

Abstract
The oocyte-to-embryo transition (OET) transforms a differentiated gamete into pluripotent blastomeres. The accompanying maternal-zygotic RNA exchange involves remodeling of the long non-coding RNA (lncRNA) pool. Here, we used next generation sequencing and de novo transcript assembly to define the core population of 1,600 lncRNAs expressed during the OET (lncRNAs). Relative to mRNAs, OET lncRNAs were less expressed and had shorter transcripts, mainly due to fewer exons and shorter 5′ terminal exons. Approximately half of OET lncRNA promoters originated in retrotransposons suggesting their recent emergence. Except for a small group of ubiquitous lncRNAs, maternal and zygotic lncRNAs formed two distinct populations. The bulk of maternal lncRNAs was degraded before the zygotic genome activation. Interestingly, maternal lncRNAs seemed to undergo cytoplasmic polyadenylation observed for dormant mRNAs. We also identified lncRNAs giving rise to trans-acting short interfering RNAs, which represent a novel lncRNA category. Altogether, we defined the core OET lncRNA transcriptome and characterized its remodeling during early development. Our results are consistent with the notion that rapidly evolving lncRNAs constitute signatures of cells-of-origin while a minority plays an active role in control of gene expression across OET. Our data presented here provide an excellent source for further OET lncRNA studies.
Keywords: lncRNA, oocyte, zygote, polyadenylation, endo-siRNA
1. Introduction
The oocyte-to-embryo transition (OET), the transformation of a differentiated oocyte into a developing embryo, involves massive reprogramming of gene expression where zygotic genome activation (ZGA) initiates production of zygotic mRNA to replace maternal RNAs in control of development. Although changes of mRNA and small RNA populations during OET were characterized in a considerable detail (reviewed in1), much less is known about composition and temporal changes of spliced long non-coding RNAs (lncRNAs). LncRNAs (reviewed for example in2–5) add another layer to the transcriptome complexity. LncRNAs are an arbitrary category adopted for spliced transcripts not encoding proteins that are longer than 200 nucleotides (nt). LncRNAs represent an assorted group of RNAs implicated in transcriptional, post-transcriptional, translational, and epigenetic regulations or, importantly, without any apparent functions. LncRNAs evolve rapidly, showing little if any sequence conservation.6 It is assumed that a relatively minor fraction of these transcripts is functional while the rest might represent transcriptional noise and/or lncRNAs that have appeared recently in evolution and have not acquired a function (reviewed in2). However, specific lncRNAs are functionally important for different processes, which include the maintenance and induction of stem cell pluripotency (reviewed in7).
Next generation sequencing (NGS)-based studies provided partial insights into some aspects of lncRNA biology during the mammalian OET. Upon single-cell RNA profiling of human preimplantation embryos, Yan et al.8 reported 2,733 novel expressed lncRNAs. Zhang et al.9 used single-cell SOLiD NGS data from OET stages and reported 5,563 novel lncRNAs. Hamazaki et al.10 studied a specific lncRNA group termed promoter associated non-coding RNAs (pancRNAs) in ovulated oocytes and two-cell zygotes. So far, the most detailed analysis of OET lncRNAs has been provided by Veselovska et al.,11 who produced de novo transcriptome assembly that included lncRNAs. However, their main research focus was the contribution of transcription to the DNA methylation landscape and not a thorough annotation and analysis of lncRNAs.
Understanding the composition of maternal and zygotic non-coding RNA pools is pre-requisite for understanding their biological roles during OET. In this study, we sought to provide a highly reliable set of de novo assembled lncRNAs present during OET (referred to as OET lncRNAs hereafter) and perform its characterization in terms of structure and expression. Accordingly, we identified, annotated, and characterized 1,600 OET lncRNA loci, including their transcriptional and post-transcriptional temporal dynamics. OET lncRNAs exhibited typical features of lncRNAs: lower expression levels than mRNAs, highly variable splicing, and restricted expression. Remarkably, the OET lncRNA expression largely falls into mutually exclusive maternal and zygotic expression patterns but rarely into the maternal-zygotic expression, which is common for mRNAs. Finally, we produced CRISPR-mediated knockouts of two maternal conserved lncRNAs without an effect on fertility.
2. Methods
2.1. RNA extraction, preparation of the NGS library and sequencing
Total RNA was extracted from 3,000 fully grown germinal vesicle (GV)-intact oocytes obtained from C57BL6/J mice, respectively, using Isogen (Nippon Gene, Tokyo, Japan), according to the manufacturer’s instructions. PolyA RNA was isolated by using mRNA purification kit (Invitrogen, Carlsbad, CA; cat no. 610.06). High-throughput sequencing of size-selected RNA (>200 nt) was performed using Genome Analyzer IIx (Illumina) and 76-nt paired-end-sequencing reads as described previously in.12 The complete set of NGS data is available in the Array Express database under accession IDs E-MTAB-2950 and E-MTAB-4775.
2.2. Analysis of lncRNA expression in oocytes and early embryos by real-time PCR
Oocytes and early embryos were obtained from C57Bl/6 mice as described previously in.13,14 Resumption of meiosis during collection of GV oocytes was prevented with 0.2 mM 3-isobutyl-1-methyl-xanthine (IBMX, Sigma). RNA from a chosen number of oocytes or early embryos was released upon incubation in water with RNase inhibitor for 5 min at 85 °C. RNA was reverse-transcribed using RevertAid First Strand cDNA Synthesis Kit (Fermentas). Maxima SYBR Green qPCR Master Mix (Fermentas) was used for qPCR. The primers and PCR conditions are shown in the Supplementary Table S5.
2.3. Production of lncRNA knockout models
LncRNA knockout models were produced in the Transgenic Unit of the Institute of Molecular Genetics ASCR, Czech Centre for Phenogenomics using Cas9-mediated deletion of lncRNA promoters (Supplementary Figs. S7 and S8).15,16 All animal experiments were approved by the Institutional Animal Use and Care Committees (project number 58-2015) and were carried out in accordance with the law.
Sequences of guide RNAs are listed in the Table S5. To produce guide RNAs, synthetic 128 nt guide RNA templates including T7 promoter, 18 nt sgRNA and tracrRNA sequences were amplified using T7 and TracrRNA primers (Supplementary Table S5). Guide RNAs were produced in vitro using the Ambion mMESSAGE mMACHINE T7 Transcription Kit, and purified using the mirPremier microRNA Isolation Kit (Sigma). The Cas9 mRNA was synthesized from pSp Cas9-puro plasmid using Ambion mMESSAGE mMACHINE T7 Transcription Kit, and purified using the Qiagen RNasy mini kit. A sample for microinjection was prepared by mixing two guide RNAs in ultra-pure water at a concentration of 25 ng/µl for each one together with Cas9 RNA (100 ng/µl) . Five picoliters of the microinjection mixture were injected into male pronuclei of C57Bl/6 zygotes and transferred into pseudopregnant recipient mice. PCR genotyping was performed on tail biopsies from 4 weeks-old animals. Primers are listed in Supplementary Table S5.
2.4. Bioinformatics analyses
2.4.1. Mapping of Illumina NGS reads on the mouse genome
Mapping of NGS data was performed as described previously in.12 Briefly, adapters were removed using the Trimmomatic software (doi: 10.1093/bioinformatics/btu170). The filtered reads were mapped onto the mm9/NCBI37 version of the mouse genome using the STAR mapper (doi: 10.1093/bioinformatics/bts635) and the genome index was constructed with the addition of the mm9 Ensembl gene annotation, downloaded on 20 September 2013 from the Ensembl database. The dynamic ranges of read counts permitted us to use counts per million normalization for downstream analyses as they did not vary significantly across experiments.12 Data were visualized in the UCSC browser by constructing bigWig tracks using the Bedtools software (10.1093/bioinformatics/btq033).
2.4.2. Transcript model assembly from NGS data
For assembling lncRNA transcript models, we used 76-nt paired end (76PE) non-directional NGS data with depths 33–58 × 106 sequence reads/sample from oocytes and early embryos (Supplementary Table S1). Except of fully grown GV-intact oocytes data, other NGS datasets were published.12
For transcript model assembly, we combined total RNA NGS datasets (Supplementary Table S1) into three sets as follows: (i) the maternal set: fully grown GV oocyte and MII egg NGS data, (ii) the ZGA (zygotic) set: two- and four-cell NGS data, and (iii) the late preimplantation (embryonic) set: morula and blastocyst NGS data. Transcript model assembly was performed for each set separately to reduce artifacts from degraded maternal RNAs and to achieve accurate assembly of overlapping sense and antisense transcripts. We tested several combinations of data pooling including pooling with published shorter single-end read NGS data (e.g.17,18). However, the aforementioned grouping of 76PE samples into three independent sets yielded by far the best results in assembling transcript models of a diagnostic set of 20 lncRNA loci. Although addition of published 35SE sets would increase the sequencing depth and reveal additional lncRNAs, it also introduced numerous artifacts into transcript models when compared with transcript assembly based on 76PE sets, which yielded the best exon-intron junction prediction.
To build transcript models from NGS data, we used Scripture,19 which performed better than Cufflinks or Stringtie in assembling the aforementioned diagnostic set of annotated and novel lncRNAs (data not shown). Transcript models generated by Scripture were refined to eliminate various artifacts. We filled introns <20 nt length and removed transcript models containing introns >250 kb, which were typically repeat-derived artifacts and disturbed assembly of transcript model clusters. We also removed single-exon transcripts and transcripts shorter than 200 nucleotides.
Refined transcript models in each developmental set were then used to build transcript clusters where each lncRNA cluster accommodated all transcript models with the same orientation sharing at least one splice site. Thus, a lncRNA cluster represents a locus containing a group of exons found in clustered transcript models. Transcript models in clusters were subsequently analysed for coding potential by CPAT20 (Supplementary Fig. S1). Building clusters and CPAT pre-filtering performed separately on the three sets was chosen because it simplified data management, allowing to work in parallel with smaller volumes of developmentally relevant data. All clusters containing any CPAT positive transcript model were removed from the lncRNA annotation in order to eliminate transcript model artifacts derived from poorly assembled mRNAs. Although one promoter could produce a shorter lncRNA and a longer mRNA (Supplementary Fig. S2), we decided to remove entire clusters ‘contaminated’ with any CPAT-positive transcript models. In any case, lncRNAs in such clusters overlap with mRNAs in sense, which represented an lncRNA category omitted from our annotation and further analysis.
Next, we merged clusters containing the same transcript models from the three NGS sets. We first merged pools from the maternal lncRNA NGS set with the ZGA lncRNAs NGS set while we removed partial ZGA transcript models matching maternal transcript models as they typically came from poor assembly of degraded maternal transcripts. Then we added transcript models from late pre-implantation NGS clusters and generated a single non-redundant set of transcript model clusters for final refining.
Transcript models in each cluster were further refined by revising terminal exon predictions since we noticed that Scripture tends to produce truncated terminal exon variants despite an apparent support from NGS or expressed sequence tag (EST) data. Thus, when Scripture predicted terminal exon variants sharing the same splice donor or splice acceptor, we calculated read densities (FPKM - fragments per kilobase of exon per million fragments mapped) for those exon variants and if they did not differ >2-fold, we collapsed shorter exon variants to retain only the longer one as the terminal exon for transcript models. We also refined predicted transcript models using mm9-annotated ESTs, which originated from oocytes and early embryos. We compared the annotated exon-intron structure of these ESTs with our transcript models and annotated lncRNAs. Truncated/partial transcript models were extended according to ESTs if both the ESTs and previously annotated lncRNAs supported such extensions. Further refining included the following filtering steps: transcript models were removed, which contained sequences of known highly abundant non-coding RNAs (e.g. rRNA, U1-U7 RNA, 7SLRNA, SSU-rRNA). We also removed all clusters where >75% of sequences were recognized by Repeatmasker.21 Although some true lncRNAs are made of >75% of repetitive sequences, repeats were causing artifacts yielding spliced transcript models (Supplementary Fig. S4). We also added a filter to eliminate strange artifacts where sequence reads derived from exons of a highly abundant mRNA were assembled into an antisense lncRNA transcript model, not following the canonical splice donor and acceptor sequence rules. Finally, refined transcript models were analysed again for coding potential using CPAT and clusters carrying CPAT positive transcript models were removed (Supplementary Fig. S1).
Then, we individually assessed all clusters which passed filtering until this point but their highest exon maximum FPKM in any of the developmental stages was <1 and we retained those, which seemed to be supported by NGS data display in the UCSC browser (476 transcript clusters were retained). The expression of each cluster was calculated as the maximum exon FPKM, after masking the parts of lncRNA exons, which overlapped exons of protein-coding genes.
The clusters (and their individual transcript models as well) were classified in six categories according to their positions in the genome (Fig. 1C and Supplementary Table S2). The classification system used a simple dichotomic annotation based on boundaries of clusters (transcript models) and their orientations relative to transcripts from protein-coding genes. If there was no overlap, clusters were considered Intergenic. When there was an overlap, we distinguished sense and antisense overlap and we further recognized whether or not an overlap formed between mature (spliced) transcripts. When one lncRNA was associated with more than gene, we opted for classifying such lncRNAs simply as Complex (in total, there were 30 such loci). Clusters from the two sense categories were omitted from the further analysis because they contained too many artifacts that were hindering the annotation effort.
Figure 1.

Overview of lncRNA analysis. (A) Samples analysed by 76PE NGS and their grouping for assembling transcript models. (B) Workflow of identification of lncRNA clusters from oocytes and early embryos NGS data. (C) Classification system used for annotating lncRNA transcript models and clusters. (D) Non-annotated lncRNA loci identified in this study. Exons from transcript models from each clusters were compared with data in ENCODE and Refseq databases. Clusters, in which none of the exon-exon junctions from transcript models matched an exon-exon junction annotated in these databases were considered novel. (E) Origin of transcript models in lncRNA clusters. The Venn diagram depicts which set produced transcript models for lncRNA clusters. For example, 19 lncRNA clusters contain transcript models assembled in all three developmental sets while 973 clusters comprise of exclusively oocyte-derived transcript models.
2.4.3. Comparison with published transcript annotations
We considered an lncRNA locus to be supported by a previous annotation9,11 if it shared at least one exon-exon junction with an annotated transcript model. For analysing exon counts and locus lengths, we grouped all the overlapping transcripts for each OET lncRNA locus and calculated the number of unique exons in the transcripts and the length of the transcript group (defined as the distance between the 5′ end of the most upstream 5′ exon in the transcript group to the 3′ end of the most downstream exon in the transcript group). We then compared the number of unique exons and length of the transcript groups to the number of unique exons and the length of our OET lncRNA loci (Supplementary Fig. S3).
2.4.4. Transcriptome analysis of Dicer and AGO2 mutant oocytes
The NGS data from Stein et al.22 were mapped to the mm9 version of the mouse genome using the STAR aligner, with the following parameters: –outFilterMultimapNmax 10, –out FilterMismatch Nover Lmax 0.2, –sjdbScore 2. The genome was indexed, prior to mapping, with the addition of the mm9 Ensembl gene annotation. For each gene we chose the transcript model with the higher number of exons and counted the reads using the SummarizeOverlaps function. The estimated expression levels per transcript were obtained by normalizing for the library size using the sizeFactors from the DESeq2 package, and the transcript width.
2.4.5. Mapping and display of 21-23 nt RNAs from oocytes
Mapping small RNAs from Tam et al.23 was performed as described previously in.24 Data were visualized in the UCSC browser by constructing bigWig tracks using the Bedtools software (10.1093/bioinformatics/btq033).
3. Results and Discussion
3.1. Identification of OET lncRNAs
We built lncRNA annotation from 76PE non-directional NGS of total non-amplified RNA from seven different OET stages in mice (Fig. 1A). Total RNA NGS enabled us to explore the entire lncRNA population including non-polyadenylated lncRNAs. Given the depth of our NGS datasets (∼4–9 × 106 mapped non-rRNA reads, Supplementary Table S1 and Supplementary Fig. S1A), we focused on annotating well-expressed lncRNAs to reduce annotation artifacts emerging when annotating as many loci as possible. The annotation pipeline (Supplementary Fig. 1B) started with grouping NGS data into three biologically relevant datasets representing distinct types of OET transcriptomes: (i) maternal, containing data (∼16.1 × 106 mapped non-rRNA reads) from ovarian GV oocytes and ovulated MII eggs. (ii) ZGA (also referred to as ‘zygotic’)—containing two- and four-cell embryo data (∼10.0 × 106 mapped non-rRNA reads), and (iii) late preimplantation embryo (also referred to as ‘embryonic’)—containing morula and blastocyst data (∼9.3 × 106 mapped non-rRNA reads). The maternal, zygotic, and embryonic datasets were used for a separate transcript model assembly, filtering, and clustering (a cluster is a group of exons from clustered transcript models from one locus). Clusters containing a transcript model with a predicted coding potential were removed. Clusters from the maternal, zygotic, and embryonic datasets were merged and refined (including a manual inspection of ∼1,200 clusters) to produce a non-redundant set of transcript model clusters. In total, we obtained 1,600 lncRNA clusters classified into four categories according to their positions relative to protein-coding genes (Fig. 1C and Supplementary Table S2). 1,142 (71%) lncRNA loci were intergenic, 30 (2%) overlapped with more than one protein-coding gene, 193 (12%) resided within introns, and 235 (15%) contained transcript models antisense overlapping with mRNA exons.
The majority of the 1,600 lncRNA clusters were assembled from the maternal NGS set (Fig. 1D), including 973 clusters assembled exclusively from the maternal set. Around 600 clusters were assembled from the ZGA set (362 clusters exclusively from the ZGA set), while mere 93 lncRNA clusters were assembled from the embryonic set (only 24 of those clusters were exclusively from the embryonic set). This result, which is discordant with the high count of lncRNAs annotated from ESCs,25,26 can be explained by sample heterogeneity and changing RNA content during early development. Abundance of lncRNAs specific for embryonic and extraembryonic lineages in morulae and blastocysts would be diluted by transcriptomes of non-expressing cells. Furthermore, the embryonic set had a lower depth than the maternal set (Supplementary Table S1 and Supplementary Fig. S1) while a blastocyst has ∼ three times more total RNA (∼1.5 ng) than an oocyte (∼0.5 ng).27 Accordingly, a transcript with an identical copy number in the blastocyst and the oocyte has a three times lower FPKM value in the blastocyst. Thus, the same FPKM cut-off value is more stringent when selecting the blastocyst-expressed genes. This would especially affect the analysis of low-level transcripts such as lncRNAs.
Of the 1,600 clusters, 350 (22%) clusters contained exons or transcript models annotated in the ENCODE or Refseq databases (Fig. 1E). A comparison with recently published 19,617 non-coding transcript models11 and 5,563 lncRNA transcripts from 3,492 loci9 showed that 24% (390 lncRNA loci) were not annotated by either of these studies (Supplementary Table S2), 44% were supported exclusively by Veselovska et al.,11 7% exclusively by Zhang et al.,9 and 25% by both datasets. Thus, almost 70% of the clusters overlapped with Veselovska et al. who used a much deeper maternal NGS set and annotated longer transcript models with more splicing variants (Supplementary Fig. S3). In contrast, lncRNA loci by Zhang et al. were shorter and had fewer exons per transcript and per lncRNA locus (Supplementary Fig. S3).
Finally, we examined expression of annotated lncRNA loci (clusters) in other available data: SOLiD NGS data by Park et al., which we used previously in,12,17 and unpublished data by Yu et al. (GSE71257) from GV oocytes, MII eggs, one-, and two-cell stages sequenced on the Illumina HiSeq 2500 platform. We found that only 34 of our lncRNA clusters produced transcript models with expression values <1 FPKM (maximum exon FPKM for each OET lncRNA locus); expression of four clusters was not detected at all in the examined datasets (Supplementary Table S2). Altogether, these data suggest that our OET lncRNA clusters form a high-quality collection of the most expressed maternal and zygotic lncRNAs.
3.2. Structure and expression of OET lncRNAs
OET lncRNA loci were stochastically distributed across the genome (Fig. 2A). The highest density of lncRNA loci was on chromosome 10 (0.98 lncRNA/Mb) while the lowest density on chromosome 17 (0.18 lncRNA/Mb), which contained just 17 lncRNAs (Fig. 2A). These densities significantly differed (Holm corrected t-test P-value < 10−6) from the mean lncRNA density across all chromosomes (0.61 lncRNA/Mb) while protein-coding gene density did not (Holm corrected t-test P-value > 0.05).
Figure 2.

Structural features of OET lncRNAs. (A) Genomic distribution of 1,600 OET lncRNA loci across the mouse genome. The color-coding indicates the highest expression (maternal, GV oocyte or MII egg; zygotic, two- or four-cell stage; late preimplantation, morula or blastocyst). (B) Number of exons in OET lncRNAs; (C) OET lncRNA locus lengths; (D) Median transcript length produced from an OET lncRNA locus; (D) Length distribution of OET lncRNA exons and introns; (F) Distribution of LTR-derived first exon sequences. (B–F) All features of OET lncRNAs (depicted in red) are compared with oocyte mRNA data (depicted in blue). (G) Contribution of retrotransposons to OET lncRNA transcriptional regulation. The graph depicts fractions of 5′ OET lncRNA exons, which contain a given type of a repetitive sequence over the putative transcription start site and 50 bp upstream. (I) Contribution of repetitive sequences to mature (spliced) OET lncRNA sequences.
When compared with maternal mRNAs, loci encoding lncRNAs were shorter and produced shorter transcripts (Fig. 2B–D); this difference could be attributed to a higher number of exons in mRNAs. OET lncRNAs had markedly shorter 5′ exons but lengths of internal and 3′ exons of mRNAs and lncRNAs were much closer to each other (Fig. 2E). Shorter 5′ exons also came from long terminal repeat (LTR) retrotransposons, which gave rise to ∼third of 5′ exons (Fig. 2G and H). The analysis of contribution of repetitive elements to lncRNA genes showed that SINE and LINE elements contributed to mature lncRNA sequences more often than to lncRNA promoters and transcription start sites. In contrast, LTR elements, especially the MaLR class, made a strong contribution to lncRNA promoters (Fig. 2G and H). A similar observation was also made by Veselovska et al.11
The most-expressed 1,600 OET lncRNAs were more than an order of magnitude less expressed than the 1,600 most expressed OET mRNAs (Fig. 3A), which is consistent with low lncRNA expression reported elsewhere.19,25,28–30 It is possible that OET lncRNAs evolved to have lower expression than mRNAs. Lower lncRNA expression could stem from different requirements for functioning than those applied to mRNAs. Alternatively, lower lncRNA expression could reflect a minimal selective pressure on high expression levels of evolving lncRNAs lacking a function. At the same time, highest expressed mRNAs may represent a derived trait, which questions whether the comparison reflects properties of highly expressed mRNAs or an lncRNA feature. If there were no major difference in control of expression between lncRNAs and mRNAs, a random set of 1,600 mRNAs would have expression similar to OET lncRNAs. Thus, we compared expression of 1,600 OET lncRNAs with 1,000 random selections of 1,600 OET mRNAs. We observed that mRNAs generally retained higher expression than lncRNAs while levels of both types of RNAs were essentially the same within the least expressed quartile (Fig. 3A56). Whether the differences in expression levels between lncRNAs and mRNAs reflect lncRNA-specific features in transcriptional or post-transcriptional regulations remains unclear. Given the arbitrary definition and functional heterogeneity of lncRNAs, it is difficult to envision some feature underlying lower lncRNA expression levels except for one: a lack of function. Expression of non-functional lncRNAs would not be maintained and would most likely decline over time due to mutations affecting transcriptional control elements.
Figure 3.
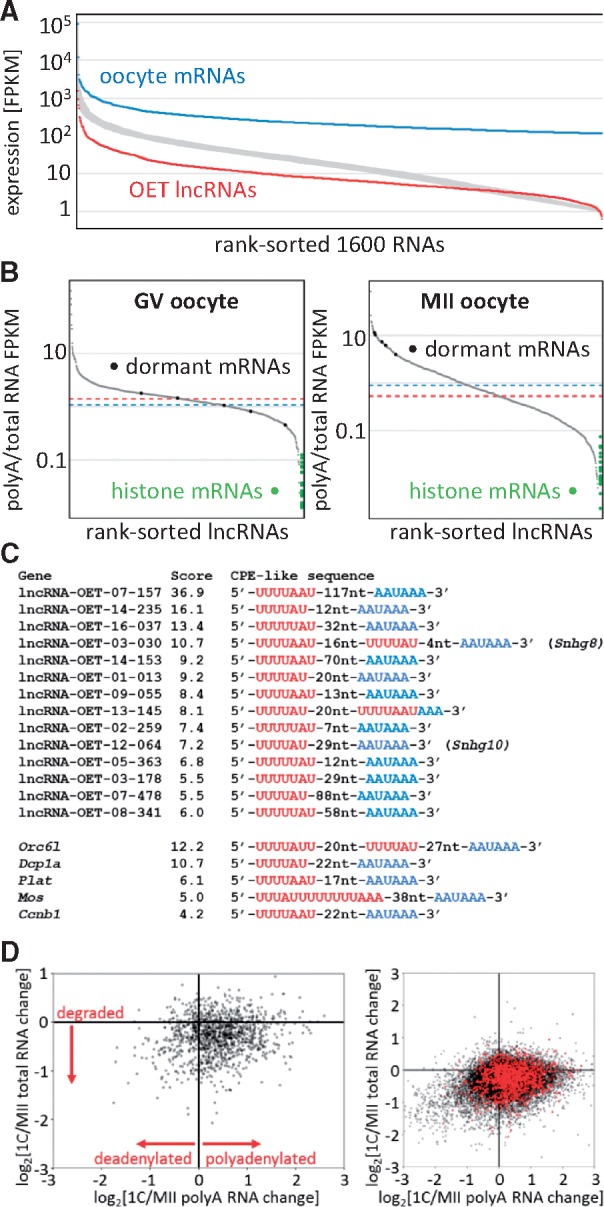
Expression features of OET lncRNAs. (A) Comparison of OET lncRNA expression levels with OET mRNAs. The Y axis shows log10 FPKM expression, the Y axis are rank-sorted RNAs as follows: blue, 1,600 most expressed maternal RNAs; red, 1,600 OET lncRNAs. The broad grey curve represents values for thousand random selections of 1,600 RNAs. The FPKM values were calculated as the maximum exon FPKM per cluster (B) Distribution of polyA scores in GV and MII oocytes. The Y axis depicts the polyA score calculated as the ratio of polyA NGS FPKM/total RNA FPKM. PolyA RNA data for GV oocytes were taken from the literature,31 for MII polyA RNA we used our own data (Supplementary Table S1). The X axis represents rank-sorted OET lncRNAs that had FPKM >0 (1131 for GV and 1273 for MII). Dashed red and blue lines represent median polyA score values for lncRNAs and mRNAs, respectively. Green points on the right site indicate polyA scores of histone mRNAS, black points on the curve indicate the rank of polyA scores of dormant maternal mRNAs. (C) Examples of putative CPE elements found among OET lncRNAs. (D) polyadenylation changes upon fertilization. The Y axis depicts relative changes of gene expression in total RNA upon fertilization (log2[1-cell/MII total RNA FPKM]), the X axis shows relative changes in polyA RNA (log2[1-cell/MII polyA FPKM]). The left scatterplot displays only OET lncRNAs, the right plot shows OET lncRNAs in red superimposed onto mRNAs (black). Each point represents expression of one gene.
3.3. Polyadenylation of OET lncRNAs
Biogenesis of lncRNAs and mRNAs is common—they are spliced polymerase II transcripts whose transcription would utilize the same set of transcription factors. One of the features, by which lncRNAs could differ, is polyadenylation of the 3′ end. Consequently, we examined whether a comparison of total RNA and polyA RNA FPKM values would be indicative of the polyadenylation status (polyA FPKM/total RNA FPKM, for simplicity referred as polyA score, Fig. 3B). GV oocytes and MII eggs are an excellent model system for testing this idea because of two possible internal controls: (i) Replication dependent histone mRNAs carrying at their 3′ ends unique stem loop structures instead of polyA tails (reviewed in32). Presence of these transcripts within polyA-selected RNA would reflect the extent of contamination with non-polyadenylated mRNA. (ii) Dormant maternal mRNAs, translationally repressed mRNAs with short polyA tails stored in the GV oocyte, which are readenylated and translated during meiotic maturation (reviewed in33). Thus, we selected 20 highly expressed replication-dependent histone genes lacking alternative polyadenylated transcript isoforms and five dormant maternal mRNAs, for which the cytoplasmic polyadenylation during meiosis was demonstrated: Mos, Plat, Cyclin B1, Orc6l, and Dcp1a.34–38
The distribution of the polyA score for lncRNAs from GV and MII stages yielded sigmoidal curves with slightly different slopes (Fig. 3B), which were reproduced with mRNA data (Supplementary Fig. S5A). Although the difference in slopes might reflect intrinsic differences of GV and MII transcriptomes, polyA scores accurately reflected the lack of polyadenylation of histone mRNAs and cytoplasmic polyadenylation of dormant maternal mRNAs during meiotic maturation (Fig. 3B and Supplementary S5A). The average polyA score of dormant maternal mRNAs, which was 1.106 in GV oocytes, increased to 7.895 in MII eggs. This difference also manifested as a shift of dormant mRNAs along the polyA score rank (Fig. 3B and Supplementary Fig. S5A). Taken together, the behavior of polyA scores appeared indicative of the polyA status. Based on this strategy, we estimated that only a minority of OET lncRNAs lacked the polyA tail. Since lncRNAs detectable in oocytes and/or two-cell zygotes (>1 FPKM) represented ∼95% of the 1,600 lncRNAs, we added normalized polyA scores (median = 0 and variance = 1) from GV oocytes, MII eggs, one-, two-, and four-cell embryos to lncRNA annotation (Supplementary Table S2). However, given the diverse origin of total RNA and polyA RNA NGS data, polyA scores should be taken as an indication rather than an annotation of the polyadenylation status.
The dynamics of polyA scores of dormant maternal mRNAs raised a question whether similar behavior could also be found among maternal lncRNAs. Remarkably, we identified 91 maternal lncRNAs with expression >1 FPKM whose polyA scores increased more than 5-fold during meiosis. Next, we analysed which of the 91 maternal lncRNAs carried putative cytoplasmic polyadenylation elements (CPEs). CPEs mediate the recruitment of dormant deadenylated mRNAs for translation. A CPE is bound by a CPE-binding protein (CPEB), which recognizes a canonical consensus site 5′-UUUUUAU-3′ art the 3′ end of RNAs. However, CPE variations such as UUUUAU, UUUUUAAU, or UUUUAACA were also reported.39 We found that at least 14 lncRNAs carried a combination of a canonical AAUAAA signal site and a CPE-like motif at their 3′ ends (Fig. 3C). lncRNAs resembling dormant maternal RNAs are remarkable because they suggest that cytoplasmic polyadenylation and dormancy could play a more general role in RNA regulation, i.e. a role that goes beyond translational control of maternal mRNAs. We hypothesize that controlled cytoplasmic polyadenylation during transcriptional quiescence could ‘activate’ stored maternal lncRNAs. Thus, putative dormant maternal lncRNAs represent excellent candidates for further functional studies of lncRNAs functioning between ovulation and ZGA.
Importantly, major changes in cytoplasmic RNA polyadenylation during OET occur also post-fertilization.40,41 In fact, cytoplasmic RNA polyadenylation in fertilized mouse eggs is so extensive that it manifests as an increase in total polyA RNA content.27 A scatter plot of relative changes of lncRNAs in polyA and total RNA NGS sets in MII eggs and one-cell embryos showed a relative enrichment in lncRNA polyadenylation upon fertilization; this was similar to changes observed for mRNAs (Fig. 3D). In contrast to mRNAs, the number of lncRNAs showing a stronger decrease in polyA RNA upon fertilization was minimal. Increased polyadenylation did not seem to be an artifact of our samples, as it also showed when other published data were used (Supplementary Fig. S5B).
Taken together, our data suggest that the bulk of OET lncRNAs are polyadenylated at their 3′ end and that maternal lncRNAs utilize the same cytoplasmic polyadenylation mechanisms as mRNAs. In case of mRNAs, cytoplasmic polyadenylation regulates translation and RNA turnover. Although lncRNAs seem to be recognized by the translation machinery,42,43 their coding capacity is highly restricted. Therefore, the interaction of lncRNAs with cytoplasmic polyadenylation and translation machinery likely regulates their functional availability and stability.
3.4. Expression of OET lncRNAs in other tissues
Since the bulk of the 1,600 OET lncRNAs appeared polyadenylated, we examined their expression across 22 tissues selected from the ENCODE polyA RNA NGS mouse tissue panel (GSE4941744). To increase the specificity of expression analysis, we included only clusters with four or more spliced reads in the tissue panel and expression >4 FPKM in at least one of the tissues. The cut-off 4 FPKM for polyA RNA was used because it is an approximate of 1 FPKM in total RNA NGS from mouse oocytes, where mRNAs make 24% of all reads (Supplementary Fig. S1). Under these filtering conditions, we obtained expression values for 356 clusters (Fig. 4 andSupplementary Table S3). The analysis revealed a small population of ubiquitously expressed lncRNA clusters (28 having expression >4 FPKM in all tissues, Supplementary Table S3). This is consistent with the notion that mammalian lncRNAs typically have a cell type-restricted expression.29 Of the 28 lncRNA clusters ubiquitously expressed >4 FPKM, 26 were annotated; they were from small nucleolar RNA host genes and other lncRNAs, such as Malat, Firre, or Rian. Remarkably, OET lncRNAs were mostly expressed in the testis. Within the tissue panel (which also included the ovary and the placenta), the testis stood out as the tissue that had the highest number of maximum expressions of clusters across tissues (121 clusters, Supplementary Table S4). The testis also yielded the highest number of expressed clusters among the tissues (202 clusters). The ovary ranked second after the testis, having the maximum expression of 19 clusters, while the total number of ovary-expressed clusters was 110 (Supplementary Table S4).
Figure 4.
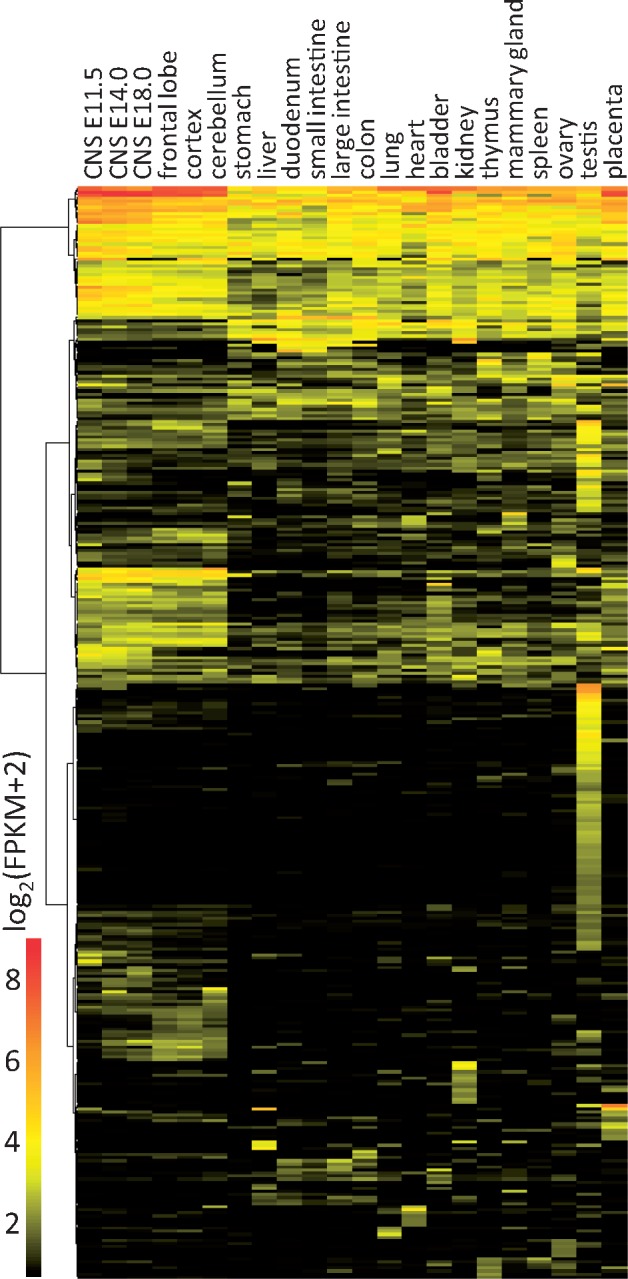
OET lncRNA expression in different tissues. The heatmap displays expression of 356 clusters with expression values >4 FPKM in at least one of 22 tissues selected from the ENCODE polyA RNA NGS mouse tissue panel (GSE49417 [37]).
LncRNAs expressed in the testis and during OET represent an interesting group of germ-line lncRNAs. We took a closer look at the transcriptional control of the 121 lncRNA clusters expressed >4 FPKMs to determine (i) if they are associated with maternal or zygotic expression, and (ii) whether they share the same promoters in the testis and OET stages or whether they have testis-specific promoters not utilized during OET. Most of the 121 lncRNA clusters during OET were highly expressed maternally (93 clusters), while 25 clusters had the highest expression in the zygotic stage and three clusters in the embryonic stage. We examined the promoters of the 121 clusters and found that approximately half of them (58 of 121 examined promoters) controlled the expression in testes and OET stages. In 37 cases, a unique non-repetitive lncRNA promoter functioned in testes, while oocytes or early embryos employed a different unique promoter (19/37) or a retrotransposon-derived one, such as a maternally active MaLR class LTR promoter (12/37). In any case, the 93 lncRNA loci expressed in oocytes and testes are prime candidates for an analysis of lncRNAs with germline-specific functions.
3.5. lncRNA dynamics during OET
Gene expression during OET can be divided into three basic classes reflecting the replacement of maternal RNAs with zygotic transcripts:
(i) maternal—concerns genes active only in oocytes whose transcripts survive until different time-points/stages of OET. They are not replaced with zygotic/embryonic transcripts. These transcripts may be important just for oocyte development or they may function during meiotic maturation or after fertilization, where they might contribute to ZGA and to the initiation of development.
(ii) zygotic—concerns genes expressed during ZGA and not active in the oocyte. Zygotic transcripts may be made just transiently during ZGA or they may remain expressed during early development. These are represented, for example, by transcripts of genes involved in the establishment and maintenance of totipotency.
(iii) maternal-zygotic—concerns genes expressed in both oocytes and early embryos. This category can be exemplified by housekeeping genes. Within this category, maternal gene expression may be much higher than that observed in early embryos or vice versa.
To characterize lncRNA dynamics during OET, we divided lncRNA clusters into the three classes of gene expression described earlier. First, we reduced the complexity of the model system by stage grouping (as it was used for transcript model assembly) into three basic expression states: maternal (M), zygotic (Z), and embryonic (E). The M level was calculated as an average lncRNA level in GV and MII oocytes; it represented lncRNA expression before fertilization. The Z level was calculated as an average lncRNA level in two- and four-cell stages; it represented the transitive period of gene expression during ZGA. The E level was calculated as an average lncRNA level in morulae and blastocysts; it represented gene expression at a later embryonic stage during which maternal RNAs were cleared up (and so was ZGA-specific expression) and replaced by zygotic/embryonic transcripts. Next, we adjusted M, Z, and E lncRNA values so that the highest value was set to one, and we displayed values for all lncRNA clusters in a single plot (Fig. 5A).
Figure 5.

OET lncRNA population dynamics during early development. (A) Overview of expression patterns of OET lncRNAs. To simplify expression pattern classification, we used average FPKM values: M, maternal (GV and MII oocytes); Z, zygotic (two- and four-cell stages); and E, late preimplantation embryo (morula and blastocyst). The plot shows dynamics of all clusters where the maximum average FPKM value of each cluster in M, Z, E was set to 1. (B) Main expression patterns of OET lncRNAs. The six panels display six basic patterns separating maternal (top left panel), zygotic (top middle and top right panels), and maternal-zygotic lncRNA (bottom panels) expression. The red lines represent the average values per each panel. (C) Expression patterns of 1,600 OET lncRNAs and 19741 mRNAs. The heatmap for lncRNAs and mRNAs was assembled from the six basic patterns (shown in (B) and schematically depicted between the heatmaps with maternal in red, zygotic in blue and maternal-zygotic in grey). Clusters with M, Z, E maxima were ordered from the left to the right and ranked based on the Z value for M and E patterns and M value for Z patterns. (D) Expression correlations estimated from reads matching different types of sequences—exons of 1,600 lncRNA and exons and introns of protein-coding genes. The color scale on the left indicates the correlation coefficient for the analysed features. Note the negative correlation for lncRNA expression between maternal and zygotic/embryonic stages reflects the apparently mutually exclusive expression patterns observed in the upper heatmap in (C). Temporal expression patterns of miR-290-295 primary precursor (E) and lncRNAs carrying antisense sequences of processed pseudogenes (F). Graphs depict expression values (FPKMs) for indicated lncRNAs. In (F), lncRNAs are labeled by gene names from which the pseudogene sequences in lncRNAs originated.
LncRNA expression patterns during OET were classified into six groups matching the maternal, zygotic, and maternal-zygotic expression types (Fig. 5B). First, we used the highest expression states to define M, Z, and E groups. Then, we defined maternal lncRNA clusters in the M group as those with a minimal E level (E values <5% of M values), while the remaining lncRNA clusters in the M group were considered maternal-zygotic lncRNA clusters. Analogically, we defined zygotic lncRNA clusters in the Z and E groups as those with a minimal M level (M values <5% of Z or E values), while the remaining lncRNA clusters in the Z and E groups were considered maternal-zygotic lncRNAs. The distribution of maximum values in M, Z, E correlated with the number of clusters defined from maternal, ZGA and embryonic sets (Fig. 1D).
To obtain a comprehensive view of temporal dynamics of OET lncRNAs, we organized all lncRNAs clusters into a heatmap based on six basic patterns while displaying expression in all sequenced stages (Fig.5C). Most clusters (1,166, displayed on the left) reached a maximum in M. The majority of those declined rapidly during ZGA, reaching low levels in the blastocyst. Of the 1,166 lncRNA cluster with the maximum in M, 993 transcripts exhibited the E value <5% of M. These represent candidates for class I—maternal lncRNA clusters. This class is clearly the most abundant one in our dataset. In total 393 and 131 lncRNA clusters had maximum values in Z and E, respectively; 251 of those had minimal maternal expression (M values <5% of Z values), thus representing class II—zygotic lncRNA clusters (Fig. 5C). Of these, 107 lncRNAs were only transiently expressed during ZGA. 446 lncRNA clusters were considered class III—maternal-zygotic transcripts. Maternal-zygotic lncRNAs could be divided into two categories: (i) those constantly present during OET, i.e. zygotic transcripts appearing before maternal ones were eliminated, and (ii) those whose maternal transcripts were strongly reduced before zygotic/embryonic transcripts emerged—there was a distinct minor group of 59 maternal-zygotic clusters whose expression reached the minimum at the two- and four-cell stages (Z value >0.05 FPKM). The dynamics of lncRNA expression differed from mRNAs (Fig. 5C bottom) mainly in the proportion of maternal and maternal-zygotic expression. Although 62% of the 1,600 OET lncRNA loci were maternal (class I), maternally expressed mRNAs made 20% of all OET mRNAs. Furthermore, maternal-zygotic lncRNAs were a minor fraction of OET lncRNAs (28%), while this class was highly abundant (68%) among mRNAs, which was not surprising considering the multitudes of housekeeping roles of encoded proteins (Fig. 5C).
Interesting results emerged from an analysis of RNA expression correlations between individual stages (Fig. 5D). We compared expression of lncRNAs and mRNA exons and introns (introns-derived reads reveal the presence of nascent transcripts, i.e. of ongoing transcription12). Remarkably, lncRNAs showed positive correlations among stages with a strong contribution of maternal RNA (GV oocyte and unfertilized/fertilized eggs) and among zygotic stages (two-, four-cell, morula, and blastocyst), but not between any two stages from the two groups. Although two-cell stage lncRNAs showed no correlation with preceding stages, the later stages even had negative correlations (Fig. 5D). This apparently reflected the extensive mutually exclusive nature of maternal and zygotic lncRNA transcriptions, which was also apparent from the expression heatmap (Fig. 5C).
In contrast, an analysis of exons of protein coding revealed good correlations between any two of the analysed stages (Fig. 5D). This was apparently due to abundant maternal-zygotic expression of protein-coding genes (68% of mRNA transcriptome in Fig. 5C). The maternal-to-zygotic transcriptome transition manifested as higher correlations among stages containing high levels of maternal transcripts (GV oocytes, unfertilized eggs, and fertilized eggs) and among stages expressing zygotic transcripts (two-cell and later). An intron-based analysis showed similar correlations within maternal and zygotic groups but their borderline was shifted to an earlier developmental stage (fertilized egg/one-cell stage), apparently reflecting the minimal amount of intron-derived reads in the maternal transcriptome and the effects of minor ZGA, which took place during the one-cell stage.12
Our results are consistent with a previous observation that lncRNA expression varies at different stages of cleavage stage embryos, suggesting cleavage stage-specific expression.9 Our results show two main expression patterns—maternal expression, which does not come back during early development, and zygotic expression dominated by a transient major ZGA expression pattern. This implies that expression of the bulk of OET lncRNA clusters is driven by oocyte- and ZGA-specific transcription factors, because ubiquitous transcription factors would control only a minority of OET lncRNA clusters. This is consistent with stereotypical observations of high numbers of cell-type-specific lncRNAs.29 But why would lncRNAs adapt their expression for tissue or stage-specific transcription factors? We speculate that lncRNAs emerge from random transcription of genic and intergenic regions and that this random expression by ubiquitous transcription factors is under stronger selective pressure than expression restricted to a specific cell type/developmental stage.
3.6. Inferring biological roles of OET lncRNAs from NGS data
The role of most of the 1,600 OET lncRNAs is unknown and it is possible that the majority of them have no function. However, several suggestive observations emerged while annotating OET lncRNA. For example, we found two novel maternally expressed lncRNA clusters located in imprinted loci, whose expressions correlate with the maternal pattern of expression. lncRNA-OET-17-106 overlaps antisense with 3′ end of Airn lncRNA, which is maternally silenced (Supplementary Fig. S6A); lncRNA-OET-12-253 is expressed just downstream of an imprinted miRNA cluster, which is expressed from the maternal allele (Supplementary Fig. S6A). We also found interesting expression patterns in several known lncRNA loci, such as Malat1, Neat1, or Cyrano (Supplementary Fig. S6B). Metastasis-associated lung adenocarcinoma transcript 1 (Malat1, reviewed in45) is among the most studied lncRNA after Xist. Malat1 is a conserved extremely abundant lncRNA non-essential for normal development.46–48Malat1 transcript levels are minimal in the oocyte relative to later preimplantation stages (Supplementary Fig. S6B). Similarly, expression of Neat1, lncRNA encoded adjacent to Malat1, starts at ZGA and a shorter Neat1 transcript isoform accumulates from the four-cell stage on (Supplementary Fig. S6A). Thus, the Malat1/Neat1 locus transcription is a zygotic component of OET. In contrast, Cyrano lncRNA, which has been implicated in embryonic development in zebrafish,30 exhibits relatively rare maternal and zygotic expression pattern while zygotic expression in the locus extents into a conserved the 3′ end region (Supplementary Fig. S6B).
Among the lncRNA types, which can be identified, are precursors for small RNAs in RNA silencing pathways, since they can be matched with small RNAs cloned from mouse oocytes and early embryos.23,49–51 We detected the primary miRNA precursor (pri-miRNA) carrying the miR-290-295 miRNA cluster (lncRNA-OET-07-048) whose expression starts at the two-cell stage and is present during early development (Fig. 5E). The miR-290-295 family is associated with pluripotency and is closely related to the miR-430 family, which functions in zebrafish embryos (reviewed in52). The miR-290-295 family most likely does not contribute strongly to maternal mRNA clearance since most maternal mRNAs become eliminated before the miR-290-295 miRNAs accumulate enough to have an impact on the cellular transcriptome. At the same time, we did not detect any pri-miRNAs of the let-7 family, the most abundant miRNA family expressed in mouse oocytes.53 This might be explained by the fact that the analysed fully grown oocytes were transcriptionally already quiescent, thus it would be possible that let-7 precursors were already processed into miRNAs. Consequently, we would not observe precursor sequences in oocyte NGS data similarly to the absence of nascent transcripts of maternally expressed genes.12
Although a miRNA precursor yields miRNA(s) with defined sequence(s), short interfering RNAs (siRNAs), which guide endonucleolytic cleavage of cognate RNAs in the RNA interference (RNAi), are produced from a precursor as a population of 21–23 nt RNAs. Mouse oocytes are unique among mammalian cells since they produce high amounts of endogenous siRNAs from double-stranded RNA (dsRNA).23,54 Endogenous dsRNA can form through (i) a transcription of an inverted repeat, (ii) a convergent transcription, and (iii) basepairing of mRNA and antisense RNA originating, e.g. from a processed pseudogene. Interestingly, of the 13 genes, for which Tam et al. predicted basepiring with transcripts from processed pseudogenes, 7 were annotated among the OET lncRNAs. All of them were maternally expressed (Fig. 5F). Antisense sequences of processed pseudogenes can be found in lncRNAs expressed elsewhere, but efficient siRNA production in mice requires a unique maternal isoform of the Dicer enzyme.14,55 Therefore, lncRNAs carrying antisense sequences of processed pseudogenes have unique functionality restricted to oocytes and early embryos, where they can be efficiently converted to endo-siRNAs.
Remarkably, two of these lncRNAs matched the predicted dormant lncRNAs shown in Figure 3C (lncRNA-OET-08-341 complementary to Ppp4r1 mRNA and lncRNA-OET-02-259 complementary to Traip mRNA). We hypothesize that cytoplasmic polyadenylation could regulate the availability of the siRNA substrate and thus control the pace of clearance of specific maternal mRNAs.
3.7. Functional analysis of two OET lncRNAs
For a functional analysis, we chose two maternal lncRNAs (lncRNA-OET-19-199 and lncRNA-OET-06-154, Fig. 6A and B), which had good expression, used a dominant promoter, showed sequence conservation among mammals, and were syntenic relative to adjacent genes. Both lncRNAs were maternally expressed and degraded after fertilization (Supplementary Figs. S7 and S8). Upon confirming lncRNA expression patterns by qPCR (Fig. 6C and E), we created mouse knockout models using RNA-guided CRISPR Cas9 system.15,16 We aimed at deleting the promoter and exon1 (Supplementary Figs. S7D and S8C) in order to suppress lncRNA transcription in the locus, not just the accumulation of mature lncRNA. We successfully produced lncRNA knockouts and confirmed the loss of lncRNA expression by a qPCR analysis (Fig. 6D and F).
Figure 6.

CRISPR-mediated lncRNA knockouts of. (A, B) Genomic structure of lncRNA-OET-19-199 and lncRNA-OET-06-154. Filled rectangles represent 5′ terminal, internal, and 3’ terminal exons. The frame over the first exon indicates the region targeted by CRISPR-mediated deletion (cleavage positions are depicted in Supplementary Figs. S7 and S8). (C, E) Relative expression of targeted lncRNA in oocytes and zygotes. RT-qPCR analysis of RNA from a constant number of oocytes/zygotes. These results are consistent with NGS (Supplementary Figs. S7B and S8B) and microarray analysis data (Supplementary Fig. S7C). (D, F) RNA expression at the targeted locus in oocytes from knockout animals. Shown is the RT-qPCR expression analysis of the targeted lncRNA and the nearest two upstream and two downstream genes (maps of the loci are available in Supplementary Figs. S7F and S8E. Error bar = SEM. (G) Characterization of lncRNA-OET-06-154 of the distal 3′ terminal exon region (corresponds to the framed region at the 3′ end of lncRN-OET-06-154 in the panel B). (H) Analysis of NGS data from Dicer and Ago2 knockout oocytes.22 The Y-scale depicts the FPKM difference in Ago2 knockouts (catalytically dead mutant22), the X-scale in Dicer knockouts. The FPKM difference was used because it better reflects the effect of suppressed RNAi on the transcriptome than the ratio, which is distorted by varying expression levels. In other words, if absence of RNAi results in stabilization of 1,000 molecules of a hypothetical mRNA, the graph will display such mRNA in the same position regardless if there is 100, 1 000, or 10 000 molecules of this mRNA in the wild-type oocyte.
Breeding of mutant mice did not reveal any effects on viability and fertility of homozygotes although breeding of lncRNA-OET-19-199 heterozygotes produced heterozygotes with a lower frequency than expected (Supplementary Figs. S7E and S8D). The basis of this effect is currently under investigation. Nonetheless, homozygous null females for each lncRNA knockout were fertile and breeding of null animals produced viable offspring in both cases. Ovarian histology of knockout animals appeared normal and normal amounts of fully grown oocytes were recovered from null females (data not shown). We also examined expression of nearest genes (Fig. 6D and F). We observed a significant difference in one of the neighbouring genes of lncRNA-OET-19-199 (Fig. 6D). However, whether this reflected the biological role of lncRNA-OET-19-199 or whether it was a consequence of the introduced DNA deletion remains unknown.
Importantly, we assigned a biological function to one of the transcript isoforms of lncRNA-OET-06-154, even though the loss expression of lncRNA-OET-06-154 had no effect on fertility (Supplementary Fig. S8D) and there was no effect on neighbouring genes (Fig. 6F). The reason was that the most downstream terminal exon of lncRNA-OET-06-154 carried an antisense sequences from the Eef1g pseudogene (Fig. 6G). The pseudogene insertion happened already in the common ancestor of mice and rats, and the pseudogene fragment was subsequently disrupted by several LTR insertions in the mouse lineage. The antisense pseudogene sequence generates an endo-siRNAs as evidenced by mapping small RNAs from NGS data23 to the locus (Fig. 6G). Small RNAs targeting Eef1g are biologically active as evidenced by Eef1g upregulation in both Dicer and Ago2 knockout oocytes (Fig. 6H). Taken together, lncRNA-OET-06-154 represents an example of a locus expressing multiple transcript isoforms that might differ in function: those carrying the most downstream 3’ terminal exon would be engaged in RNAi-mediated repression of Eef1g in the oocyte, while others might have another function (or no function at all). This lncRNA example also shows that functional siRNAs may have originated from a pseudogene insertion more than 40 million years old as suggested by molecular dating of the common ancestor of mice and rats based on 658 nuclear genes.56
Supplementary Material
Acknowledgements
We thank Radek Malik, Radek Jankele, Martin Moravec, Helena Fulkova, Josef Pasulka, and other members of our laboratories for discussions and assistance, the Mediterranean Institute for Life Sciences for hosting the data mining sessions, and the Transgenic and Archiving Module of the Czech Centre for Phenogenomics, Institute of Molecular Genetics ASCR whose work was supported by LM2011032 and LM2015040 (MEYS) and the BIOCEV European Regional Development Fund CZ.1.05/1.1.00/02.0109.
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
The main support for this research was provided through a Marie Curie Initial Training Network (project no. 607720, RNATRAIN) funding for S.G. Research of PS lab was further supported by the Czech Science Foundation grant GACR P305/12/G034, and Ministry of Education, Youth, and Sports project NPU1 LO1419. The IMG institutional support was provided by RVO: 68378050. R.K., V.F., and K.V. were supported through the European Commission Seventh Framework Program (Integra-Life; grant 315997 to KV), and Croatian Science Foundation (grant IP-2014-09-6400 to KV). The work of FA was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (no. 20062002, no. 25252054).
References
- 1. Svoboda P., Franke V., Schultz R.M.. 2015, Sculpting the transcriptome during the oocyte-to-embryo transition in mouse. Curr. Top. Dev. Biol., 113, 305–49. [DOI] [PubMed] [Google Scholar]
- 2. Marques A.C., Ponting C.P.. 2014, Intergenic lncRNAs and the evolution of gene expression. Curr. Opin. Genet. Dev., 27, 48–53. [DOI] [PubMed] [Google Scholar]
- 3. Guttman M., Rinn J.L.. 2012, Modular regulatory principles of large non-coding RNAs. Nature, 482, 339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mercer T.R., Mattick J.S.. 2013, Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol., 20, 300–7. [DOI] [PubMed] [Google Scholar]
- 5. Ulitsky I., Bartel D.P.. 2013, lincRNAs: genomics, evolution, and mechanisms. Cell, 154, 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kutter C., Watt S., Stefflova K., et al. 2012, Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet., 8, e1002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ng S.Y., Stanton L.W.. 2013, Long non-coding RNAs in stem cell pluripotency. Wiley Interdiscip. Rev. RNA, 4, 121–8. [DOI] [PubMed] [Google Scholar]
- 8. Yan L., Yang M., Guo H., et al. 2013, Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol., 20, 1131–9. [DOI] [PubMed] [Google Scholar]
- 9. Zhang K., Huang K., Luo Y., Li S.. 2014, Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genomics, 15, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamazaki N., Uesaka M., Nakashima K., Agata K., Imamura T.. 2015, Gene activation-associated long noncoding RNAs function in mouse preimplantation development. Development, 142, 910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Veselovska L., Smallwood S. A., Saadeh H., et al. 2015, Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol., 16, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abe K., Yamamoto R., Franke V., et al. 2015, The first murine zygotic transcription is promiscuous and uncoupled from splicing and 3' processing. EMBO J., 34, 1523–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagy A. 2003, Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY. [Google Scholar]
- 14. Flemr M., Malik R., Franke V., et al. 2013, A retrotransposon-driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell, 155, 807–16. [DOI] [PubMed] [Google Scholar]
- 15. Cong L., Ran F.A., Cox D., et al. Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen B., Gilbert L. A., Cimini B. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell, 155, 1479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park S.J., Komata M., Inoue F., et al. 2013, Inferring the choreography of parental genomes during fertilization from ultralarge-scale whole-transcriptome analysis. Genes Dev., 27, 2736–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xue Z., Huang K., Cai C., et al. 2013, Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature, 500, 593–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guttman M., Garber M., Levin J. Z., et al. 2010, Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol., 28, 503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W.. 2013, CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res., 41, e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tarailo-Graovac M., Chen N.. 2009, Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, Chapter 4, Unit 4 10. [DOI] [PubMed] [Google Scholar]
- 22. Stein P., Rozhkov N.V., Li F., et al. 2015, Essential Role for endogenous siRNAs during meiosis in mouse oocytes. PLoS Genet., 11, e1005013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tam O.H., Aravin A.A., Stein P., et al. 2008, Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature, 453, 534–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flemr M., Ma J., Schultz R. M., Svoboda P.. 2010, P-body loss is concomitant with formation of a messenger RNA storage domain in mouse oocytes. Biol. Reprod, 82, 1008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guttman M., Amit I., Garber M., et al. 2009, Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature, 458, 223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelley D., Rinn J.. 2012, Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol, 13, R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Piko L., Clegg K.B.. 1982, Quantitative changes in total Rna, total Poly(a), and ribosomes in early mouse embryos. Dev. Biol., 89, 362–78. [DOI] [PubMed] [Google Scholar]
- 28. Cabili M.N., Trapnell C., Goff L., et al. 2011, Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev., 25, 1915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Derrien T., Johnson R., Bussotti G., et al. 2012, The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res., 22, 1775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ulitsky I., Shkumatava A., Jan C. H., Sive H., Bartel D.P.. 2011, Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell, 147, 1537–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smallwood S.A., Tomizawa S., Krueger F., et al. 2011, Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet., 43, 811–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marzluff W.F., Gongidi P., Woods K.R., Jin J., Maltais L.J.. 2002, The human and mouse replication-dependent histone genes. Genomics, 80, 487–98. [PubMed] [Google Scholar]
- 33. Richter J.D., Lasko P.. 2011, Translational control in oocyte development. Cold Spring Harb. Perspect. Biol., 3, a002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gebauer F., Richter J.D.. 1997, Synthesis and function of Mos: the control switch of vertebrate oocyte meiosis. Bioessays, 19, 23–8. [DOI] [PubMed] [Google Scholar]
- 35. Huarte J., Belin D., Vassalli A., Strickland S., Vassalli J.D.. 1987, Meiotic maturation of mouse oocytes triggers the translation and polyadenylation of dormant tissue-type plasminogen activator mRNA. Genes Dev., 1, 1201–11. [DOI] [PubMed] [Google Scholar]
- 36. de Vantery C., Stutz A., Vassalli J.D., Schorderet-Slatkine S.. 1997, Acquisition of meiotic competence in growing mouse oocytes is controlled at both translational and posttranslational levels. Dev. Biol., 187, 43–54. [DOI] [PubMed] [Google Scholar]
- 37. Murai S., Stein P., Buffone M.G., Yamashita S., Schultz R.M.. 2010, Recruitment of Orc6l, a dormant maternal mRNA in mouse oocytes, is essential for DNA replication in 1-cell embryos. Dev. Biol., 341, 205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma J., Flemr M., Strnad H., Svoboda P., Schultz R.M.. 2013, Maternally recruited DCP1A and DCP2 contribute to messenger RNA degradation during oocyte maturation and genome activation in mouse. Biol. Reprod., 88, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Charlesworth A., Cox L.L., MacNicol A.M.. 2004, Cytoplasmic polyadenylation element (CPE)- and CPE-binding protein (CPEB)-independent mechanisms regulate early class maternal mRNA translational activation in Xenopus oocytes. J. Biol. Chem., 279, 17650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alizadeh Z., Kageyama S., Aoki F.. 2005, Degradation of maternal mRNA in mouse embryos: selective degradation of specific mRNAs after fertilization. Mol. Reprod. Dev., 72, 281–90. [DOI] [PubMed] [Google Scholar]
- 41. Sakurai T., Sato M., Kimura M.. 2005, Diverse patterns of poly(A) tail elongation and shortening of murine maternal mRNAs from fully grown oocyte to 2-cell embryo stages. Biochem. Biophys. Res. Commun., 336, 1181–9. [DOI] [PubMed] [Google Scholar]
- 42. Carlevaro-Fita J., Rahim A., Guigo R., Vardy L.A., Johnson R.. 2016, Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA, 22, 867–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ruiz-Orera J., Messeguer X., Subirana J.A., Alba M.M.. 2014, Long non-coding RNAs as a source of new peptides. Elife, 3, e03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yue F., Cheng Y., Breschi A., et al. 2014, A comparative encyclopedia of DNA elements in the mouse genome. Nature, 515, 355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gutschner T., Hammerle M., Diederichs S.. 2013, MALAT1 – a paradigm for long noncoding RNA function in cancer. J. Mol. Med. (Berl), 91, 791–801. [DOI] [PubMed] [Google Scholar]
- 46. Eissmann M., Gutschner T., Hammerle M., et al. 2012, Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol., 9, 1076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang B., Arun G., Mao Y. S., et al. 2012, The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep., 2, 111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakagawa S., Ip J. Y., Shioi G., et al. 2012, Malat1 is not an essential component of nuclear speckles in mice. RNA, 18, 1487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watanabe T., Totoki Y., Toyoda A., et al. 2008, Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature, 453, 539–43. [DOI] [PubMed] [Google Scholar]
- 50. Garcia-Lopez J., Alonso L., Cardenas D.B., et al. 2015, Diversity and functional convergence of small noncoding RNAs in male germ cell differentiation and fertilization. RNA, 21, 946–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garcia-Lopez J., Hourcade Jde D., Alonso L., Cardenas D.B., del Mazo J.. 2014, Global characterization and target identification of piRNAs and endo-siRNAs in mouse gametes and zygotes. Biochim. Biophys. Acta, 1839, 463–75. [DOI] [PubMed] [Google Scholar]
- 52. Svoboda P., Flemr M.. 2010, The role of miRNAs and endogenous siRNAs in maternal-to-zygotic reprogramming and the establishment of pluripotency. EMBO Rep., 11, 590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ma J., Flemr M., Stein P., et al. 2010, MicroRNA activity is suppressed in mouse oocytes. Curr. Biol., 20, 265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Watanabe T., Cheng E.C., Zhong M., Lin H.. 2015, Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome Res., 25, 368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nejepinska J., Malik R., Filkowski J., Flemr M., Filipowicz W., Svoboda P.. 2012, dsRNA expression in the mouse elicits RNAi in oocytes and low adenosine deamination in somatic cells. Nucleic Acids Res., 40, 399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kumar S., Hedges S.B.. 1998, A molecular timescale for vertebrate evolution. Nature, 392, 917–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.