (GO:0005488) were significantly enriched with a FDR adjusted q-value < 0.05. The total number of expressed genes is at least 4,000 fewer in latex than in other tissues (Supplementary Table S5).
Figure 2.

(a) Numbers of tissue-specific and co-expressed genes in RRIM 600 latex, bark, petiole and leaf. (b) Numbers of clone-specific and co-expressed genes in PB 350, RRIM 901 and RRIM 600 latexes. We defined expressed genes with FPKM > 0.
In Malaysia rubber has been improved by conventional breeding programmes, which have created cultivars with high and low latex yields. We have analyzed two other cultivars, RRIM 901 that has a high latex yield and PB 350 that yields a moderate amount of latex but has improved disease resistance and high timber production. Our results show that 24,081 genes were commonly expressed and 800-1,100 genes were specifically expressed in these cultivars (Fig. 2b).
We looked into whether the 745 latex-specific genes in RRIM 600 are also expressed in other cultivars. Although 96.7% of the expressed genes in RRIM 600 were shared with RRIM 901 and/or PB 350 (Fig. 2b), only 50.5% of the latex-specific genes in RRIM 600 were shared with RRIM 901 and/or PB 350. In addition, although we expected that the highly expressed latex-specific genes in RRIM 600 would be similar in all the cultivars, we did not find that this was the case (Supplementary Table S6). It is unlikely that essential genes for latex synthesis are different between cultivars. Tissue-specificity might be less important to elucidate complete pathway of latex synthesis.
To search for the key factors that confer higher latex yield in RRIM 901, we examined genes whose expression in latex is over 10 times higher in RRIM 901 than in RRIM 600 (Supplementary Table S7). At this threshold, most of the genes are categorized as having unknown function. When we considered genes whose expression is five times higher in RRIM 901 compared with RRIM 600 we found they belonged to several GO classes: growth (GO:0040007), cellular homeostasis (GO:0019725), lipid binding (GO:0008289), biological process (GO:0008150) and protein metabolic process (GO:0019538). These genes are candidates for the factor or factors that confer high latex production in RRIM 901.
3.7. Expression profile of isoprenoid biosynthesis genes
Natural rubber is a biopolymer consisting of cis-1,4-polyisoprene which is formed from isopentenyl diphosphate (IPP). IPP is synthesized by the cytosolic mevalonate (MVA) pathway from acetyl-CoA.40 As an alternative route, the plastidic 2-C-methyl-D-erythritol-4-phosphate (MEP) pathway from glyceraldehyde 3-phosphate (G3P) and pyruvate has also been demonstrated.41 Using RNA-Seq technology, we have confirmed the expression of 37 genes in the MVA pathway and 21 in the MEP pathway, and they are defined in our previous genome article5 (Fig. 3). As a comparison of their expression in RRIM 600 latex, genes in the MVA pathway (the average FPKM is 80.3) are more highly expressed than the genes in the MEP pathway (the average FPKM is 10.0). Also, the average FPKM value of CMK (4-(cytidine 5-diphospho)-2-C-methyl-D-erythritol kinase) in the MEP pathway was only 0.2 in the latex. This result is the same in both the PB 350 and the RRM 901 latex. Our data suggest that MVA is the main IPP synthetic pathway in latex. Also, the expression levels of the genes in the MEP pathway are higher in non-latex samples (leaf, petiole, and bark) compared with latex. This high expression in non-latex samples may be associated with non-latex terpenoid biosynthesis. IPP isomerase is known as a key rate-limiting step of terpenoid biosynthesis.42 Our data indicate that the expression level of the IPP isomerase gene in latex is also low and so, in order to enhance latex production, this gene can be an ideal candidate for modification of expression and activity.
Figure 3.

Latex biosynthesis pathway and heat maps of scaled expression values (log10(FPKM + 1)) for genes associated with latex biosynthesis. The lines of each heat map represent duplicate genes and the columns represent samples: PB 350 latex, RRIM 901 latex, RRIM 600 latex, RRIM 600 bark, RRIM 600 petiole and RRIM 600 leaf from the left. 37 MVA pathway, 21 MEP pathway and 18 prenyl-PP biosynthetic genes, 7 CPTs, 1 CPTL, 9 REFs and 8 SRPP genes are defined in our draft genome article.5 In the heat maps, genes were sorted by the expression value in RRIM 600 latex. These figures were drawn by an R package of gplots. Abbreviations in the MVA pathway: PD, pyruvate dehydrogenase; PDC, pyruvate dehydrogenase complex; DLD, dihydrolipoamide dehydrogenase; AACT, acetyl-CoA acetyltransferase; HMGS, hydroxymethyglutaryl coenzyme A synthase; HMG-CoA, hydroxymethylglutaryl coenzyme A; HMGR, hydroxymethyglutaryl coenzyme A reductase; MK, mevalonate kinase; mevalonate-OP, mevalonate monophosphate; PMK, phosphomevalonate kinase; mavalonate-OPP, mevalonate diphosphate; MDC, diphosphomevalonate decarboxylase; in the plastidic MEP pathway: DXS, 1-deoxy-D-xylulose 5-phosphate synthase; DXP, 1-deoxy-D-xylulose-5-phosphate; DXR, 1-deoxy-D-xylulose 5-phosphate reductoisomerase; MEP, 2-C-methyl-D-erythritol 4-phosphate; MCT, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; CDP-ME, 2-C-methyl-D-erythritol-4-phosphate; CMK, 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase; CDP-ME2P, 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol; MDS, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; MEcPP, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate; HDS, 4-hydroxy-3-methylbut-2-enyl diphosphate synthase; HMB-PP, 4-hydroxy-3-methyl-but-2-en-1-yl diphosphate; HDR, 4-hydroxy-3-methylbut-2-enyl diphosphate reductase. Corresponding gene IDs are as follow: PD#1:Hb_001638_290, PD#2:Hb_006420_080, PD#3:Hb_132840_110, PD#4:Hb_001500_040, PD#5:Hb_116349_120, PD#6:Hb_001123_160, PD#7:Hb_027506_040, PD#8:Hb_003397_030, PDC#1:Hb_000800_090, PDC#2:Hb_005306_180, PDC#3:Hb_002477_290, PDC#4:Hb_000110_310, PDC#5:Hb_003411_090, PDC#6:Hb_000207_300, PDC#7:Hb_002876_240, DLD#1:Hb_001723_060, DLD#2:Hb_002217_100, DLD#3:Hb_000645_180, DLD#4:Hb_012305_100, AACT#1:Hb_000613_200, AACT#2:Hb_000184_040, AACT#3: Hb_000377_090, AACT#4:Hb_000795_060, HMGS#1:Hb_003207_110, HMGS#2:Hb_000107_440, HMGS#3:Hb_004032_440, HMGS#4:Hb_001153_150, HMGS# 5:Hb_014497_120, HMGS#6:Hb_142382_010, HMGS#7:Hb_000116_530, MK#1:Hb_035273_030, MK#2:Hb_127194_010, MK#3:Hb_002042_080, PMK#1:Hb_002759_250, PMK#2:Hb_116349_100, MDC#1:Hb_00 0539_030, MDC#2:Hb_000721_030, DXS#1:Hb_003185_080, DXS#2:Hb_062135_030, DXS#3:Hb_019 153_040, DXS#4:Hb_ 002473_110, DXS#5:Hb_021596_070, DXS#6:Hb_000046_120, DXS#7:Hb_005628_050, DXS#8:Hb_012796_050, DXS#9:Hb_124247_030, DXR#1:Hb_007520_020, DXR#2:Hb_000922_160, MCT#1:Hb_029142_030, MCT#2:Hb_007576_140, CMK#1:Hb_000453_240, CMK#2:Hb_001975_040, MDS#1:Hb_177215_030, MDS#2:Hb_ 002529_090, HDS#1:Hb_000029_350, HDS#2:Hb_000008_410, HDR#1:Hb_159809_090, HDR#2:Hb_000963_090, IPPI#1:Hb_005686_130, IPPI#2:Hb_000608_190, DMAT#1:Hb_001842_100, DMAT#2:Hb_000428_050, DMAT#3:Hb_000130_260, FPPS#1:Hb_000072_070, FPPS#2:Hb_100414_010, FPPS#3:Hb_011224_160, GGPPS# 1:Hb_1 15919_020, GGPPS#2:Hb_000371_100, GGPPS#3:Hb_134789_010, GGPPS#4:Hb_020367_050, GGPPS#5:Hb_000114_150, GGPPS#6:Hb_002768_080, GGPPS# 7:Hb_000009_180, GGPPS#8:Hb_010672_040, GGPPS#9:Hb_009780_060, GGPPS#10:Hb_001948_040.
3.8. Expression profile of rubber biosynthesis genes
Latex is an emulsion of rubber serum and rubber particles produced from laticifer cells localized in the outer layers of the vascular bundle of the bark. A rubber particle is composed of a lipid monolayer membrane and it is suggested that there are three major rubber-related proteins (CPTs, REFs and SRPPs) localized on the surface of this particle. We confirmed the expression of 7 CPTs, 1 CPTL, 9 REFs and 8 SRPPs (Fig. 3). We found that CPT1, CPT2, CPTL, REF1, REF2 and SRPP1 were expressed at least 20 times more in latex than in leaf. The newly identified REF8 from the latest RRIM 600 draft genome sequence also shows a similar expression pattern to REF1 and REF2. This protein sequence has a REF domain and shows 59% identity with 90/117 amino acid alignment to SRPP1 with blastp. Its FPKM value is 11,221 and its level is 250 times higher in latex than in leaves. Therefore, REF8 is a candidate for further experimental validation to allow more characterization of its function.
When we calculated the correlation coefficients of all FPKM values between each sample, latex is more highly correlated in bark (correlation coefficient is 0.82) than in the petiole and leaf (correlation coefficient is 0.75–0.76) (Supplementary Fig. S3). Unlike the overall trend, many of the well-studied latex polymerization-related factors, such as CPTL, CPT2, REF1, REF2 and SRPP1, showed high expression in latex and petioles, but not in bark (Fig. 3). Since the petiole can produce a small amount of latex such a correlation may explain this observation.
3.9. Candidate TFs that regulate rubber biosynthesis genes
Little is known about the transcriptional regulation of rubber biosynthesis genes. We calculated the FPKM value for the 3,126 predicted TFs and selected 39 whose average FPKM value of three latex samples was more than five times higher than the average FPKM value of other tissues (Supplementary Table S8). We carried out gene set enrichment analysis against TF families and four of them were significant with a FDR adjusted q-value < 0.05 (Table 3). The C2C2-Dof,43 TAZ44 and AP245 families are related to the stress response. Since latex is exuded after tissue injury, these TFs are important candidate regulators. The cis-element of AP2 is known in A.thaliana and we confirmed partially similar motifs in the promoters of the CPT genes (Table 3).
Table 3.
Gene set enrichment analysis against TF families that are significantly highly expressed in latex.
| TF family | No. of TFs | Highly expressed TFs in latex | q-value | Function | cis-element | |
|---|---|---|---|---|---|---|
| C2C2-Dof | 49 | 7 | 0.009 | plant specific | Plant-specific phenomena including light, phytohormone and defense responses, seed development and germination | — |
| TAZ | 7 | 2 | 0.027 | plant specific | Responses to different stress stimuli | — |
| AP2 | 28 | 4 | 0.032 | plant specific | A variety of biological processes and response to biotic and environmental stress | 5′-gCAC(A/G)N(A/T) TcCC(a/g)ANG(c/t)-3′ |
3.10. Expression profile for disease-resistance genes
Corynespora leaf fall disease caused by infection with Corynespora cassiicola is one of the major fungal diseases in rubber plantations and it can cause a severe reduction in rubber latex production.46 We performed cultivar-specific RNA-Seq analysis using the C. cassiicola resistant cultivar PB 350 and the susceptible cultivars RRIM 600 and 901. At first we focused on 483 disease-resistance genes (R genes) that play important roles in the response against the pathogen. In normal or uninfected tissue, the expression of the R genes in PB 350 was the same or slightly lower than RRIM 600 and 901 (Supplementary Fig. S4). We also examined genes whose expression in PB 350 is over 10 times higher than in RRIM 600 and detected 41 genes (Supplementary Table S9). 40% (17 out of 41) of these are enriched with heat shock proteins (HSPs). Since HSPs are known to be critical for the plant defense response and some function as chaperones of R proteins for effector-triggered immunity,47 HSPs are likely to be important regulatory factors for the pathogen-stress response in rubber and contribute to the higher disease resistance of PB 350.
4. Conclusion
This study is the first large-scale collection of FL-cDNAs in H. brasiliensis in which 19,487 FL-cDNA clones harboring genetic information for 7,704 known genes and 22 new genes were obtained. The FL-cDNAs also provide 9,486 TSSs but the current coverage is not enough to carry out comprehensive TSS analysis. For better understanding, Cap Analysis of Gene Expression profiling or other genome-wide expression analyses will be necessary.
In this study, we used our newly sequenced rubber draft genome as a reference for transcriptome analysis.5 The genome sequence offers a more robust analysis of the generated RNA-Seq data in turn giving a more accurate expression profile analysis as the gene models will be more precise. Our findings reveal the tissue-specific expression patterns of the rubber biosynthesis genes and list candidate transcriptional regulators.
Our FL-cDNA sequences and RNA de novo assembly will be useful information for future improvement of the rubber tree and also as a basis for comparative analysis with other economically important Euphorbiaceae, such as cassava and the castor oil plant.
Data Availability
Our FL-cDNA sequence data and RNA-Seq data are available at DDBJ/EMBL/Genbank BioProject under accession PRJDB4387. We also provide the genome browser at http://matsui-lab.riken.jp/rubber/. Rubber FL-cDNA clones are available from the RIKEN BioResource Center (BRC).
Supplementary Material
Acknowledgements
We thank Setsuko Shimada (RIKEN Center for Sustainable Resource Science) for her constructive discussions and Shunsuke Imai (Sumitomo Riko Co.) for his advice on Figure 3.
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This research was conducted under the research programme of the RIKEN Biomass Engineering Programme.
References
- 1. Cornish K., 2001, Similarities and differences in rubber biochemistry among plant species. Phytochemistry, 57, 1123–34. [DOI] [PubMed] [Google Scholar]
- 2. van Beilen J.B., Poirier Y.. 2007, Establishment of new crops for the production of natural rubber. Trends Biotechnol., 25, 522–9. [DOI] [PubMed] [Google Scholar]
- 3. Mooibroek H., Cornish K., 2000, Alternative sources of natural rubber. Appl. Microbiol. Biotechnol. 53, 355–65. [DOI] [PubMed] [Google Scholar]
- 4. Rahman A.Y., Usharraj A.O., Misra B.B., et al. 2013, Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics, 14, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lau N.S., Makita Y., Kawashima M., et al. 2016, The rubber tree genome shows expansion of gene family associated with rubber biosynthesis. Sci. Rep., 6, 28594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tang C., Yang M., Fang Y., et al. 2016, The rubber tree genome reveals new insights into rubber production and species adaptation. Nat. Plants, 2, 16073. [DOI] [PubMed] [Google Scholar]
- 7. Han K.H., Shin D.H., Yang J., Kim I.J., Oh S.K., Chow K.S.. 2000, Genes expressed in the latex of Hevea brasiliensis. Tree Physiol. 20, 503–10. [DOI] [PubMed] [Google Scholar]
- 8. Asawatreratanakul K., Zhang Y.W., Wititsuwannakul D., et al. 2003, Molecular cloning, expression and characterization of cDNA encoding cis-prenyltransferases from Hevea brasiliensis. A key factor participating in natural rubber biosynthesis. Eur. J. Biochem., 270, 4671–80. [DOI] [PubMed] [Google Scholar]
- 9. Yeang H.Y., Cheong K.F., Sunderasan E., et al. 1996, The 14.6 kd rubber elongation factor (Hev b 1) and 24 kd (Hev b 3) rubber particle proteins are recognized by IgE from patients with spina bifida and latex allergy. J. Allergy Clin. Immunol., 98, 628–39. [DOI] [PubMed] [Google Scholar]
- 10. Berthelot K., Lecomte S., Estevez Y., et al. 2012, Rubber elongation factor (REF), a major allergen component in Hevea brasiliensis latex has amyloid properties. PLoS One, 7, e48065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Epping J., van Deenen N., Niephaus E., et al. 2015, A rubber transferase activator is necessary for natural rubber biosynthesis in dandelion. Nat. Plants, 1, 15048. [Google Scholar]
- 12. Chow K.S., Wan K.L., Isa M.N., et al. 2007, Insights into rubber biosynthesis from transcriptome analysis of Hevea brasiliensis latex. J. Exp. Bot., 58, 2429–40. [DOI] [PubMed] [Google Scholar]
- 13. Triwitayakorn K., Chatkulkawin P., Kanjanawattanawong S., et al. 2011, Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res., 18, 471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salgado L.R., Koop D.M., Pinheiro D.G., et al. 2014, De novo transcriptome analysis of Hevea brasiliensis tissues by RNA-seq and screening for molecular markers. BMC Genomics, 15, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lembaga Getah Malaysia, 2009, Chapter 2: development of rubber clones Rubber Plantation and Processing Technologies, pp. 21-68. Malaysian Rubber Board (MRB; ), Malaysia. [Google Scholar]
- 16. Deng L.H., Luo M.W., Zhang C.F., Zeng H.C.. 2012, Extraction of high-quality RNA from rubber tree leaves. Biosci. Biotechnol. Biochem. 76, 1394–6. [DOI] [PubMed] [Google Scholar]
- 17. Carninci P., Kvam C., Kitamura, et al. 1996, High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics, 37, 327–36. [DOI] [PubMed] [Google Scholar]
- 18. Carninci P., Shibata Y., Hayatsu N., et al. 2001, Balanced-size and long-size cloning of full-length, cap-trapped cDNAs into vectors of the novel lambda-FLC family allows enhanced gene discovery rate and functional analysis. Genomics, 77, 79–90. [DOI] [PubMed] [Google Scholar]
- 19. Huang X., Madan A.. 1999, CAP3: A DNA sequence assembly program. Genome Res., 9, 868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martin M., 2011, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, 17, 10–2. [Google Scholar]
- 21. Blattner F.R., Plunkett G., Bloch C.A., et al. 1997, The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–62. [DOI] [PubMed] [Google Scholar]
- 22. Langmead B., Trapnell C., Pop M., Salzberg S.L.. 2009, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grabherr M.G., Haas B.J., Yassour M., et al. 2011, Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol., 29, 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haas B.J., Delcher A.L., Mount S.M., et al. 2003, Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res., 31, 5654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu T.D., Watanabe C.K.. 2005, GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics, 21, 1859–75. [DOI] [PubMed] [Google Scholar]
- 26. Kent W.J. 2002, BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trapnell C., Pachter L., Salzberg S.L.. 2009, TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dobin A., Davis C.A., Schlesinger F., Drenkow J., et al. 2013, STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trapnell C., Roberts A., Goff L., et al. 2012, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc., 7, 562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benjamini Y., Hochberg Y.. 1995, Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser., B57:289–300. [Google Scholar]
- 31. Blake J.A., Dolan M., Drabkin H., et al. 2013, Gene Ontology annotations and resources. Nucleic Acids Res., 41, D530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Makita Y., Shimada S., Kawashima M., Kondou-Kuriyama T., Toyoda T., Matsui M.. 2015, MOROKOSHI: transcriptome database in Sorghum bicolor. Plant Cell Physiol., 56, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimada S., Makita Y., Kuriyama-Kondou T., et al. 2015, Functional and expression analyses of transcripts based on full-length cDNAs of Sorghum bicolor. DNA Res., 22, 485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lamesch P., Berardini T.Z., Li D., et al. 2012, The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res., 40, D1202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sato S., Hirakawa H., Isobe S., et al. 2011, Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res., 18, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chan A.P., Crabtree J., Zhao Q., et al. 2010, Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol., 28, 951–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lander E.S., Linton L.M., Birren B., et al. 2001, Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 38. Consortium I.H.G.S. 2004, Finishing the euchromatic sequence of the human genome. Nature, 431, 931–45. [DOI] [PubMed] [Google Scholar]
- 39. Engström P.G., Steijger T., Sipos B., et al. 2013, Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods, 10, 1185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kekwick R.G.O. 1989, The formation of isoprenoids in Hevea latex In: d’Auzac J., Jacob J.L., Chrestin L., editors. Physiology of Rubber Tree Latex, pp. 145–164. Boca Raton, FL: CRC Press. [Google Scholar]
- 41. Chow K.S., Mat-Isa M.N., Bahari A., et al. 2012, Metabolic routes affecting rubber biosynthesis in Hevea brasiliensis latex. J. Exp. Bot. 63, 1863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berthelot K., Estevez Y., Deffieux A., et al. 2012, Isopentenyl diphosphate isomerase: a checkpoint to isoprenoid biosynthesis. Biochimie, 94, 1621–34. [DOI] [PubMed] [Google Scholar]
- 43. Yanagisawa S. 2002, The Dof family of plant transcription factors. Trends Plant Sci., 7, 555–60. [DOI] [PubMed] [Google Scholar]
- 44. Du L., Poovaiah B.W.. 2004, A novel family of Ca2+/calmodulin-binding proteins involved in transcriptional regulation: interaction with fsh/Ring3 class transcription activators. Plant Mol. Biol., 54, 549–69. [DOI] [PubMed] [Google Scholar]
- 45. Nole-Wilson S., Krizek B.A.. 2000, DNA binding properties of the Arabidopsis floral development protein AINTEGUMENTA. Nucleic Acids Res., 28, 4076–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Narayanan C., Mydin K.K.. 2012, Breeding for disease resistance in Hevea spp. - Status, potential threats, and possible strategies. General Technical Report. PSW-GTR-240. U.S. Department of Agriculture. pp. 240–51. [Google Scholar]
- 47. Lee J.H., Yun H.S., Kwon C.. 2012, Molecular communications between plant heat shock responses and disease resistance. Mol. Cells, 34, 109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our FL-cDNA sequence data and RNA-Seq data are available at DDBJ/EMBL/Genbank BioProject under accession PRJDB4387. We also provide the genome browser at http://matsui-lab.riken.jp/rubber/. Rubber FL-cDNA clones are available from the RIKEN BioResource Center (BRC).