

Abstract
Alternative splicing (AS) is a key control mechanism influencing signal response cascades in different developmental stages and under stress conditions. In this study, we examined heat stress (HS)-induced AS in the heat sensitive pollen tissue of two tomato cultivars. To obtain the entire spectrum of HS-related AS, samples taken directly after HS and after recovery were combined and analysed by RNA-seq. For nearly 9,200 genes per cultivar, we observed at least one AS event under HS. In comparison to control, for one cultivar we observed 76% more genes with intron retention (IR) or exon skipping (ES) under HS. Furthermore, 2,343 genes had at least one transcript with IR or ES accumulated under HS in both cultivars. These genes are involved in biological processes like protein folding, gene expression and heat response. Transcriptome assembly of these genes revealed that most of the alternative spliced transcripts possess truncated coding sequences resulting in partial or total loss of functional domains. Moreover, 141 HS specific and 22 HS repressed transcripts were identified. Further on, we propose AS as layer of stress response regulating constitutively expressed genes under HS by isoform abundance.
Keywords: tomato pollen, RNA-seq, alternative splicing, heat stress, transcriptome, assembly
1. Introduction
The response of sessile lifeforms like plants to environmental alterations like heat stress (HS) is dependent on a complex network of distinct and interconnected pathways to maintain protein homeostasis and minimize cellular damage. Heat shock proteins (Hsps) are examples for HS-induced genes in various plant species,1–6 which contribute to thermotolerance as shown by genetic studies.7–10 The majority of these HS-induced genes are under the control of HS transcription factors (Hsfs). Although metazoans and fungi contain only one, plant genomes code for a large repertoire of Hsfs, which are of major importance due to their ability to rapidly activate the transcription of essential genes for stress response and survival.11
In addition to transcriptional changes, pre-mRNA alternative splicing (AS) is another important mechanism involved in heat stress response (HSR).12 Splicing is a central process for pre-mRNA maturation of intron-containing genes in all eukaryotic organisms.13,14 In response to environmental or developmental stimuli, AS of a single pre-mRNA can lead to various transcripts that may code for different functional protein variants.15 AS is a key mechanism for eukaryotes to increase proteome diversity from a defined number of pre-mRNAs.16,17 Consequently, AS is a common regulatory mechanism and the enhanced transcriptome diversity is required for stress adaptation18 and definition of plant tissue identity.19 In line, in plants about 61% of multi-exon genes are alternative spliced.20
At stage, at least four different modes of AS are annotated as intron retention (IR), exon skipping (ES), alternative acceptor (AA) and alternative donor (AD) site splicing.20 IR occurs when an intron is not removed from the pre-mRNA and remains in the mature mRNA. ES describes the event where an exon is spliced out of the pre-mRNA and AA and AD describe the usage of alternative 3′ and 5′ splice sites by the spliceosome, respectively.21 The various modes of AS can lead to premature termination codons (PTCs) resulting either in degradation of the mRNA through the nonsense-mediated decay (NMD) pathway or translation of a truncated protein.22,23 In addition, AS can result in changes or complete removal of functional domains, which may lead to proteins with novel functions.24 The prevalent AS event in Arabidopsis thaliana is IR that leads to PTCs.25,26
Therefore, the analysis of genome-wide AS especially in crop plants is of major interest to describe the mechanisms involved in stress response27 and pollen development.28 Based on this, we analysed the transcriptome of tomato (Solanum lycopersicum) pollen as model for a reduced tissue (two or three celled structure)29. Pollen development is characterized by a remarkable sensitivity to high temperatures.30,31 For example, plants exposed to HS show decreased pollen viability,32 which can cause sterility and in an agricultural aspect lead to crop damage.33
We used pollen from two different tomato cultivars, namely Moneymaker and Red Setter to analyse the changes induced by AS in response to HS using RNA-seq datasets. The application of two cultivars enabled us to exclude genotype specific AS and focus on HS induced AS. We focused on changes induced by IR, ES, AA and AD. Additionally, HS-dependent accumulation and reduction of AS events in specific genes was observed and these genes used to identify biological processes affected by AS under HS. In the end, transcriptome assembly led to the identification of 141 HS-specific and 22 HS-repressed transcripts. HS-specific and repressed putative protein isoforms were analysed concerning length differences in general and functional changes due to loss, gain or change of functional domains. We observed transcripts with steady expression levels under control conditions and HS showing a clear difference in the occurrence of specific isoforms partially or completely lacking functional domains. The latter exemplifies that post transcriptional AS leads to synthesis of transcripts putatively encoding for alternative protein isoforms, which might be required for HSR.
2. Materials and methods
2.1. Plant material, stress treatment and pollen analysis
Tomato plants (S.lycopersicum cv. Moneymaker and Red Setter) were grown under controlled greenhouse conditions (24 °C/18 °C, day/night temperature; 60% humidity). Eight-week old flowering plants were transferred to a climate chamber and exposed to a 39°C HS treatment for 1 h and then allowed to recover till anthesis. Flowers that received the stress 9–12 days before anthesis, corresponding to pollen meiosis and tetrads stages of development were analysed for pollen viability and number as previously described.34 In total 15–20 plants were used for the analysis for each genotype and treatment, and the experiment was repeated three independent times.
For RNAseq analysis flowering plants received the same treatment and buds were collected directly after HS or allowed to recover for 1.5 h at 24 °C. Untreated/control plants were kept at 24°C for the same time. Anthers from four flower buds were sampled in 500 µl of germination solution (2 mM boric acid, 2 mM calcium nitrate, 2 mM magnesium sulphate, 1 mM potassium nitrate) and squeezed open with slight mechanical stress, vortexed and filtered through muslin cloth. The collected pollen grains were harvested by centrifugation at 10,000× g for 15 min at 4 °C. The pelleted pollen were washed in 200 µl germination solution and centrifuged at 1,000× g for 1 min at 4 °C. This step was repeated a couple of times to remove the unwanted material. In total, more than 100 flower buds were used for pollen isolation. Purity of pollen was inspected under a light microscope. After isolation of pollen, all developmental pollen stages (microspore mother cell, tetrads, microspores and mature pollen) were pooled together for each of the four conditions: cv. Moneymaker-CO, cv. Moneymaker-HS, cv. Red Setter-CO and cv. Red Setter-HS. For each condition, two biological replicates were used for RNA-seq analysis, and another three independent biological replicates were used for qPCR analysis.
2.2. RNA isolation and mRNA sequencing
Total RNA was extracted from pollen of all eight samples using the E.Z.N.A. Plant RNA Kit (Omega Bio-Tek, Norcross, GA, USA). Total RNA was used for the preparation of mRNA-seq libraries (GenXPro GmbH, Frankfurt Main, Germany) by performing rRNA depletion and enrichment of mRNA via polyadenylation (polyA) selection and purification.35 Constructed libraries from each replicate of cv. Moneymaker and cv. Red Setter were strand-specific sequenced (paired-end) with a read length of 135 bases on a HiSeq2500 (GenXPro GmbH, Frankfurt Main). The mRNA-seq datasets discussed in this publication have been deposited in NCBI Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE80556 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE80556).
2.3. Read alignment and identification of AS events
The eight FASTQ files were aligned in paired-end mode to the S. lycopersicum genome (version ITAG2.4, cv. Heinz) available from the Sol Genomics Network.36 The alignment of the RNA-seq reads onto the reference genome was done via TopHat2 (version 2.0.13)37 using the following alterations of default parameters: –max-multihits 1; –min-intron-length 36; –max-intron-length 50,000; –mate-inner-dist 300; –mate-std-dev 100. The parameters were chosen due to the fact that the wild form of tomato Solanumpimpinellifolium has only a nucleotide divergence of 0.6%38 and by this our cultivars should have no more divergence.
Next, the Pearson correlation coefficient of read counts was computed pairwise between all samples based on computed read counts of all genes. Read counts were obtained by counting the number of reads (NuR) aligned on a specific gene. Due to the high correlation (>0.99) and small biological variation between biological replicates, the TopHat2 alignments of biological replicates were merged into a single alignment file resulting in four final alignment files (Moneymaker CO, Moneymaker HS, Red Setter CO and Red Setter HS).
Next, TopHat2 alignments were used for the identification of AS events, namely IR, ES, AA and AD. For this purpose, the coordinates of all annotated introns and exons were extracted from the ITAG2.4 corresponding Generic feature format version 3 (GFF3) file of S. lycopersicum. Introns were assumed to be retained (IR) if all positions of the intron were covered by at least one read. The other three exon specific events (ES, AA and AD) were considered to be present if the NuR witnessing the particular event exceeded the average genome coverage per position of each library.
2.4. FPKM calculation and verification
As cDNA fragments were sequenced from both ends (paired-end reads), quantification was done by counting paired reads as one fragment. Assignment of fragments was preceded to annotated exons of gene models (gene fragments). The assignment of fragment counts was performed with htseq-count of the High-Throughput Sequencing python framework (HTSeq).39 Afterwards, for each gene the number of gene fragments was normalized by the size of the library and the length of all exons within the gene. Finally, multiplication by one billion (to counteract the division by kilobase and by million mapped reads) resulted in FPKM (fragments per kilo base of exon per million fragments mapped) values for genes.
FPKM calculation was verified by qRT-PCR. cDNA synthesis was performed with 1 μg of total RNA using the Revert Aid reverse transcriptase (Thermo Scientific) following the manufacturer’s instructions. The expression of selected genes was investigated by real-time PCR on a Stratagene Mx3000P cycler (Agilent Technologies). The qRT-PCR reaction (10 μl) included gene specific primers (Supplementary Materials S1), PerfeCTa SYBR Green FastMix Low ROX™ (Quanta Biosciencies) and the appropriate cDNA template. Thermal cycling conditions were 95°C/3 min followed by 95°C/15 s, 60°C/30 s, 72°C/30 s for 40 cycles. Gene primers (Supplementary Materials S1) were designed using PRIMER3 (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3.cgi/; September 2016). Data were analysed by standard methods40 and presented as relative levels of gene expression using ubiquitin (UBI; Solyc07g064130) and EF1α (Solyc06g005060) genes as internal standards. Each qRT-PCR reaction was repeated three times on the basis of three independent biological replicates. The RNA used for qRT-PCR analysis was independent from the RNA used for RNA-seq.
2.5. Detection of HS-dependent IR and ES events
At first, the frequency of all IR and ES was determined by normalizing the NuR witnessing the event to the size of the respective library. For determination of HS-dependent accumulation or reduction of the IR and ES events, the frequencies of the events were first normalized to the expression of the underlying gene by dividing it by the FPKM value of the gene. Next, log2 fold changes between the ratios of the HS libraries and the CO libraries were computed for each cultivar (Formula 1):
Afterwards, genes were discriminated by this in sets of (I) HS-dependent accumulation of AS and (II) HS-dependent reduction of AS. Regarding the first set (I), IR and ES events of the upper 5% quantiles of the log2 fold change distributions and exclusively occurring under HS were determined. In the next step IR and ES events were assigned to genes. Finally, only genes showing HS-dependent accumulation of at least one common AS event (IR or ES) in both cultivars were extracted. Regarding the second set (II), in a similar approach genes showing HS-dependent reduction of AS were identified by extracting those IR and ES events of the lower 5% quantile and those exclusively present under CO.
2.6. Gene set enrichment analysis
Gene ontology (GO)41 terms were assigned to all annotated genes (ITAG 2.4) via InterProScan (version 5.14-53.0).42 Next, genes alternatively spliced under HS and CO conditions were analysed for enrichment of GO terms related to the biological process category. The enrichment analysis was performed with the Cytoscape (version 3.2.1)43 plugin BiNGO (version 3.0.3)44 by using the Benjamini and Hochberg false discovery rate (FDR) correction45 and applying an adjusted P-value cut-off of 0.01 as threshold. In addition, visual inspection of enriched GO terms was performed to exclude unspecific terms (Supplementary Materials S2; bold terms). Terms were excluded if their function could be more precisely defined by a child-term (e.g. transport and intracellular transport).
2.7. HS-dependent transcript assembly
All mapped reads located in a range from 1 kilobase pair (kbp) downstream to 1 kbp upstream of genes showing HS-dependent accumulation or reduction of AS were extracted from TopHat2 Sequence Alignment/Map files of all four runs and used as input for reference based transcript assembly via Cufflinks (version 2.2.1).46 Cufflinks was executed with default parameters except for the following modifications: –min-isoform-fraction 0.05; –pre-mRNA-fraction 0.05; –small-anchor-fraction 0.05.
Transcripts assembled by Cufflinks were afterwards assigned to annotated tomato genes of the ITAG2.4 and renamed concerning the identifier (e.g. SolycXXgYYYYYY) of the gene. Next, all transcripts assigned to the same gene identifier were compared between HS and CO condition and those present only under HS for both cultivars determined as HS-specific transcripts and those only present under CO for both cultivars as HS-repressed transcripts. Transcripts were assumed to be similar if all underlying exon coordinates were identical except for the start coordinate of the first and the end coordinate of the last exon as such differences are expected to lead to alternative untranslated regions (UTRs) without an effect on the coding sequence (CDS).
2.8. Identification of AS events in HS-specific and repressed transcripts
HS-specific and -repressed transcripts were compared with the transcripts annotated in the GFF3 of the ITAG2.4. In a first step, transcripts, whose exterior exons were not overlapping the exterior exons of the ITAG2.4 (inconsistent length) were excluded. In a second step, transcripts covering more than one annotated tomato gene (multisolyc transcripts) or containing no stop codon (transcripts with no CDS) were excluded. Next, the remaining transcripts were sub-divided into five groups based on the presence and absence of AS events: transcripts with only IR (i, only IR), transcripts with only ES (ii, only ES), transcripts with mixtures of IR and ES (iii, IR + ES), transcripts with other events than only IR and ES like AA and AD events as well as not annotated introns and exons (iv, others) and transcripts showing no difference in their splicing pattern to the ITAG2.4 annotation (v, perfectly).
2.9. Confirmation of HS-specific isoforms via RT-PCR
Transcripts assembled via Cufflinks and TopHat2 based on RNA-seq data were verified via RT-PCR. One microgram of total RNA was used for cDNA synthesis with Revert Aid reverse transcriptase (Thermo Scientific) following the manufacturer’s protocol. Splicing of selected genes was monitored by RT-PCR using specific primers (Supplementary Materials S1). Four transcripts were chosen, one serves as control with no AS isoform in all four samples (Solyc08g068350). The other three transcripts contain HS-specific IR (Solyc09g074680), ES (Solyc03g121800) or both (Solyc10g084400) in the HS datasets.
2.10. Functional domain analysis
As a first step, the CDS within the HS-specific and repressed transcripts was determined. The CDS search was performed by taking the start codon annotated in the ITAG2.4 as starting point and searching for the first stop codon (TAA, TAG or TGA). The obtained CDSs of all transcripts were then translated into protein sequences and together with the corresponding annotated protein sequences of tomato ITAG2.4 analysed for functional domains. For domain detection the protein families database (PFAM)47 and HMMER348 were used executing hmmscan with default parameters. After that, the predicted domains were compared between the predicted and annotated proteins to identify gain, loss and changes in the domain architecture.
2.11. Large datasets
The mRNA-seq datasets discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus49 and are accessible through GEO Series accession number GSE80556 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE80556). Sixteen paired-end fastq files for eight samples are deposited. These eight samples are from tow tomato cultivars (Moneymaker and Red Setter) and treated with HS (39 °C) or under control conditions (25 °C) in two replicates each.
3. Results
3.1. AS events in tomato pollen in response to HS
To analyse the effect of HS on the transcriptional fate of tomato pollen, total RNA of pooled developmental stages of pollen was extracted from Moneymaker and Red Setter tomato plants. Phenotypic analysis revealed that the short (1 h) HS at 39°C did not affect the viability of Moneymaker pollen but caused a 21% reduction in Red Setter (Supplementary Materials S3). The analysis was done for the earlier developmental stages, which are considered as more sensitive to elevated temperatures. Subsequently, the selection of these two cultivars allowed us to analyse changes occurring in two genotypes with slight pollen thermotolerance differences. In a similar manner, for genome-wide transcriptome analysis, flowering plants were exposed to 39 °C for 1 h and pollen was isolated directly after the treatment as well as after recovery of flowers at 25 °C for 1.5 h. Both pollen samples were combined. By this way we were able to monitor the AS events occurring during HS and during the recovery from HS, as both represent important phases of stress response with distinct transcriptional signatures.50 To analyse the HSR, pollen from plants kept at 25 °C were isolated and analysed. This sample is annotated as control (CO).
RNA-seq libraries from two biological replicates were constructed for HS and CO conditions for both cultivars. After sequencing, all libraries were aligned to the tomato genome of cultivar Heinz with the spliced aligner TopHat2.37 In contrast to de novo or reference based transcriptome assembly,35 the alignment to a reference genome allows the expression analysis and the detection of AS events for annotated genes. A high Pearson correlation between the read counts of biological replicates (Supplementary Materials S4), allowed us to merge their alignments into a single file leading to four samples to be analysed (Moneymaker CO, Moneymaker HS, Red Setter CO and Red Setter HS) with a total amount of more than 280 million reads each (Table 1). The rate of read alignment slightly differed between the four libraries ranging from 42% for Moneymaker-CO to 61% for Red Setter-HS. Reads witnessing an IR, ES, AA or AD event were traced in the alignment files.
Table 1.
Statistics of mRNA-Seq libraries
| Moneymaker |
Red Setter |
|||
|---|---|---|---|---|
| CO | HS | CO | HS | |
| NuR | 285,468,115 | 329,165,337 | 280,177,153 | 351,372,546 |
| AR | 120,969,297 | 192,550,586 | 160,831,268 | 213,563,554 |
| % | 42.4 | 58.5 | 57.4 | 60.8 |
Given is the total number of reads (NuR), of aligned reads (AR) and the percentage ARs (%) for Moneymaker and Red Setter HS and CO libraries.
We compared the alignment position with the exon-intron structures of the gene annotation in the S.lycopersicum cv. Heinz genome (ITAG2.4; http://solgenomics.net/; cv. Heinz 1706) (Fig. 1A). An IR was assumed to be present if the respective intron was covered at each position by at least one read. An ES was assigned if a read was aligned to two exons excluding another exon or even more exons in between. In the same line, we annotated an AA and AD in case that a read was aligned to two exons, but either the donor or acceptor site differed from that deposited one in the database. An ES, AD and AA event was considered significant when the NuR witnessing the AS event exceeded the average genome coverage of the library.
Figure 1.

AS events in pollen of different tomato cultivars exposed to HS. (A) Schematic representation for detection of AS events using reads witnessing a particular event. Detected events are IRs, ESs, AD sites and AA sites. (B and C) representing the number of genes showing particular AS events (IR, ES, AA and AD) under CO, HS or both conditions (Overlap) for the cultivars Moneymaker (B) and Red Setter (C).
For each of the four AS events the number of genes showing this particular event was determined, where multiple occurrences of the event in a single gene were counted only once. The number of genes with a particular IR and ES event is clearly different between the CO and HS conditions. We observed 8,415 genes with IR under HS, and 4,578 genes with IR under CO in Moneymaker (Fig. 1B). About 87% of the genes with IR found at CO also showed an IR under HS (Fig. 1B, grey bar). In Red Setter we observed only a slight increase of genes with IR at HS (8,795 genes) when compared with CO (8,394 genes), while the total number of genes with IR under HS conditions is rather comparable in both cultivars. We realized that IR is by far the most common AS event (Fig. 1B and C). Furthermore, an increase of genes with ES from 684/612 under CO to 1,225/901 genes under HS was observed in Moneymaker and Red Setter, respectively. In contrast the number of genes with AA and AD did not increase upon HS in the two cultivars. Remarkably, 54 and 61% of all genes with ES under CO and HS occur in both cultivars. Further, 81/89% of all genes with IR in Moneymaker were also present in Red Setter under control or HS conditions. These findings demonstrate that AS is a general temperature-dependent response, while cultivar-specific AS exists depending on the AS event and condition (Supplementary Materials S5).
3.2. HS-dependent IR and ES events in tomato pollen
The increase of genes with IR and ES events in tomato pollen upon HS might indicate heat-sensitive IR and ES. Next, we checked whether the different AS events are related with the abundance of expression of the respective genes. For this purpose we determined the FPKM normalized expression of all genes and categorized them with respect to their AS behavior, meaning AS under both, one or none of the conditions (Supplementary Materials S6). In total, 23,868 (Moneymaker) and 25,314 (Red Setter) genes showed an FPKM > 1 in both conditions (CO and HS), respectively. For the majority of these genes (58.7 and 56.8%) we did not identify reads that would indicate AS events. In turn, for 22.3 and 28.8% of all genes reads were identified, which suggest the existence of alternatively spliced transcripts under both conditions in Moneymaker and Red Setter, respectively. Differences between cultivars were observed for genes showing AS under a single condition. For Moneymaker we found with 15.7% a five times higher amount of genes with alternatively spliced transcripts under HS compared with CO (3.25%). For Red Setter we realized nearly similar amounts of alternatively spliced transcripts under HS (7.6%) and CO (6.7%). Notably, the identification of AS was not dependent on FPKM expression of a certain gene (Supplementary Materials S6). For verification of FPKM expression levels, these were correlated with expression levels obtained from qPCR. Results revealed for both Moneymaker and Red Setter correlation higher than 0.97 (Supplementary Materials S7). Furthermore, we aimed to rule out a potential dependency of detected IR events on the number of introns comprised in a gene. Therefore, genes were examined for their intron density and the amount of retained introns (Supplementary Materials S8). Although a small number of individual genes showed a higher degree of retained introns with up to 12 IR events (Supplementary Materials S8, white scatter plot), the general tendency revealed an accumulation of IR between zero and three IR events. This observation was irrespective of the number of introns contained in a gene, where Moneymaker CO showed the lowest abundance with the majority of genes having only up to one IR event (Supplementary Materials S8, contour plot). In addition, we compared these observations to a model supporting a dependency between the amount of IR events and the number of introns in a gene (Supplementary Materials S8, red scatter plot), but no relationship could be detected. These results confirm that detection of AS is neither dependent on the expression of the underlying gene nor on the composition of the gene and by this shows that AS is a controlled mechanism and no side effect.
Consequently, we aimed to quantify the alteration of IR and ES between the two conditions at intron and exon resolution, respectively. For this purpose, the IR frequency of each intron and the ES frequency of each exon were normalized to the library size and the expression of the underlying gene (Formula 1). The log2 value of the normalized frequency under HS divided by the normalized frequency under CO serves as a measure of HS-dependent accumulation (positive log2 ratio) or reduction (negative log2 ratio) for IR and ES (Fig. 2A and B). Interestingly, the fold change distribution of IR (light and dark blue) and ES frequency (light and dark orange) for both cultivars represents a normal distribution. In total, 4,331/8,275 IR and 553/618 ES events could be detected in both conditions for Moneymaker (light color) and Red Setter (dark color), respectively. Thus, we defined for each cultivar the IR and ES events of the upper (accumulation) and lower (reduction) 5% quantile of the respective IR and ES distribution as HS-dependent (Fig. 2A and B). In addition, 10,882 and 7,334 IR events as well as 1,178 and 735 ES events solely occur under HS in Moneymaker or Red Setter, respectively (Fig. 2C). These events were added to the respective set of IR and ES events accumulating under HS (in total: 11,099 IR events in Moneymaker and 7,748 IR events in Red Setter; 1,206 ES events in Moneymaker and 766 ES events in Red Setter). Similarly, the 1,625/5,224 IR and 309/243 ES events exclusively found under CO condition in Moneymaker or Red Setter were added to the respective set of IR and ES events reduced under HS (in total: 1,842 IR events in Moneymaker and 5,638 IR events in Red Setter; 337 ES events in Moneymaker and 274 ES events in Red Setter). This demonstrates that induction of AS events like IR and ES is a more abundant regulatory mode in HSR than the reduction of AS events, irrespective of the cultivar analysed.
Figure 2.
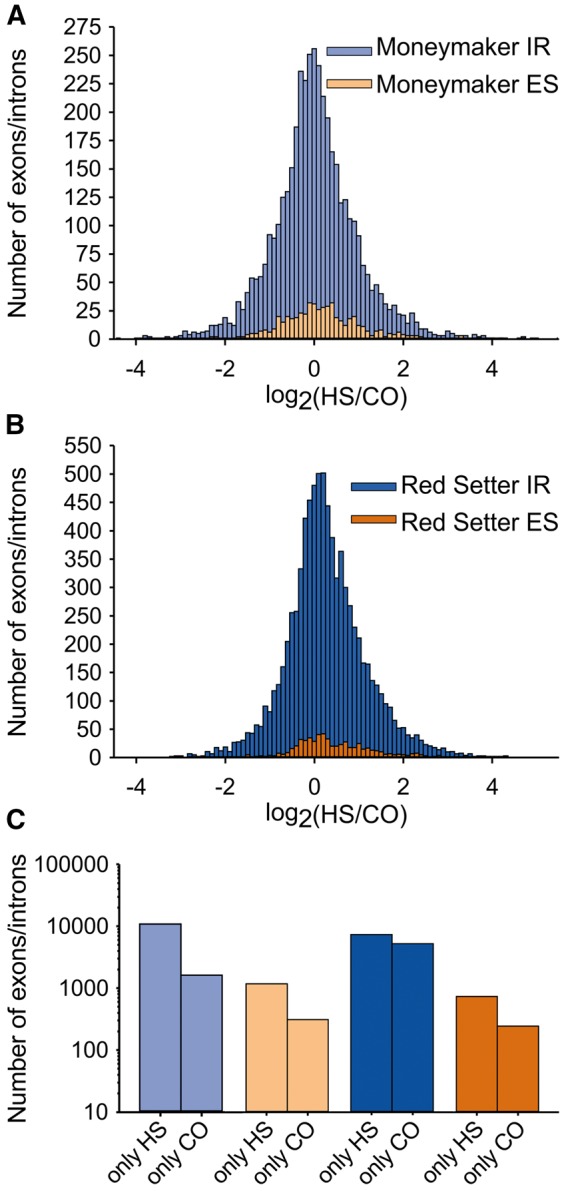
Detection of IR and ES events accumulated or reduced under HS. (A and B) show the distribution of HS-dependent accumulated (log2 ratio > 0) and HS-dependent reduced (log2 ratio < 0) IR (light and dark blue) and ES (light and dark orange) events. The log2 ratio is based on normalized event frequencies between HS and CO samples in Moneymaker (A) and Red Setter (B). Given in C is the number of IR and ES solely detected under HS or CO condition for both Moneymaker and Red Setter.
3.3. Processes affected by HS-dependent IR and ES events in tomato pollen
After the global analysis of splicing events we aimed at the definition of the global network of genes regulated by AS under HS irrespective of genotype specificities. Thus, we searched for genes regulated by the same type of AS event (IR or ES) in both cultivars under HS or CO conditions, respectively. To this end, we collected all genes for which at least one IR or ES event shows HS-dependent accumulation or HS-dependent reduction and determined the overlap between both cultivars. Approximately 7,500 genes with HS-dependent accumulation of IR and ES were detected (Fig. 3A). For ∼3,400 genes at least one IR or one ES event occurred in both cultivars (Fig. 3A; white numerals). The majority of these genes (2,731) contained only IR events in both cultivars. HS-dependent reduction of IR and/or ES was found in 4,846 genes (Fig. 3B). Most genes contained an IR solely in Red Setter (2,974 genes). In contrast, only 19% of the genes shared at least the same AS event type in both cultivars (Fig. 3B; white numerals). Thus, we postulate that the HS-dependent accumulation of IR and ES appears to be more global, while the reduction seems to be more cultivar specific (Fig. 3A and B).
Figure 3.
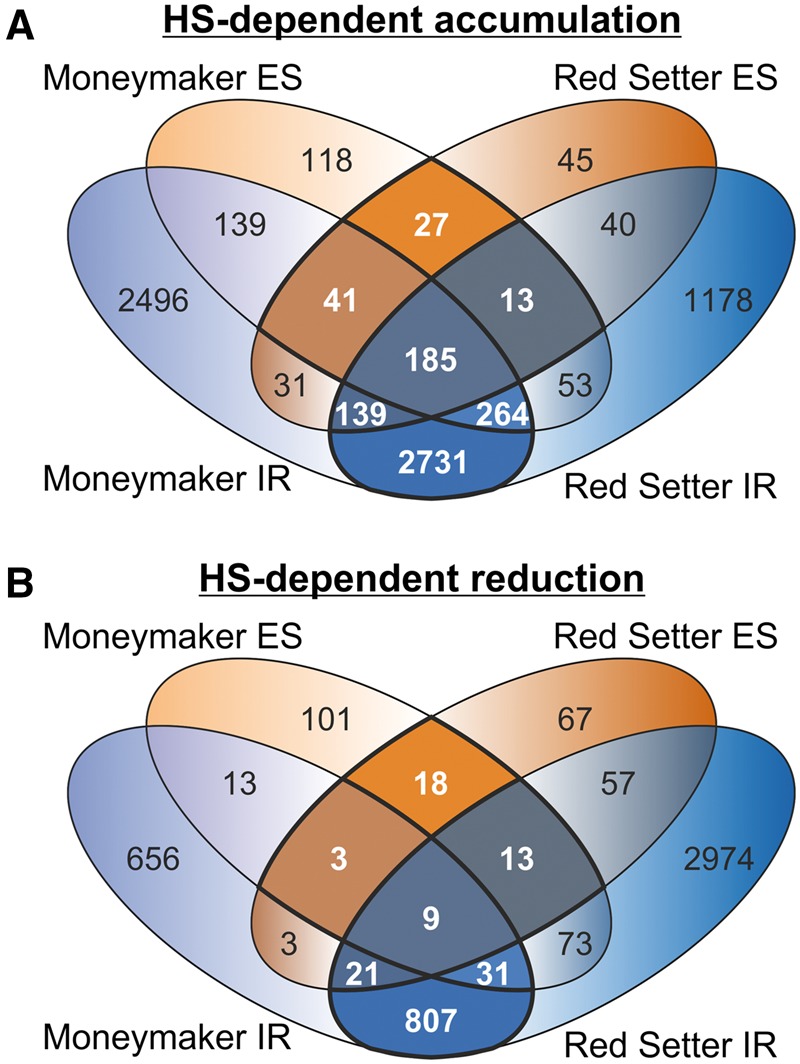
Genes showing HS-dependent accumulation or reduction of AS in pollen. (A) and (B) display Venn diagrams representing the overlap of IR and ES containing genes showing HS-dependent accumulation (A) or reduction (B). Genes featuring IR (light and dark blue) and/or ES (light and dark orange) for the cultivars Moneymaker and Red Setter are shown. Numbers in white and bold lines indicate genes showing at least one common IR or ES event in both cultivars.
Finally, we inspected for all genes found to be modulated by IR or ES whether at least one event targets the same intron or exon in both cultivars. In total, we observed 2,343 genes for which the same AS event shows HS-dependent accumulation and 451 genes with the same HS-dependent AS reduction event in both cultivars. Subsequently, enrichment of GO terms was performed to identify the cellular functions that are affected by alterations of IR or ES events in response to HS (Fig. 4). Interestingly, the analysis of the 451 genes with HS-dependent IR or ES reduction did not lead to the enrichment of any biological category annotated in the GO term database.
Figure 4.

Enriched biological processes due to HS-dependent accumulation of AS. Enriched GO terms (biological process) within the 2,343 genes showing HS-dependent accumulation of AS. Character sizes are dependent on the adjusted P-values (Benjamini and Hochberg FDR correction with a < 0.01 significance level) of the enrichment analysis, whereby bigger character sizes indicate lower p-values. Coloring of the biological process terms is related to their affiliation to functional categories, namely heat response (orange); gene expression (red), biosynthetic processes (green) as well as transport and localization (blue).
HS-dependent IR or ES accumulation was found to be enriched in genes coding for proteins represented by 33 GO terms (Fig. 4), which fall in four general functional categories: (i) gene expression, RNA modification, translation and protein folding (Fig. 4, red), (ii) localization and transport (Fig. 4, blue), (iii) heat response (Fig. 4, orange) and (iv) metabolic related processes (Fig. 4, green). One of the most enriched GO terms is protein folding and gene expression. In the category protein folding we identified genes coding for Hsps, namely seven genes coding for Hsp90, four for Hsp70, eight for HSP60, seven for Hsp40 and three for HSP10 (Table 2). With this, we found IR or ES events for all Hsp90 coding genes, 19% of all Hsp70 coding genes, 44% of all HSP60 coding genes, 7% of all Hsp40 coding genes and 75% of all HSP10 coding genes.2 The category gene expression comprised many transcription factors including six Hsfs (Table 2).11 The latter is remarkable, as 27 Hsf coding genes exist in the genome of tomato11 and thus, for 22% a HS-dependent accumulation of a splice variant is found. This observation suggests that AS is involved in the complex regulatory network of HSR.12
Table 2.
Genes involved in protein folding and gene expression altered by HS-dependent IR or ES accumulation
| GO-term | Protein family | Protein | Gene acc. number |
|---|---|---|---|
| protein folding | Hsp90 | Hsp90-1 | Solyc12g015880 |
| Hsp90-2 | Solyc07g065840 | ||
| Hsp90-3 | Solyc06g036290 | ||
| Hsp90-4 | Solyc03g007890 | ||
| Hsp90-5 | Solyc05g010670 | ||
| Hsp90-6 | Solyc07g047790 | ||
| Hsp90-7 | Solyc04g081570 | ||
| Hsp70 | Hsp70-9 | Solyc11g020040 | |
| Hsp70-11 | Solyc01g103450 | ||
| Hsp70-12 | Solyc01g106210 | ||
| Hsp70-13 | Solyc01g106260 | ||
| HSP60 | Cpn60 (1) | Solyc05g053470 | |
| Cpn60 (2) | Solyc09g091180 | ||
| Cpn60 (3) | Solyc03g121640 | ||
| Cpn60-α (2) | Solyc11g069790 | ||
| Cpn60-β (1) | Solyc01g028810 | ||
| Cpn60-β (2) | Solyc03g120850 | ||
| CCT-γ | Solyc05g056310 | ||
| CCT-ε | Solyc05g013990 | ||
| Hsp40 | DnaJ-A | Solyc01g105340 | |
| DnaJ-A | Solyc01g086740 | ||
| DnaJ-A | Solyc11g071830 | ||
| DnaJ-A | Solyc03g118500 | ||
| DnaJ-A | Solyc01g090550 | ||
| DnaJ-B | Solyc02g077670 | ||
| DnaJ-B | Solyc01g079610 | ||
| HSP10 | Cpn10-P | Solyc01g088610 | |
| Cpn10 (1)-M | Solyc05g056390 | ||
| Cpn21-P | Solyc07g042250 | ||
| transcription factor | Hsfs | HsfA1b | Solyc03g097120 |
| HsfA2 | Solyc08g062960 | ||
| HsfA3 | Solyc09g009100 | ||
| HsfA7 | Solyc09g065660 | ||
| HsfB2a | Solyc03g026020 | ||
| HsfB1 | Solyc02g090820 |
3.4. Occurrence of HS-related alternative transcripts
So far, AS events were analysed independently for each intron and exon without drawing conclusions concerning putative transcripts. Therefore, we performed reference based transcriptome assembly for the 2,343 and 451 genes showing HS-dependent accumulation or reduction of IR and ES events, respectively, in both cultivars (see ‘Materials and methods’ section). In brief, reads aligned to the genomic sequence in the range of 1 kb downstream to 1 kb upstream of the respective genes were collected for all four RNA-seq libraries (Moneymaker CO & HS and Red Setter CO & HS). These reads were used for each individual library as input for the transcriptome assembly. All assembled transcripts were annotated according to the annotation of the Heinz genome (ITAG2.4). For the 451 genes with HS-dependent reduction of AS about 1,400–1,900 transcripts were assembled in the various pools. More than 90% of these transcripts are assigned to gene identifiers (Table 3). The remaining transcripts without assignment to gene identifiers result from reads aligned in proximity to the genes of interest, but are not derived from these. For genes with HS-dependent accumulation of AS ∼6,900 to 9,100 transcripts were established, and again >90% of the transcripts could be assigned to at least one gene of the reference genome (Table 3).
Table 3.
Transcriptome assembly descriptive statistics
| Gene set | HS-dependent reduction |
HS-dependent accumulation |
||||||
|---|---|---|---|---|---|---|---|---|
| Library | Moneymaker | Red setter | Moneymaker | Red setter | ||||
| CO | HS | CO | HS | CO | HS | CO | HS | |
| Assembled transcripts | 1,868 | 1,639 | 1,597 | 1,481 | 8,347 | 9,088 | 6,910 | 8,057 |
| Transcripts with SolycID | 1,746 | 1,552 | 1,522 | 1,400 | 7,868 | 8,652 | 6,636 | 7,716 |
Given is the number of assembled transcripts and assembled transcripts with assigned SolycID for the Moneymaker and Red Setter CO and HS libraries with HS-dependent reduction or accumulation of alternative spliced transcripts.
In depth analysis yielded 332 transcripts occurring only after HS (HS-specific transcripts) and 70 transcripts only under CO conditions (HS-repressed transcripts) in both cultivars. At first we confirmed that all of those transcripts are assigned to at least one gene of the reference genome. Second, we removed transcripts with inconsistent length, lack of CDS or assignment to more than one identifier as described in methods. Removal was performed due to the fact that these transcripts could comprise non-fully assembled transcripts and might represent misannotations in the ITAG2.4, which were not in the focus of our study. 58 and 69% of all HS-specific and HS-repressed transcripts, respectively, were excluded by this procedure from further analyses. The remaining 141 HS-specific and 22 HS-repressed transcripts were classified in five categories according to the AS event observed. Category ‘i’ unifies transcripts with IR only (i, only IR), category ‘ii’ transcripts with ES only (ii, only ES) and category ‘iii’ transcripts with IR and ES only (iii, IR + ES). Transcripts with other AS events in addition to or even in the absence of IR or ES (iv, others) were annotated as category ‘iv’ and transcripts without AS when compared with the ITAG2.4 as category ‘v’ (v, perfectly; Fig. 5). The latter two occurred if other events than IR or ES arise (i.e. AA, AD, not annotated introns or not annotated exons) or if the transcript annotated in the ITAG2.4 is only detected after HS and CO, respectively. Most transcripts show only IR (i: 36% HS-specific, 50% HS-repressed), while another large fraction represents perfectly spliced transcripts (v: 26% HS-specific, 23% HS-repressed). Interestingly, 26% of the HS-specific and of the 23% HS-repressed transcripts show modifications of the transcript distinct from IR or ES (iv). The results of the reference based transcriptome assembly were confirmed by RT-PCR analysis of three randomly selected genes with HS-specific transcripts and one gene constitutively spliced under all conditions (Supplementary Materials S9).
Figure 5.

CDS length alterations of transcripts due to AS under HS in pollen. Shown are changes in CDS length for HS-repressed (dark grey; left) and HS-specific transcripts (light grey; right) due to AS. Transcripts were classified in five categories with respect to the AS events observed: Only IR events (only IR); only ES events (only ES); IR and ES events (IR + ES); other AS events than IR and ES (others); transcripts perfectly spliced (perfectly). Numbers above the bars indicate the amount of transcripts assigned to the different categories.
Afterwards the length of the CDS represented by the HS-specific and repressed transcripts was compared with the annotated CDSs (ITAG2.4). As expected no alterations of CDS length was observed for all perfectly spliced transcripts (perfectly; Fig. 5). In turn, the CDS encoded by the transcripts of category ‘i’–‘iv’ showed generally a length reduction (Fig. 5). Although this was expected for transcripts with ES, the shortened CDS for transcripts of category ‘i’ and ‘iv’ suggests the occurrence of PTCs. Therefore, alterations in the architecture of functional domains can be expected as well as the occurrence of PTCs.
3.5. Transcripts coding for proteins with altered domain architecture upon HS
We analysed the domain architecture of the proteins putatively translated from the CDS of the HS-repressed or specific transcripts. Each CDS was translated into a protein sequence followed by a domain search with HMMER (hmmsearch).48 Predicted domains were compared with domains annotated for the proteins encoded by the CDS of the ITAG2.4. For both, the HS-repressed and specific proteins, at least 54% of the proteins had the same domain architecture as predicted for the annotated gene (No change; Table 4). Only five HS-specific proteins showed a gain of functional domains (Gain and Gain + Loss; Table 4). Nevertheless, only for the F-box/LRR-repeat protein (Solyc10g080020) a new F-box domain was detected going along with loss of a Fist_C domain. The remaining proteins showed either an AS-induced domain split, which led to the detection of two sub-domains, or an AS-induced transition to a similar domain. For example, the Calpain-2 catalytic subunit (Solyc11g068460) showed an IR induced switch from EF_hand_5 to left-hand domain. In turn, loss of domains constitutes for all of the alterations of the HS-repressed proteins and 88% of the alterations of the HS-specific proteins (Loss, Table 4). To analyse this dominant effect of domain loss in more detail, all proteins with partial domain loss and translated from transcripts containing only IR and ES were inspected in more detail (Table 5).
Table 4.
Domain alterations of HS-repressed and specific proteins
| Gain | Loss | Gain+Loss | No change | ||
|---|---|---|---|---|---|
| HS avoi. proteins | only ES | — | 1 | — | — |
| only IR | — | 7 | — | 4 | |
| IR+ES | — | — | — | — | |
| others | — | 2 | — | 3 | |
| perfectly | — | – | — | 5 | |
| HS occ. proteins | only ES | 1 | 7 | — | 6 |
| only IR | — | 17 | 2 | 32 | |
| IR+ES | — | 3 | — | 1 | |
| others | — | 12 | 2 | 21 | |
| perfectly | — | — | — | 37 |
Given is the number of HS-repressed and specific proteins with alterations in domain architecture; either domain gain, loss, gain and loss, or no changes in domain architecture with respect to predicted domains for annotated ITAG2.4 proteins.
Table 5.
Proteins with partial domain loss encoded by alternatively spliced isoforms
| Solyc | Function | CDS | Domain alterations | ||
|---|---|---|---|---|---|
| HS avoioded proteins | IR | Solyc10g079360.5 | transcription initiation factor IIB | 49.67 |  |
| Solyc10g081600.1 | Rhomboid protease GlpG | 64.54 |  |
||
| Solyc12g098930.1 | pyruvate dehydrogenase kinase | 72.82 |  |
||
| HS-specific prtoteins | IR | Solyc01g065490.1 | Putative Aldo/keto reductase | 45.00 |  |
| Solyc04g078050.2 | transcription initiation factor | 26.63 |  |
||
| Solyc05g053190.1 | Putative tRNA pseudouridine synthase | 37.19 |  |
||
| Solyc08g081070.3 | Mitochondrial carrier family Mitoferrin-1 | 62.13 |  |
||
| Solyc09g074680.1 | Putative CRL complex subunit | 83.53 |  |
||
| Solyc11g069490.1 | Serine/threonine-protein phosphatase 6 regulatory subunit 3 | 43.98 |  |
||
| ES | Solyc10g009070.2 | RNA helicase DEAD32 | 65.52 |  |
Listed are the category (HS-repressed/specific), the AS process leading to the isoform, the accession number, the putative function of the protein, percentage of CDS length compared with the ITAG2.4 and domains present (black), completely (grey) or partially absent (black to grey color gradient).
We detected three HS-repressed transcripts that coded for proteins with the loss of at least one functional domain. We observed a transcript for the transcription initiation factor IIB (Solyc10g079360.5) coding for the protein without the two TFIIB repeats (TFIIB). The rhomboid protease GlpG (Solyc10g081600.1) encoded by the AS transcript does not contain the ubiquitin associated domain. Moreover, the AS containing transcript for pyruvate dehydrogenase kinase (Solyc12g098930.1) does not code for the ATPase domain (HATPase_c).
In turn, seven proteins translated from HS-specific transcripts with at least one completely lost domain were detected. For four of the seven proteins (Solyc01g065490.1; Solyc05g053190.1; Solyc08g081070.3; Solyc11g069490.1) lost only repeated domains or motifs due to IR or ES. The remaining three proteins are the putative cullin-RING ubiquitin ligase (CRL) complex subunit (Solyc09g074680.1), which is lacking its neddylation site, a transcription initiation factor (Solyc04g078050.2) missing the TAF4 domain and the RNA helicase DEAD32 (Solyc10g009070.2) lacking the DBP10CT domain. The partial domain loss observed for these proteins raises the question whether the proteins would still be functional with either reduced or alternative function or if the underlying transcripts are triggered for NMD.
4. Discussion
4.1. The degree of AS in response to HS is cultivar specific
AS is a central regulatory mechanism to adjust transcript abundance or expand the diversity of the protein content.15–17 The observed number of AS events and the ratio between IR, ES, AD and AA events in tomato pollen (Fig. 1) are in agreement with recent studies on young tomato fruits.19 Furthermore, similar AS profiles have been observed in cereals27 and different tissues of Zea mays.51 Both studies demonstrated that IR is the most dominant AS event, followed by AA, AD and ES, which is the general tendency in plants.20 Moreover, we report an increase of the number of genes for which transcripts with IR or ES are identified under HS conditions (Fig. 1). Although this observation stands in contrast to the reported AS alteration in response to HS in Physcomitrella patens,18 our findings are in line with the observed enhanced number of IR events in response to salt and cold stress in A.thaliana and S.lycopersicum.52,53 Consequently, these studies and our findings suggest that IR is a tissue independent general stress response mechanism.
In pollen, AS is a general regulatory process, independent of transctipt abundance or regulation under HS (Supplementary Materials S6), or even with the number of introns of a gene (Supplementary Materials S8). The amount of observed IR and ES events after HS application is comparable between both tested cultivars (Fig. 1) and many HS-dependent accumulated or reduced IR and ES events were commonly detected in both cultivars (Supplementary Materials S5). For example, about 80% of all genes with transcripts carrying a HS dependent accumulated IR were found in both, Moneymaker and Red Setter. However, the number of genes with HS-dependent accumulated AS (2,343) exceeds the number of genes with HS reduced AS by a factor of five (451). This difference might be due to the reduced functionality of the spliceosome under HS, which is supported by the high number of HS-induced IR events.54,55 However, the process of stress regulated AS is at least in parts specific. On the one hand, while many of alternative spliced transcripts carry NMD features, it was observed in A. thaliana that this does not necessarily lead to NMD.22 On the other hand, we observed a decline of specific AS events in response to HS (Fig. 3), which might be related to a reprogramming of splicing. The large overlap of observed AS events after HS between the two cultivars might even suggest that HS-induced AS is global regulatory mechanism rather than a cultivar specific event.
Although the degree of AS after HS is comparable between both cultivars (Fig. 1), differences in the amount of IR and ES events were observed in the transcriptome of non-stressed pollen (Fig. 1). Although the number of AS events is largely increased in pollen of Moneymaker after HS treatment, a high overlap (78%) of genes with transcripts with IR found under CO and HS conditions was observed for Red Setter. One possible interpretation could be that Red Setter exhibits higher sensitivity against HS than Moneymaker and HS-induced alteration already occur at conditions used as control. However, irrespective of the reason for the difference found for Red Setter and Moneymaker, due to the high amount of IR under control conditions Red Setter seems to have a lower capacity for alterations of the transcriptome by AS under HS than Moneymaker.
4.2. HS-related AS targets genes involved in HSR
Commonly identified HS induced AS events in both cultivars mainly target genes coding for factors involved in protein folding and gene expression among other processes (Fig. 4). Within these functional categories, we realized an enrichment of AS events in HS related genes like Hsps and Hsfs exclusively under HS (Table 2). Therefore, we aimed to analyse these two protein families in more detail with respect to HS-dependent accumulation of IR and ES events as not all members of these families were represented in the protein folding and gene expression category (Supplementary Materials S10). Within the set of Hsfs we observed up to four HS accumulated AS events in at least one cultivar, whereby the majority of these Hsfs were up-regulated upon HS like for example HsfA2 (Solyc08g062960) for both Moneymaker and Red Setter. In Hsp families 90, 70, 60 and 40 on the other side, the majority of Hsps were constitutively expressed but both HS induced and constitutively expressed Hsps showed a high degree of HS accumulated AS. For example, the two HS induced Hsp90s (Solyc06g036290 and Solyc03g007890) showed both two IR events and the constitutively expressed Hsp70-12 (Solyc01g106210) four IR events in both cultivars. A similar pattern was observed for the Hsp100 family, where the HS induced ClpB3 (Solyc02g088610) comprised seven HS accumulated IR events in Red Setter and the constitutively expressed ClpC1 (Solyc03g118340) two IR events in both cultivars.
The AS of Hsfs has been described in a variety of plant species, such as Medicago sativa, A.thaliana or Oryza sativa56,57 and for different members of this family, including the HS induced HsfA2 in A.thaliana.12,58 Many of these Hsf splice variants were shown to be subjected to NMD; however, for the HsfA2-III splice variant of A.thaliana a truncated translation product was detected.57 In contrast, for Hsps less about AS events is known. But for instance in human and rat, splice variants of heat-shock cognate protein 70 were identified, which are translated into truncated proteins lacking about 150 amino acid residues of the full-length protein.59,60 In addition, AS of mouse HSP47 was shown to be induced by heat shock leading to an extension of the 5′UTR by 169 nucleotides and by this to higher translation efficiency.61 The HS accumulated IR events we observed for the different constitutively expressed and HS induced Hsps were all introducing PTCs. One possible reason might be that although we excluded a global dependency of AS detection from expression of the underlying gene (Supplementary Materials S6), some of the IR events and especially the low abundant might be related to noisy splicing introduced by errors in the splicing process instead of a targeted process.62–64 On the other hand, the PTCs could lead to degradation of Hsp mRNAs by the NMD pathway if the mRNAs are NMD-sensitive.22 As we used pooled samples extracted directly after HS and after a recovery period, the IR events observed for the constitutively expressed Hsps might be associated to the sample extracted directly after HS, where transcript levels of these Hsps should be reduced with the purpose of a higher translation rate of HS induced Hsps.65 Similarly, IR events of HS induced Hsps might be related to the recovery period, where HS induced Hsps are no longer required and reduced by degradation of the respective PTC containing mRNAs.66 This mechanism has been described in mammals where targeted introduction of PTCs by retained introns led to degradation of transcripts not required for the physiology of the cell.53
4.3. HS-related AS results in alteration of protein sequences
By reference based transcriptome assembly we detected various isoforms of pre-selected genes containing AS events accumulated or reduced dependent on HS compared with the annotation of tomato cultivar Heinz (ITAG 2.4).38 Within the 141 HS-specific and the 22 with HS-repressed transcripts, we observed an AS-induced length reduction of the CDS mainly due to PTC occurrence (Fig. 5). Although most of these transcripts carry classical NMD features, it was recently shown that at least for some genes IR transcripts possessing NMD features are not sensitive to NMD.22 This suggests that at least 36% of HS-specific transcripts (Fig. 5, only IR) are putatively translated and not degraded. Among these proteins translated, six show a loss of at least one functional domain. The lack of one or more functional domains was shown to be a negative regulatory mechanism, where truncated isoforms may lack important domains but still form nonfunctional dimers that may compete with functional dimers.24,67 This negative regulation was for example shown for different transcription factors68 and in our study we could also detect two transcription initiation factors (Solyc10g079360 and Solyc04g078050) having a putative competing isoform under HS. In addition, the observed C-terminal truncation of protein domains has been described recently.69 Such terminal domain truncation, also known as domain atrophy, may be introduced by PTCs but contrary to the expectation leaves the domain often functional, albeit reduced. Next to the proteins showing AS-induced domain loss, 26% of HS-specific transcripts were perfectly spliced (Fig. 5; perfectly). This means that under HS the normal protein function is maintained, whereas under CO either an alternative functionality of the protein or a degradation of the transcripts may occur.
Up to now, the main focus of studies dealing with cellular response to HS was focused on genes showing expression changes upon HS70,71 as these are likely candidates with important roles in stress-regulatory networks.68 However, this procedure may have underestimated the importance of genes with constant expression. These genes may not contribute to the cellular stress response by expression changes but AS transcripts, which offers a new layer of stress response. To verify whether such a regulation exists, we first extracted HS-specific transcripts (Table 4) and divided them in two pools comprised of (i) 14 transcripts containing an AS event inducing domain gain and/or loss (Supplementary Materials S11, red) and (ii) 37 perfectly spliced transcripts (Supplementary Materials S11, green). Second, expression levels of the underlying genes were compared for HS and CO of both cultivars and by this constantly expressed genes under both conditions could be identified (Supplementary Materials S11). The general tendency of expression levels was comparable between CO and HS for both pools supporting evidence for the proposed new layer of stress response (Fig. 6). Beside HSR regulated by up- or down-regulation of specific genes (Fig. 6; 1 and 3) we propose HSR by AS in the case of constant gene expression (Fig. 6; 2). The AS driven HSR can take place either under HS (Fig. 6; 2A) leading to a competing isoform or under CO (Fig. 6; 2B), at which the constitutively spliced isoform is expressed under HS. The transcript levels of selected genes supporting this new regulatory concept were verified by qPCR (Supplementary Materials S12), which revealed constant expression between CO and HS samples for both Moneymaker and Red Setter.
Figure 6.

New putative regulatory pathways via AS due to HSR in pollen. The model shows two stages of regulating HSR: I. Transcript levels may be altered by reduction (1) or induction (3) of expression.; II. Independent of expression (2). For II. the HSR is regulated by AS creating isoforms leading to reduced/altered functionality. The concept of this regulation for HS (2A; Solyc09g074680; Solyc11g069490) and CO (2B; Solyc09g064920; Solyc06g069370) dependent AS is shown. Gene models for the annotated ITAG2.4 isoform and the HS or CO specific isoforms are provided as well as the domain architecture of the translated protein.
To exemplify regulation under HS by AS, we chose two candidates of the first pool (i), namely the ubiquitin ligase complex subunit Cullin 1B (Solyc09g074680) and the serine/threonine-protein phosphatase 6 regulatory subunit 3 (PP6R3, Solyc11g069490). Cullin proteins are evolutionary conserved proteins acting as scaffold in multi-subunit CRL complexes required for ubiquitin-dependent protein degradation.72 Cullin proteins are composed of a conserved cullin domain and a neddylation site.73 Conjugation of the ubiquitine-like molecule Nedd8 to the neddylation site leads to activation of the CRL complex.72 In addition, CRL complexes were shown to be involved in plant abiotic stress pathways including first indications for their involvement in temperature stress response.74 The isoform only detected under HS revealed retention of intron 17 leading to a PTC and a C-terminal truncation of 16.47%. The truncation comprised the entire neddylation site (Cullin_Nedd8) and a small portion of the cullin domain (Cullin). A possible effect of the truncation might be that the CRL complex is not fully assembled or remains in an inactive state due to the inability for neddylation. This assumption is supported by results in Xaneopus laevis, where C-terminally truncated Cullin-1 constructs were shown to have negative effects on ubiquitination by titrating out endogeneous components into inactive complexes, leading to an increase of degradation targets.75 The second protein exemplifying response by AS under HS is PP6R3. PP6R3 forms together with the catalytic subunit PP6c and an ankyrin repeat-containing protein the heterotrimeric PP6 holoenzyme complex, which is required for regulation of different signaling pathways.76 The involvement of serine/threonine phosphatases in HSR was shown by overexpression of the AtPP5, which led to enhanced heat shock resistance of A.thaliana.77 The HS-specific PP6R3 isoform showed retention of intron 12 comprising a PTC leading to a truncation including about 51% of the SAPS domain. If the truncated SAPS domain is still functional, like in the case of domain atrophy,69 the binding to PP6c might still be feasible, as the SAPS domain is sufficient for binding. However, the interaction of PP6R3 with the ankyrin-repeat subunit would not be feasible anymore as the C-terminal region is required for interaction.78 This may suggest that like in the case of Cullin 1B, the observed PP6R3 isoform leads to inactive PP6 complexes having a contrary effect on signal transduction. In the end, these results exemplify our proposed model, at which the transcript abundance is constant but different AS isoforms serve as alternative layer of HSR.
Supplementary Material
Acknowledgements
We would like to thank Daniela Bublak for excellent technical assistance and members of the SPOT-ITN consortium for helpful comments during the manuscript preparation. We are grateful to Holger Schranz and Gerald Kircher for maintenance of plants in the greenhouse. We acknowledge the contribution of all SPOT-ITN members in discussion of the results. The article is dedicated to two of our wonderful colleagues that have contributed to the field of HSR of tomato of the last four decades, a work that has inspired us in our research: Prof Dr Lutz Nover and Dr Klaus-Dieter Scharf.
Conflict of interests
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
The work was supported by grants of the Deutsche Forschungsgemeinschaft (Schleiff: CRC-SFB902 B9; EXC-Cluster of excellence macromolecular complexes) and from the EU/Marie Curie (project: Pollen thermotolerance and crop fertility, acronym: SPOT-ITN, grant agreement No. 289220).
References
- 1. Finka A., Mattoo R. U., Goloubinoff P.. 2011, Meta-analysis of heat- and chemically upregulated chaperone genes in plant and human cells. Cell Stress Chaperones, 16, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fragkostefanakis S., Simm S., Paul P., Bublak D., Scharf K. D., Schleiff E.. 2015, Chaperone network composition in Solanum lycopersicum explored by transcriptome profiling and microarray meta-analysis. Plant Cell Environ., 38, 693–709. [DOI] [PubMed] [Google Scholar]
- 3. Jung K.H., Ko H.J., Nguyen M.X., Kim S.R., Ronald P., An G.. 2012, Genome-wide identification and analysis of early heat stress responsive genes in rice. J. Plant Biol., 55, 10. [Google Scholar]
- 4. Larkindale J., Hall J.D., Knight M.R., Vierling E.. 2005, Heat stress phenotypes of Arabidopsis mutants implicate multiple signaling pathways in the acquisition of thermotolerance. Plant Physiol., 138, 882–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Y.F., Wang Y., Tang Y., Kakani V.G., Mahalingam R.. 2013, Transcriptome analysis of heat stress response in switchgrass (Panicum virgatum L.). BMC Plant Biol., 13, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarkar N.K., Kim Y.K., Grover A.. 2014, Coexpression network analysis associated with call of rice seedlings for encountering heat stress. Plant Mol. Biol., 84, 125–143. [DOI] [PubMed] [Google Scholar]
- 7. Zhou Y., Chen H., Chu P., et al. 2012, NnHSP17.5, a cytosolic class II small heat shock protein gene from Nelumbo nucifera, contributes to seed germination vigor and seedling thermotolerance in transgenic Arabidopsis. Plant Cell Rep., 31, 379–89. [DOI] [PubMed] [Google Scholar]
- 8. Sung D. Y., Guy C. L.. 2003, Physiological and molecular assessment of altered expression of Hsc70-1 in Arabidopsis. Evidence for pleiotropic consequences. Plant Physiol., 132, 979–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Montero-Barrientos M., Hermosa R., Cardoza R.E., Gutierrez S., Nicolas C., Monte E.. 2010, Transgenic expression of the Trichoderma harzianum hsp70 gene increases Arabidopsis resistance to heat and other abiotic stresses. J. Plant Physiol., 167, 659–65. [DOI] [PubMed] [Google Scholar]
- 10. Cazale A. C., Clement M., Chiarenza S., et al. 2009, Altered expression of cytosolic/nuclear HSC70-1 molecular chaperone affects development and abiotic stress tolerance in Arabidopsis thaliana. J. Exp. Bot., 60, 2653–64. [DOI] [PubMed] [Google Scholar]
- 11. Scharf K. D., Berberich T., Ebersberger I., Nover L.. 2012, The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim. Biophys. Acta, 1819, 104–19. [DOI] [PubMed] [Google Scholar]
- 12. Guerra D., Crosatti C., Khoshro H.H., Mastrangelo A.M., Mica E., Mazzucotelli E.. 2015, Post-transcriptional and post-translational regulations of drought and heat response in plants: a spider’s web of mechanisms. Front. Plant Science, 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kornblihtt A.R., Schor I.E., Allo M., Dujardin G., Petrillo E., Munoz M.J.. 2013, Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol., 14, 153–65. [DOI] [PubMed] [Google Scholar]
- 14. Moore M. J., Proudfoot N. J.. 2009, Pre-mRNA processing reaches back to transcription and ahead to translation. Cell, 136, 688–700. [DOI] [PubMed] [Google Scholar]
- 15. Staiger D., Brown J. W.. 2013, Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell, 25, 3640–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reddy A.S. 2007, Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol., 58, 267–94. [DOI] [PubMed] [Google Scholar]
- 17. Wang Z., Burge C.B.. 2008, Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA, 14, 802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang C. Y., Lin W. D., Tu S. L.. 2014, Genome-wide analysis of heat-sensitive alternative splicing in Physcomitrella patens. Plant Physiol., 165, 826–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun Y., Xiao H.. 2015, Identification of alternative splicing events by RNA sequencing in early growth tomato fruits. BMC Genomics, 16, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu R., Loraine A.E., Dickerson J.A.. 2014, Comparisons of computational methods for differential alternative splicing detection using RNA-seq in plant systems. BMC Bioinform., 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Syed N.H., Kalyna M., Marquez Y., Barta A., Brown J.W.. 2012, Alternative splicing in plants–coming of age. Trends Plant Sci., 17, 616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kalyna M., Simpson C.G., Syed N.H., et al. 2012, Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res., 40, 2454–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mastrangelo A.M., Marone D., Laido G., De Leonardis A.M., De Vita P.. 2012, Alternative splicing: enhancing ability to cope with stress via transcriptome plasticity. Plant Sci., 185-186, 40–49. [DOI] [PubMed] [Google Scholar]
- 24. Reddy A.S., Marquez Y., Kalyna M., Barta A.. 2013, Complexity of the alternative splicing landscape in plants. Plant Cell, 25, 3657–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Filichkin S.A., Priest H.D., Givan S.A., et al. 2010, Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res., 20, 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ner-Gaon H., Halachmi R., Savaldi-Goldstein S., Rubin E., Ophir R., Fluhr R.. 2004, Intron retention is a major phenomenon in alternative splicing in Arabidopsis. Plant J., 39, 877–85. [DOI] [PubMed] [Google Scholar]
- 27. Min X.J., Powell B., Braessler J., Meinken J., Yu F., Sablok G.. 2015, Genome-wide cataloging and analysis of alternatively spliced genes in cereal crops. BMC Genomics, 16, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loraine A.E., McCormick S., Estrada A., Patel K., Qin P.. 2013, RNA-seq of arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol., 162, 1092–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dai S., Li L., Chen T., Chong K., Xue Y., Wang T.. 2006, Proteomic analyses of Oryza sativa mature pollen reveal novel proteins associated with pollen germination and tube growth. Proteomics, 6, 2504–29. [DOI] [PubMed] [Google Scholar]
- 30. Bokszczanin K.L., Solanaceae Pollen Thermotolerance Initial Training Network, C. Fragkostefanakis S. 2013, Perspectives on deciphering mechanisms underlying plant heat stress response and thermotolerance. Front. Plant Sci., 4, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giorno F., Wolters-Arts M., Mariani C., Rieu I.. 2013, Ensuring reproduction at high temperatures: the heat stress response during anther and pollen development. Plants, 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pressman E., Peet M.M., Pharr D.M.. 2002, The effect of heat stress on tomato pollen characteristics is associated with changes in carbohydrate concentration in the developing anthers. Ann. Bot., 90, 631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rutley N., Twell D.. 2015, A decade of pollen transcriptomics. Plant Reprod., 28, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fragkostefanakis S., Mesihovic A., Simm S., et al. 2016, HsfA2 controls the activity of developmentally and stress-regulated heat stress protection mechanisms in tomato male reproductive tissues. Plant Physiol., 170, 2461–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paul P., Chaturvedi P., Selymesi M., et al. 2016, The membrane proteome of male gametophyte in Solanum lycopersicum. J. Proteomics, 131, 48–60. [DOI] [PubMed] [Google Scholar]
- 36. Bombarely A., Menda N., Tecle I.Y., et al. 2011, The Sol Genomics Network (solgenomics.net): growing tomatoes using Perl. Nucleic Acids Res., 39, D1149–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. 2013, TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tomato Genome C. 2012, The tomato genome sequence provides insights into fleshy fruit evolution. Nature, 485, 635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anders S., Pyl P.T., Huber W.. 2015, HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Livak K.J., Schmittgen T.D.. 2001, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402–8. [DOI] [PubMed] [Google Scholar]
- 41. Ashburner M., Ball C.A., Blake J.A., et al. 2000, Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet., 25, 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones P., Binns D., Chang H.Y., et al. 2014, InterProScan 5: genome-scale protein function classification. Bioinformatics, 30, 1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shannon P., Markiel A., Ozier O., et al. 2003, Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res., 13, 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maere S., Heymans K., Kuiper M.. 2005, BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics, 21, 3448–9. [DOI] [PubMed] [Google Scholar]
- 45. Benjamini Y., Hochberg Y.. 1995, Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Methods, 57, 289–300. [Google Scholar]
- 46. Trapnell C., Roberts A., Goff L., et al. 2012, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc., 7, 562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Finn R.D., Bateman A., Clements J., et al. 2014, Pfam: the protein families database. Nucleic Acids Res., 42, D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson L.S., Eddy S.R., Portugaly E.. 2010, Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics, 11, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Edgar R., Domrachev M., Lash A.E.. 2002, Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Higashi Y., Okazaki Y., Myouga F., Shinozaki K., Saito K.. 2015, Landscape of the lipidome and transcriptome under heat stress in Arabidopsis thaliana. Sci. Rep., 5, 10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thatcher S.R., Zhou W., Leonard A., et al. 2014, Genome-wide analysis of alternative splicing in Zea mays: landscape and genetic regulation. Plant Cell, 26, 3472–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen H., Chen X., Chen D., Li J., Zhang Y., Wang A.. 2015, A comparison of the low temperature transcriptomes of two tomato genotypes that differ in freezing tolerance: Solanum lycopersicum and Solanum habrochaites. BMC Plant Biol., 15, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Braunschweig U., Barbosa-Morais N.L., Pan Q., et al. 2014, Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res., 24, 1774–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bond U. 2006, Stressed out! Effects of environmental stress on mRNA metabolism. FEMS Yeast Res., 6, 160–70. [DOI] [PubMed] [Google Scholar]
- 55. Utans U., Behrens S.E., Luhrmann R., Kole R., Kramer A.. 1992, A splicing factor that is inactivated during invivo heat-shock is functionally equivalent to the [U4/U6.U5] Triple Snrnp-Specific Proteins. Genes Dev., 6, 631–41. [DOI] [PubMed] [Google Scholar]
- 56. Cheng Q., Zhou Y., Liu Z., et al. 2015, An alternatively spliced heat shock transcription factor, OsHSFA2dI, functions in the heat stress-induced unfolded protein response in rice. Plant Biol., 17, 419–29. [DOI] [PubMed] [Google Scholar]
- 57. Liu J., Sun N., Liu M., et al. 2013, An autoregulatory loop controlling Arabidopsis HsfA2 expression: role of heat shock-induced alternative splicing. Plant Physiol., 162, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sugio A., Dreos R., Aparicio F., Maule A.J.. 2009, The Cytosolic protein response as a subcomponent of the wider heat shock response in Arabidopsis. Plant Cell, 21, 642–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tsukahara F., Yoshioka T., Muraki T.. 2000, Molecular and functional characterization of HSC54, a novel variant of human heat-shock cognate protein 70. Mol. Pharmacol., 58, 1257–263. [DOI] [PubMed] [Google Scholar]
- 60. Yamada M., Yamada M., Kiuchi Y., et al. 1999, Identification of a novel splice variant of heat shock cognate protein 70 after chronic antidepressant treatment in rat frontal cortex. Biochem. Biophys. Res. Commun., 261, 541–5. [DOI] [PubMed] [Google Scholar]
- 61. Takechi H., Hosokawa N., Hirayoshi K., Nagata K.. 1994, Alternative 5’ splice site selection induced by heat shock. Mol. Cell. Biol., 14, 567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dvinge H., Bradley R.K.. 2015, Widespread intron retention diversifies most cancer transcriptomes. Genome Med., 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pickrell J.K., Pai A.A., Gilad Y., Pritchard J.K.. 2010, Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet., 6, e1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Warnecke T., Hurst L.D.. 2011, Error prevention and mitigation as forces in the evolution of genes and genomes. Nat. Rev. Genet., 12, 875–81. [DOI] [PubMed] [Google Scholar]
- 65. Šamaj J., Thelen J.J.. 2007, Plant Proteomics. Springer: Berlin; New York. [Google Scholar]
- 66. Volkov R.A., Panchuk II, Schoffl F.. 2003, Heat-stress-dependency and developmental modulation of gene expression: the potential of house-keeping genes as internal standards in mRNA expression profiling using real-time RT-PCR. J. Exp. Bot., 54, 2343–9. [DOI] [PubMed] [Google Scholar]
- 67. Staudt A.C., Wenkel S.. 2011, Regulation of protein function by ‘microProteins’. EMBO Rep., 12, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seo P.J., Hong S.Y., Kim S.G., Park C.M.. 2011, Competitive inhibition of transcription factors by small interfering peptides. Trends Plant Sci., 16, 541–9. [DOI] [PubMed] [Google Scholar]
- 69. Prakash A., Bateman A.. 2015, Domain atrophy creates rare cases of functional partial protein domains. Genome Biol., 16, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Swindell W.R., Huebner M., Weber A.P.. 2007, Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics, 8, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bita C.E., Zenoni S., Vriezen W.H., Mariani C., Pezzotti M., Gerats T.. 2011, Temperature stress differentially modulates transcription in meiotic anthers of heat-tolerant and heat-sensitive tomato plants. BMC Genomics, 12, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sarikas A., Hartmann T., Pan Z.Q.. 2011, The cullin protein family. Genome Biol., 12, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pan Z.Q., Kentsis A., Dias D.C., Yamoah K., Wu K.. 2004, Nedd8 on cullin: building an expressway to protein destruction. Oncogene, 23, 1985–97. [DOI] [PubMed] [Google Scholar]
- 74. Guo L., Nezames C.D., Sheng L., Deng X., Wei N.. 2013, Cullin-RING ubiquitin ligase family in plant abiotic stress pathways(F). J. Integr. Plant Biol., 55, 21–30. [DOI] [PubMed] [Google Scholar]
- 75. Voigt J., Papalopulu N.. 2006, A dominant-negative form of the E3 ubiquitin ligase Cullin-1 disrupts the correct allocation of cell fate in the neural crest lineage. Development, 133, 559–68. [DOI] [PubMed] [Google Scholar]
- 76. Dai M., Terzaghi W., Wang H.. 2013, Multifaceted roles of Arabidopsis PP6 phosphatase in regulating cellular signaling and plant development. Plant Signal. Behav., 8, e22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Park J.H., Kim W.Y., Chae H.B., Kim M.G., Lee S.Y.. 2012, Serine/threonine protein phosphatase 5 (PP5) interacts with substrate under heat stress conditions and forms protein complex in Arabidopsis. Plant Signal. Behav., 7, 535–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guergnon J., Derewenda U., Edelson J.R., Brautigan D.L.. 2009, Mapping of protein phosphatase-6 association with its SAPS domain regulatory subunit using a model of helical repeats. BMC Biochem., 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.