

Abstract
The genus Ruellia (Wild Petunias; Acanthaceae) is characterized by an enormous diversity of floral shapes and colours manifested among closely related species. Using Illumina platform, we reconstructed the draft genome of Ruellia speciosa, with a scaffold size of 1,021 Mb (or ∼1.02 Gb) and an N50 size of 17,908 bp, spanning ∼93% of the estimated genome (∼1.1 Gb). The draft assembly predicted 40,124 gene models and phylogenetic analyses of four key enzymes involved in anthocyanin colour production [flavanone 3-hydroxylase (F3H), flavonoid 3′-hydroxylase (F3′H), flavonoid 3′,5′-hydroxylase (F3′5′H), and dihydroflavonol 4-reductase (DFR)] found that most angiosperms here sampled harboured at least one copy of F3H, F3′H, and DFR. In contrast, fewer than one-half (but including R. speciosa) harboured a copy of F3′5′H, supporting observations that blue flowers and/or fruits, which this enzyme is required for, are less common among flowering plants. Ka/Ks analyses of duplicated copies of F3′H and DFR in R. speciosa suggested purifying selection in the former but detected evidence of positive selection in the latter. The genome sequence and annotation of R. speciosa represents only one of only four families sequenced in the large and important Asterid clade of flowering plants and, as such, will facilitate extensive future research on this diverse group, particularly with respect to floral evolution.
Keywords: anthocyanin, Asterid, evolution, Ka/Ks, phylogenetic
1. Introduction
The Asterid clade is one of the most species-rich lineages of flowering plants, containing over a quarter of all known species of angiosperms (∼80,000 of 300,000). In this clade are four of the 10 most diverse families of flowering plants: Asteraceae (Sunflowers: ∼24,000 spp.), Rubiaceae (Coffee Family: ∼11,200 spp.), Lamiaceae (Mint Family: ∼7,200 spp.), and Acanthaceae (Acanthus Family: 4,200 spp.). Asterids are of tremendous economic and ecological significance, with cultivated and economically important members including coffee, mints (basil, oregano, rosemary, sage, thyme, lavender, teak), tomatoes and relatives (potatoes, tobacco, chilies, peppers, petunias), and carrots and relatives (carrots, cumin, cilantro, dill, fennel, parsley, celery), as well as blueberries, olives, sunflowers, sweet potatoes, snapdragons, dogwoods, ashes, milkweeds, and hollies.1 Members of the Asterid clade furthermore dominate ecosystems worldwide.2
Despite a revolution in sequencing plant genomes over the last decade, there exists only a handful of plants in the Asterid clade with nuclear reference genomes (http://www.plantgdb.org/).3 These genomes derive primarily from the tomato family (Solanaceae) in which nine species have been sequenced (Solanum, four species: ∼838–900 Mb;4–7Nicotiana, three species: ∼3 Gb;8,9Capsicum, three species: ∼3.48 Gb;10,11and Petunia, two species: ∼1.4 Gb12). Beyond this, there exists only two additional reference genomes in the Asterid clade, only one of which is >100 Mb in size: the monkeyflower Mimulus guttatus, ∼430 Mb;13 the other derives from the insectivorous bladderwort Utricularia gibba, which is only ∼81.9 Mb.14 Thus, only three of 100+ families that comprise Asterids have representative nuclear genomes.
To build genomic resources for Asterids, we here contribute new nuclear genome reference from a fourth family in this clade: Acanthaceae. The genus Ruellia (Wild Petunias) contains ∼400 species that are distributed primarily in the Neotropics and Paleotropics. With very few exceptions, all species of Ruellia (∼50 species counted to date) are thought to be diploid and share a somatic chromosome count of 2n = 34 (reviewed by Tripp15 and Tripp16). Ruellia is marked by tremendous diversity in both flower shape and colour, with very closely related species (e.g. sister taxa) often divergent in floral form (Fig. 1).17 Flowers of Ruellia range from blue to purple, pink, red, green, yellow, and white and are pollinated by bees, butterflies, hawkmoths, bats, hummingbirds, and sunbirds.15,18 Owing to the floral diversity and species richness in the genus, Ruellia has potential as a future model system for floral evolution in angiosperms.
Figure 1.

Floral colour and floral shape diversity present within Ruellia. (A) R. speciosa. (B) Ruellia macrantha. (C) Ruellia longipedunculata. (D) Ruellia hirsuto-glandulosa. (E) Ruellia bourgaei. (F) Ruellia saccata. (G) Ruellia biolleyi. (H) R. breedlovei. (I) Ruellia californica. (J) Ruellia noctiflora.
Ruellia speciosa (Beautiful Wild Petunia) is a pale yellow-flowered species native to a narrow portion of the southern Sierra Madre Oriental of Mexico and is nearly extinct in the wild due to habitat destruction.16 Because of its tiny population sizes and isolation from related species, the genome is expected to be highly homozygous and suitable for sequencing as a reference.
2. Materials and methods
2.1. DNA isolation, library preparation, and sequencing
Plant material for this study was collected in the wild by E. Tripp and S. Acosta (voucher # 175, housed at the Duke University Herbarium; in living cultivation in the University of Colorado Greenhouses), in a small canyon of a mountain that lies within the city limits of Oaxaca City, Oaxaca, Mexico. Total genomic DNA was extracted from young leaf tissue of R.speciosa using the DNeasy Plant Mini Kit (QIAGEN) following the manufacturer’s instructions. Presence of high molecular weight DNA was confirmed by 1% agarose-gel electrophoresis stained with SYBR Safe (Invitrogen). DNA concentration was quantified using a Qubit 2.0 Fluorimeter spectrophotometer (Life Technology). DNA that passed quality control was used for mate-pair library preparation followed by whole-genome shotgun sequencing. Small fragment libraries with insert sizes of 250, 350, 450, and 550 bp were prepared using a TruSeq DNA PCR-Free Library Prep Kit (Illumina). Two mate-pair libraries with average insert sizes of 2 and 5 kbp were also built using a Nextera Mate Pair Library Preparation Kit (Illumina). Final libraries were quantified using a Qubit and qualities were assessed using a Bioanalyzer. Libraries were sequenced on an Illumina HiSeq2500 using either 2 × 125 bp paired-end or 2 × 150 bp paired-end chemistry at the Genomics and Microarray Core, University of Colorado–Anschutz Medical Campus.
2.2. Flow cytometry
Prior to sequencing, we conducted flow cytometry to estimate the genome size of R.speciosa. Samples were transferred to the Iowa State University Flow Cytometry Facility and analysed on a BD Biosciences FACSCanto Flow Cytometer using Solanum lycopersicum as an internal standard. All cytometry protocols followed in house methods at that facility. The software package BD FACSDiva v.6.1.319 was used to analyse the data.
2.3. De novo nuclear genome assembly
De novo assembly of the R.speciosa genome into contigs was conducted using MaSuRCA v3.1.0.20 All raw untrimmed sequenced reads were used as input for MaSuRCA as instructed in the manual and run with default settings except the memory usage option (NUM_THREADS and JF_SIZE). Gap closing was conducted via BGI’s GapCloser v1.6 of SOAPdenovo.21 Following this, we used the package SSPACE v2.0 (scaffolding pre-assemblies after contig extension)22 to conduct the final round of scaffolding. Reads trimmed with Trimmomatic v0.3323 (ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50) were also assembled with SOAPdenovo221 (‘Multi-kmer’ method was used, kmer ranges from 65 to 80) and Abyss v1.9.024 (k = 71, b = 1,000, P = 0.95 and s = 500). Assembly generated by MaSuRCA was used for all downstream analysis as it yielded the best statistics based on scaffold N50. Completeness of genome assembly was evaluated using BUSCO v1.2.25
2.4. Ploidy estimation and heterozygosity analysis
To estimate ploidy of R.speciosa, we used an approach similar to that employed by Kentaro Yoshida’s group.26 After quality trimming and error correction, reads were mapped backed to the assembled genome using BWA v0.7.1327 with default settings allowing two gaps and without seeding. Samtools was used to convert the sam file containing the alignment information into bam format and the resulting bam file was sorted and indexed.28 SNP calling was conducted using both samtools mpileup and bcftools.28 To reliably calculate base and minor allele frequencies to enable identification of heterozygous alleles, only positions with read coverages ≥ ×50 (approximately one-half of the estimated overall read coverage) and minor allele frequencies ≥ 0.2 were considered. Ploidy is determined by the ratio of alleles at heterozygous positions. In a diploid genome, a ratio of 1:1 is expected for the majority of the heterozygous alleles; in triploid and tetraploid genomes, these ratios should ∼1:2 and 1:3, respectively. Using Marker-predicted genes as input, prediction of recent or ancient polyploidization was secondarily tested by plotting the distribution of ages of duplicated sequences using DupPipe.29 Synonymous divergence (Ks) was estimated using PAML v4.9c and the F3X4 model of evolution.30 As polyploidy or whole-genome duplication (WGD) yields a large and enduring signature in such plots, significant features (α = 0.05) in age distributions were identified with SiZer v0.1-4.31 To estimate genome heterozygosity, the number of distinct k-mers in our reads was calculated using Jellyfish v2.2.5.32 k-mers were plotted as frequencies, and the number of peaks was used as an indicator of genome heterozygosity.
2.5. Genome annotation with MAKER
Genome annotation was conducted using the MAKER-P annotation pipeline and its dependent programmes.33 Two rounds of running the pipeline were completed to yield the final annotation file. Prior to annotation of the protein-coding genes, a R.speciosa-specific repeat library was constructed following the tutorial included in MAKER-P tool kit33 and RepBase v21.09.34 This library served to mask repeat elements in the assembled genome using RepeatMasker v3.2.0.35 The repeat-masked assembled genome of R. speciosa (scaffold length ≥ 1,000 bp) was then processed through the MAKER annotation pipeline. Gene prediction was conducted using SNAP36 and AUGUSTUS v3.2.2,37 trained on Arabidopsis thaliana and S.lycopersicum as models. RNA-seq data obtained from corolla and leaf samples of R. speciosa (Zhuang and Tripp, in rev.; SRA accession: SRP075855) served as EST evidence to refine gene models. High confidence annotation of protein sequences of 31 dicot species were retrieved from the PLAZA database v3.038 to serve as protein evidence for refining gene predictions. The annotation file from the first-round run was used to create the HMM training file for SNAP. The resulting HMM training file together with first-round GFF annotation file were further processed by MAKER-P to generate a final annotation file. Reads generated from previous RNA-seq study (Zhuang and Tripp, in rev.) were mapped against the R. speciosa genome assembly using BWA27 with default parameters. Paired-end reads where each end mapped to gene-containing regions of different scaffolds were then used to combine the fragments of a single gene.39 All predicted protein sequences were further validated following methods described by Upadhyay et al.40
2.6. SSR identification and maker predication
We used GMATo (genome-wide microsatellite analysing tool; https://sourceforge.net/projects/gmato/files/)41 to characterize the masked SSR loci. Masked SSR sequences were extracted using an in-house Perl script prior to analysis with GMATo. To enable comparison of our SSR statistics to those reported from other species,42 we employed similar GMATo parameters to those used in prior studies. These included retaining SSR motifs that were 2–8 bp in length and had ≥4 repeats with a minimum of 10 bp length (−r 5 −m 2 −× 10 −s 0).
2.7. Gene Ontology database annotation
Functional annotation of predicted gene models was conducted using the Trinotate pipeline v3.0.0 (http://trinotate.sourceforge.net/), with a blast e-value threshold of 1 × 10−5. To assign function annotations to the predicted genes, local NCBI-BLAST v2.2.3143 was used to search for homologies between the gene and SwissProt,44 UniProt,45 and eggNOG/Gene Ontology (GO) databases.46 In addition, all green plant entries integrated into UniProtKB/TrEMBL (Taxonomy: Viridiplantae)47 were also retrieved to serve as a custom database for blast searches. To enable comparison and visualization of GO annotations in R.speciosa to other plants, the annotated GO terms of Ruellia were combined with GO annotation files of A.thaliana and Solanum tuberosum, which were downloaded from AgriGO (http://bioinfo.cau.edu.cn/agriGO/dowload).48 This combined file was analysed using BGI WEGO (http://wego.genomics.org.cn)49 to classify GO terms into Level-2 sub categories.
2.8. Phylogenetic analysis of genes involved in the anthocyanin biosynthesis pathway
To enable phylogenetic investigation of key structural genes involved in the production of anthocyanin pigments that occur in flowers, fruits, and leaves, amino acid sequences representing genes from several angiosperm species were downloaded from PLAZA38 and OrthoDB v9.50 Sequences from Pinus taeda were also download from PINREFSEQ (http://pinegenome.org) to facilitate tree rooting.51 In total, four structural genes from 25 dicots plus three monocots were downloaded then subject to phylogenetic analysis along with orthologues retrieved from R.speciosa and P.taeda (ntax = 30 in total). Based on Trinotate annotation, genes annotated as flavanone 3-hydroxylase (F3H), flavonoid 3′-hydroxylase (F3′H), flavonoid 3′,5′-hydroxylase (F3′5′H), and dihydroflavonol 4-reductase (DFR) were extracted, and CD-HIT52 was used to retrieve non-redundant copies (at a 99% similarity cutoff). To accurately estimate copy numbers of family genes studied, SNPs located within target gene-containing regions identified from polyploidy analysis were further examined; the gene was considered to have unidentified paralogues if the percentage of SNPs were ≥90% of gene length and averaged ≥10 reads mapped to the SNP-containing region. We searched for orthologues of these genes in queried plants using the package Proteinortho v5.15.53 Identified sequences were parsed and extracted with an in-house Perl script. Two alignment matrices were constructed: the first containing all F3H, F3′H, and F3′5′H sequences and the second containing all DFR sequences. Sequence alignment was conducted using the ‘linsi’ option of MAFFT v7.305.54 Resulting alignments were truncated to exclude regions of extremely high sequence divergence (namely: autapomorphic divergence), which can interfere with phylogenetic signal owing to phenomena such as long branch attraction.55 Evolutionary histories were inferred using maximum likelihood methods, employing the LG+ F56 amino acid model as the best-fitting model determined by MEGA6,57 and final resulting phylogenetic trees were rooted using P.taeda. Branch support was assessed via 200 ML bootstrap replicates. To test selection on and assess potential functional divergence of duplicated genes, ratios of non-synonymous (Ka) to synonymous (Ks) nucleotide substitutions were estimated using the Ka/Ks web service at the computational biology unit, hosted at The University of Bergen.58 Phylogenetic trees from the earlier analyses were used as reference trees for the Ka/Ks analysis.
2.9. Genome-wide characterization of MYB3R and R2R3 type MYB gene family
To characterize the MYB gene family in R.speciosa, protein sequences of 125 typical R2R3-MYB proteins and six atypical MYB proteins (AtMYBCDC5, AtMYB3R-like, and AtMYB4R1) were downloaded from the Arabidopsis information resource (https://www.arabidopsis.org/browse/genefamily/MYB.jsp).59 Three Petunia MYBs regulate anthocyanin biosynthesis were also downloaded from NCBI database (GI:673536265, GI:321688260, GI:321688230) to facilitate function annotation. Proteinortho was used to conduct homologue searches of Arabidopsis MYBs against protein sequence of all gene models annotated by Maker-P with default parameters. Proteins without homologues among Arabidopsis MYBs were removed from subsequent analysis. The resulting MYB proteins were then assessed for the presence of Myb domains using Pfam v26.0 on the Pfam server with default parameters (http://pfam.sanger.ac.uk).60 Proteins containing Myb domains were extracted and subjected to phylogenetic analysis using methods as earlier employing the LG + G56 amino acid model as the best-fitting model. Function assignments were conducted based on known functions of Arabidopsis MYBs.61–78 MYBs with bootstrap values ≥ 70% were considered to belong to the same subgroup.
2.10. Gene expression analysis
To measure expression levels of selected anthocyanin genes and identified MYBs, reads from previous tissue-specific RNA-seq libraries (Zhuang and Tripp, in rev.; SRA accession: SRP075855) were trimmed with Trimmomatic v0.3323(parameters used: ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36) and mapped back to Maker-P predicted gene sequences using BWA with default parameters. eXpress v1.5.179 was used to measure gene abundance with default parameters. The ‘fpkm’ column of tissue-specific eXpress output was extracted and used for building expression heatmaps using the heatmap.2 program within the gplots80 package for R.
2.11. Reference-based chloroplast assembly
We additionally used resulting reads to assemble the R.speciosa chloroplast genome. First, we retrieved all available plant chloroplast sequences that were 100,000–200,000 bp in size from NCBI. Blat was used to compared our contigs generated from MaSuRCA20 to these reference chloroplast sequences to find nucleotide sequences with high similarity.81 Five contigs (length ≥ 10,000 bp; query coverage ≥ 80%) were identified. We used Blast82 to compare these five contigs against reference chloroplasts and identified two with the highest e-values and query coverages: Lindenbergia philippensis (Accession: HG530133) and Andrographis paniculata (Accession: KF150644). These, together with a chloroplast reference from closed related species Ruellia breedlovei (Accession: KP300014) were used as references for our R. speciosa assembly. Quality trimmed paired-end reads were aligned to the references using Bowtie v2-2.2.3.83 Reads with either end mapped to the references were extracted using Samtools and an in-house Perl script. Mapped reads were randomly subsampled from ×100 to ×500 coverage of the estimated chloroplast genome size; reads with an apparent depth under ×5 were discarded using BBmap v36.02.84 The resulting aligned reads were assembled using SPAdes v3.5.085 and scaffolded using SSPACE v2.022 against all sequenced libraries. Scaffolds were gap closed using Gapcloser v1.6.21 The best assemblies containing three scaffolds ≥ 300 bp were selected and subjected to second-round scaffolding and gap closing, thereby generating a circular chloroplast genome. The chloroplast sequence of the closely related R.breedlovei was used to correct the orientation of the region between the inverted repeats (IRs). CpGAVAS86was used for chloroplast annotation and visualization.
3. Results and discussion
3.1. Genome sequencing and assembly
From a whole-genome perspective, the successful sequencing, assembly, and annotation of the R.speciosa genome contributes important new data about one of the largest yet understudied clades of flowering plants—the Asterids. Our targeting Ruellia initiates the process of building a new model system for studies of floral evolution. We herein estimate the genome size of R.speciosa to be ∼1.2 Gb based on flow cytometry or ∼1.1 Gb based on Kmer analysis (Fig. 2B). For the nuclear genome assembly, nine paired-end libraries with insert sizes ranging from 250 to 550 bp and two mate-pair libraries with insert sizes of 3–6 kbp were constructed. After quality trimming, we generated >118.9 Gb of sequence data representing >×104 coverage of the predicted genome size (Supplementary Table S1). Before scaffolding, short reads were assembled into 808,710 contigs with an N50 of 1,551 bp that totaled 733 Mb (spanning 67.25% of the predicted genome size; Table 1). Additional MP reads improved the N50 by ∼10 and the additional gap closing step increased genome coverage to ∼90.07% of its predicted size without Ns (Table 1). The success of a draft genome assembly is strongly dependent on the genetic complexity of the specific organism and its genome size.87 Of the two methods we used for genome size estimation, the Kmer protocol estimated a smaller genome size, which may suggest a substantial repeat fraction in the genome that is difficult to account for using this type of analysis.88 Our repeat DNA analysis suggested that ∼70% of the genome is repetitive (Table 2). Repeat sequences are difficult to assemble because high-identity reads can derive from different portions of the genome, generating gaps, ambiguities, and collapses in alignment and assembly.89,90 This high repeat DNA content is likely to explain the relatively low scaffold N50 (17,908 bp), even though the sequencing depth exceeds ×100. Nonetheless, our scaffold N50 is comparable to those derived from genome assemblies of Cannabis sativa (N50: 16.7 kb),91Conyza canadensis (N50: 33.5 kb, repeat content 6.25%),92Ocimum tenuiflorum (N50: 27.1 kb; repeat content: 42.9%),40Lemna minor (N50: 23.6 kb, repeat content: 61.5%),93Solanum commersonii (N50: 44.3 kb, repeat content: 44.5%),4Hevea brasiliensis (N50: 3 kb, repeat content: 72%),94Aegilops tauschii (N50: 57.6 kb, repeat content: 66%),95 and Triticum urartu (N50: 63.7 kb, repeat content: 67%).96 Although data from additional sequencing libraries or a combination of existing and additional sequencing technologies may be needed to make further improvements to the quality of the R. speciosa genome for purposes of whole-genome synteny analyses, our current assembly enables us to characterize numerous aspects of this genome ranging from repeat elements, gene content, gene annotation, and phylogenetic history of specific pathways of interest.
Figure 2.

(A) Ploidy analysis of assembled R. speciosa genome. A major peak around 0.5 supports hypothesis of diploidy in R. speciosa. (B) Twenty-one k-mer depth distribution of whole-genome Illumina reads; the single peak observed indicates high level of genome homozygosity.
Table 1.
Summary of the de novo genome assembly of R.speciosa
| Category | Contigs | Scaffolds (all) | Scaffold (>1 k) |
|---|---|---|---|
| Number of contigs | 808,710 | 341,168 | 84,681 |
| N50 (bp) | 1,551 | 17,908 | 19,156 |
| L50 | 92,735 | 14,124 | 10,074 |
| Largest contig/scaffolds (bp) | 34,954 | 345,229 | 345,229 |
| GC content (%) | 39.27 | 38.80 | 38.77 |
| N’s percentage | 3.62 | 3.82 | |
| Total span (M) | 733 | 1,021 | 892 |
| Estimated coverage (%) | 67.25 | 93.69 | 81.89 |
| Number of gene models | 40,124 | ||
| Mean transcript length | 1,131 bp | ||
| Number of genes annotated | 37,848 (94.32%) |
Table 2.
De novo identification of sequence repeats in the genome of R.speciosa
| Class | Number | Total length (bp) | Percentage of genome |
|---|---|---|---|
| Retrotransposons | |||
| LINE | 29,246 | 11,469,068 | 1.23 |
| SINE | 276 | 14,095 | 1.50e−05 |
| LTR Copia | 53,812 | 23,369,721 | 2.50 |
| LTR Gypsy | 342,944 | 158,185,560 | 16.92 |
| LTR other | 26,556 | 9,772,864 | 1.05 |
| DNA transposons | 113,878 | 35,584,790 | 3.81 |
| Tandem repeats | |||
| Satellite | 1,213 | 176,347 | 0.02 |
| Low complexity | 64,404 | 3,277,475 | 0.35 |
| Simple repeats | 352,081 | 19,601,079 | 2.10 |
| Unknown | 1,158,125 | 384,916,724 | 41.17 |
| Total | 2,142,535 | 646,367,723 | 69.13 |
3.2. Ploidy and heterozygosity of R. speciosa
Polyploidization is rampant in flowering plants and is an important contributor to genomic diversity and function; as such, polyploidization can facilitate novel ecological transitions and substantially impact evolutionary trajectories.97 As discussed by Yoshida et al.,26 for diploid species, we expect a single peak at 0.5 for the mean of read counts at heterozygous positions. To characterize the ploidy of R. speciosa, the distribution of read counts of biallelic SNPs was calculated from high coverage genomic regions. Figure 2A depicts a major peak around 0.5, supporting a hypothesis of diploidy in R. speciosa. This ploidy was further confirmed by Ks analysis using DupPipe: peaks of gene duplication serve as evidence of ancient WGDs, but none was identified through Sizer analysis in our Ks plot (Supplementary Fig. S1) thus supporting our earlier conclusion that R. speciosa genome is diploid.
Kmer analysis (Fig. 2B) revealed the presence of only a single peak, consistent with our prediction that R.speciosa is highly homozygous. A very minor peak immediately adjacent to the major peak could be detected only when Kmer size ≤ 31, which represents regions with higher copy numbers such as repeats. To further explore heterozygosity and polymorphism,we calculated the percentage of total insertions/deletions and SNPs using SAMtools and BCFtools.28 We found a total of 2,431,330 SNPs (∼0.20%) with relatively relaxed search parameters (depth ≥ 3) or 299,334 SNPs (0.02%) of high confidence (depth ≥ 30), further supporting our hypothesis that the R. speciosa genome is highly homozygous.
3.3. Genome annotation and quality assessment
Of the 84,681 scaffolds (length > 1,000 bp) used for gene annotation, 25,256 (29.82%) were found to contain annotated genes. Our final Maker-P33 annotation predicted 40,124 protein-coding genes with an average length of 1,131 bp. This number exceeds the number of genes annotated for two relatively closely related species that have comparable genome sizes: tomato (gene number: 34,727, genome size ∼900 Mb)6 and potato (gene number: 35,004, genome size ∼844 Mb).7 To assess completeness of our R. speciosa genome assembly, 966 genes present as single-copy orthologues in at least 90% of plant species in OrthoDB were compiled. Using BUSCO, we retrieved orthologues for 741 (77.52%) conserved genes in Ruellia, suggesting a relatively high level of completeness for draft plant genomes generated only from Illumina reads. Additionally, mapping reads from previous RNA-seq experiment on R. speciosa (Zhuang and Tripp, in rev.) back to our genome assembly demonstrated that 76.12% of the quality trimmed reads could be successfully mapped using Tophat298 with default parameters. We interpret this as further evidence of the completeness of our genome assembly given this number is on par with expected mapping rates for RNA-seq data from very well assembled genomes such as the human genome, which has a 70–90% RNA-seq read mapping rate.99
Gene annotation databases are commonly used to evaluate functional properties of experimentally derived gene sets.100 Comparison of completed sets of genes from different genomes helps to reveal the genetic basis of biological traits as well as differences among species.101 Using the Trinotate pipeline, we assigned GO functional terms to 30,852 (76.89%) genes. Genome-wide comparative analyses among R. speciosa, S. tuberosum, and A. thaliana revealed distinct patterns of enriched Level 2 GO terms (Supplementary Fig. S2). Although potato is more closely related to R. speciosa than is Arabidopsis, gene enrichment patterns were more similar between R. speciosa and Arabidopsis than between R. speciosa and S. tuberosum for two of the three categories [cellular component and biological process categories (BP)]. GO terms categorized under molecular function were more consistent among the three species. Across all functions, fewer GO terms were enriched in S. tuberosum compared with in R. speciosa and A. thaliana, with the exception of GO terms involved metabolic processes (BP).
3.4. Analysis of repetitive elements
Repetitive DNA elements generally compose the majority of nuclear genomes in plants,102 but this content varies tremendously, ranging from 3% (U.gibba)14 to 80% of the total genome (Triticum aestivum).103 In this study, a combination of homologue-based searching and de novo analyses identified 417,698 (2.47%) tandem repeats in our assembled R. speciosa genome, which can be parsed into 352,081 simple repeats (2.1%), 64,404 low-complexity elements (0.35%), and 1,213 satellite elements (0.02%). Various types of retrotransposons comprising ∼25.51% of the whole assembled genome were also recovered in this analysis; these include 29,246 (1.23%) long interspersed nuclear elements (LINE), 276 (1.50e−05%) short interspersed nuclear elements (SINE), 424,312 (20.47%) long terminal repeats (LTR), and 113,878 (3.81%) DNA transposons. Overall, using homologue-based and de novo approaches, we estimate that ∼70% of the R. speciosa genome is composed of repetitive DNA (Table 2). However, because repeat elements are difficult to assemble, our use of assembly dependent methods for repeat identification may still underestimate total repeat content in the genome. Repetitive sequences possess high sequence homogeneity but, over the course of evolution, accumulate variations in both sequence and copy numbers.104 Because of this, repetitive elements are very useful for markers for many downstream applications, ranging from plant population genetics to breeding studies.105 In particular, molecular makers developed from simple sequence repeats (or microsatellites) have been utilized most extensively as these can be readily amplified by PCR and contain a large amount of allelic variation at each locus.106 We identified 181,793 SSR loci using GMATo. These repetitive elements are depicted via motif type in a frequency distribution plot shown in Fig. 3. The most abundant repeat motifs are AT and TA, together accounting for ∼53.37% of the total SSRs identified (Fig. 3A). Dinucleotide repeat motifs were the greatest in number (Fig. 3B), consistent with results from SSR study in holy basil107 but differing from patterns in most other plants including tomato, potato, cucumber, and rice where trinucleotide SSRs represent the majority (not including mononucleotide SSRs)42 of repeat type. Taken together, these data suggest that SSRs constitute a relatively unique and important aspect of the R. speciosa genome.
Figure 3.

Simple sequence repeat (SSR or microsatellite) analysis. (A) Bar plot of top 15 repeat motifs with highest abundance. (B) Pie chart showing distribution of identified SSRs based on motif type.
3.5. Analysis of genes involved in the anthocyanin biosynthesis pathway
Flower colour is among the key traits that serve to signal rewards to pollinators and thus helps determine reproductive success of both plants and pollinators.108–112 The anthocyanin biosynthesis pathway (ABP), largely responsible for flower colour, has furthermore been extensively studied for its beneficial contributions to human health.113 Using data from the R.speciosa genome as well as orthologues in other plants with annotated nuclear reference genomes, we reconstructed evolutionary histories of four structural enzymes in the ABP. In R.speciosa, we found two putative copies of F3H, eight of F3′H, one of F3′5′H, and three of DFR. These enzymes were present in variable duplicated copy numbers in other reference genomes as well as in Ruellia (Fig. 4, Supplementary Figs S3 and S4; see also ref. 114), although studies in other systems have reported these enzymes as single copy (12 species of Penstemon).114
Figure 4.

Summary phylogenies for (A) F3H, F3′H, and F3′5′H and (B) DFR. Phylogenetic analyses incorporated all copies of these four genes available from published angiosperm genomes (these collapased into clades depicted with triangles) as well as all copies from one gymnosperm, P. taeda. (A) Analyses recovered two major clades of F3H, three of F3′H, and one of F3′5′H in angiosperms (A) as well as three major clades of DFR in angiosperms (B). All Pinus copies depicted as individual lineages. Numbers of copies of genes recovered in R. speciosa genome shown in brackets. Numbers above branches indicate maximum likelihood bootstrap support (only values at or above 70% shown except for DFR-2). Full phylogenetic detail provided in Supplementary Figs S3 and S4.
Phylogenetic reconstruction yielded two clades of F3H, three of F3′H, one of F3′5′H, and three of DFR, and strong bootstrap support (≥70%) was recovered for some but not all key branches in our topologies (Fig. 4, Supplementary Figs S3 and S4). Our trees in part contradict those in Campanella et al.,115 which depicted clades of F3′H and DFR that correspond specifically to monocots vs. dicots (Supplementary Figs S3 and S4). Instead, Fig. 4 suggests the evolution of different copies of F3H, F3′H, F3′5′H, and DFR predates the divergence of monocots from Eudicots.1 With a given gene genealogy in our analyses (e.g. Fig. 4A), there has been subsequent lineage-specific duplication. Thus, ABP genes are marked by a history of early duplication as well as subsequent duplication events, products of which have in some cases facilitated the evolution of tissue-specific functions.116 Consistent with this, our expression analysis of putative F3′H genes showed tissue-specific expression patterns: among 5 putative F3′H copies with detectable expression levels (fpkm > 1), two copies appeared to be leaf specific and two were corolla specific (Fig. 5). However, Ka/Ks analysis of R.speciosa copies yielded ratios consistently <1, suggesting an overall signature of purifying selection or constraint on these copies (Fig. 6; Supplementary Table S2). Our phylogenetic results also suggest that F3′5′H is more closely related to one of the three clades of F3′H than it is to F3H, corroborating Seitz et al.117 who documented multiple evolutionary origins of F3′5′H from an F3′H ancestor in Asteraceae. F3′5′H is an enzyme required for production of blue delphinidins and, across all land plants, is evolutionarily younger than F3H and F3′H.115 Our finding that only one-half of all reference genomes sampled harbour a copy of this enzyme corroborates observations that blue anthocyanins are less common in plants than are purple, pink, and red cyanidins and pelargonidins that derive from other branches of the ABP pathway. Finally, DFR plays a crucial role in production of all three branches of the ABP pathway and was traditionally thought of as a substrate generalist, having the capacity to metabolize precursors to all three branches.118 However, substrate specificity has evolved in numerous groups of plants, in some cases driven by a single amino acid change.118–121 We recovered three copies of DFR in the R.speciosa genome, each resolved in three separate clades (Fig. 4B). RNA-seq analysis detected expression of two of these three copies: DFR.1 and DFR.3 as labelled in Fig. 4B. Although both copies were expressed in leaf as well as corolla tissue, DFR.1 was up-regulated in leaves (a 2.56-fold increase) whereas DFR.3 was up-regulated in corollas (a 3.50-fold increase) (Fig. 5). In contrast to F3′H copies, Ka/Ks analysis of DFR copies present in R.speciosa yielded ratios > 1 in one of the three copies (Fig. 6; Supplementary Table S3). These data suggest positive selection and are consistent with a hypothesis of adaptive evolution of this copy but functional assays are needed to understand the role of duplicated ABP loci in R.speciosa.
Figure 5.

Heatmap showing expression patterns of putative structural genes functional in ABP and MYBs identified in Ruellia. Only genes with detectable expression level (fpkm > 1) were shown.
Figure 6.
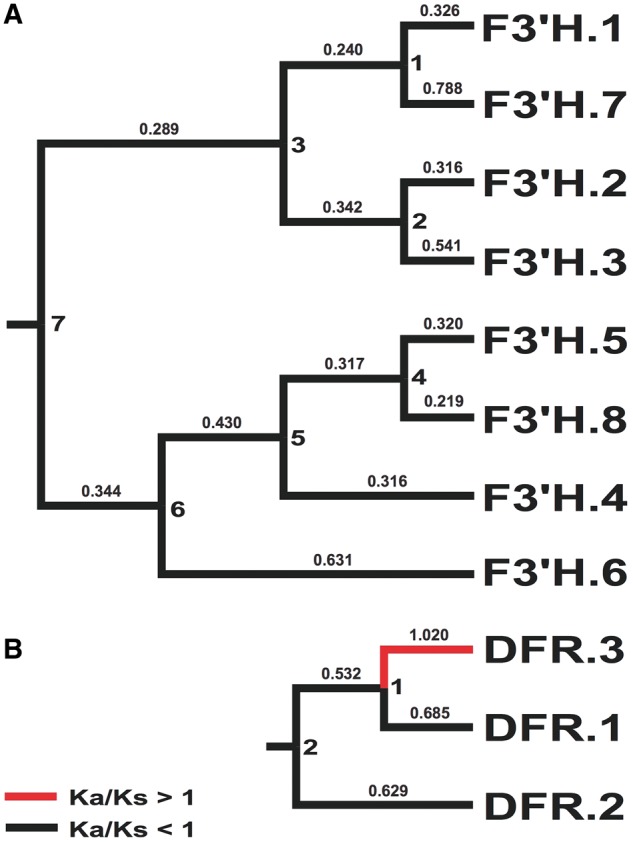
Phylogenetic hypothesis for F3'H (A) and DFR (B) identified in R speciosa. Ka/Ks values shown above branches (values >1 in red). Node IDs (1 through 7 for each gene) reflect Ka and Ks values in Supplementary Tables 3 & 4.
3.6. Phylogenetic analysis of MYB gene family
MYB transcription factors, marked by a conserved DNA-binding domain,72 comprise the largest transcription factor family in plants.78 R2R3 type MYBs are specific to the plant kingdom and are involved in the transcriptional control of plant-specific processes.71 Based on homologue searches to known MYB3R and R2R3 type MYBs in A.thaliana, we identified 96 homologous MYBs in the R.speciosa genome then assigned putative functions to 90 of them based on 87 function-annotated MYBs in Arabidopsis (Fig. 7). Our phylogenetic tree is generally consistent with prior data from Arabidopsis alone.66 Among classified MYBs, of particular interest are three subgroups that function in flavonoid biosynthesis (highlighted in blue in Fig. 7), a large pathway that includes the ABP branch. In most species, the anthocyanin branch is controlled by a ternary complex of MYB-bHLH-WD40 transcription factors and the specificity of each function appears to be the result of a particular R2R3-MYB protein that joins the complex.71–73,78 A total of nine Ruellia MYBs (RsMYBs; all highlighted in red in Fig. 7) belonging to two of the three flavonoid subgroups were identified (Fig. 7). Phylogenetic analyses resolved RsMYB71 in the same clade as several flavonoid MYBs from Arabidopsis as well as the R2R3-MYB members anthocyanin2, deep purple, and purple haze of Petunia, which are responsible for full petal colour, flower tube venation/bud-blush, and vegetative pigmentation.122–124 Transcriptome data demonstrated corolla-specific expression of RsMYB71 in Ruellia (Fig. 5), suggesting that this regulatory factor may be a key candidate in regulating flower colour in Ruellia. In contrast, our transcriptome data failed to detect any expression of the remaining eight RsMYBs, all of which belong to a second subgroup (Figs 5 and 7). Comparative analysis of these MYBs across multiple species of Ruellia followed by functional assays are needed to advance knowledge of flavonoid biosynthesis and flower colour production in this group.
Figure 7.

Phylogenetic analysis of MYB genes present in Ruellia speciosa, Petunia hybrida, and Arabidopsis thaliana. MYBs of R. speciosa are in red text, MYBs of A. thaliana are in blue text, and MYBs of P. hybrida are in black text. Functional annotations based on experimental confirmation in Arabidopsis are marked by asterisks. Branches supported by ML bootstrap values > 70% are labeled to the right of the supported node. Clades highlighted in orange and yellow depict genes involved in non-flavonoid functions; clades highlighted in blue depict genes involved in flavonoid biosynthesis.
3.7. De novo assembly of chloroplast
Chloroplasts are essential photosynthetic organelles of plant cells that manufacture energy in the presence of sunlight.125 Because the chloroplast genome is small, relatively easily sequenced and assembled, and generally uniparentally inherited, this molecule can provide abundant molecular information to support comparative evolutionary research, especially for species without a whole nuclear genome reference available. Furthermore, nucleotide substitution rates of chloroplast are relatively slow and therefore provide an appropriate window of resolution to study plant phylogeny at deep evolutionary time scales.126 In angiosperms, most assembled chloroplast genomes range from 120 to 160 kb in length and exist as circular molecules.127 Employing reference-based assembly methods, a subset of chloroplasts reads were extracted from our whole-genome shotgun dataset and successfully assembled into a circular contig with a length of 148,941 bp. Function and structure annotations conducted using CPGAVAS86 show that the obtained R.speciosa chloroplast genome has a standard quadripartite organization (Fig. 8), comprising two copies of IRs, a large single copy region, and a small single copy region, typical of other plants.125,127,128 In total, five genes involved in photosynthesis and 38 genes involved in self-replication were identified in the R. speciosa chloroplast genome, these with an average intron length of 659 bp (Fig. 8). The whole chloroplast genome assembled in this study will be useful to future phylogenetic research both in Acanthaceae and across angiosperms.
Figure 8.

Gene organization of the R. speciosa chloroplast genome. Genes drawn inside the circle are transcribed clockwise while those drawn outside are transcribed counterclockwise. Different gene functional groups are colour coded. The map was drawn using CpGAVAS.
4. Conclusion
Asterids contain a quarter of all known species of flowering plants and four of the 10 most diverse families of flowering plants. We have contributed a new nuclear genome sequence that serves as only the third family represented by such a sequence within this important clade. By building new genomic resources for R.speciosa, this study facilitates future investigation of Asterid evolution, particular with respect to flower colour production. Additionally, this new resource may facilitate future investigation of the genomic architecture of evolutionary intermediates (sensu Stebbins129), as R.speciosa is member to a clade that demonstrates a clear transition from bee to hummingbird to bat pollination and has features intermediate between the latter two stages.18
Availability
Raw nucleotide sequence data are available in the NCBI sequence read archive database (BioProject PRJNA326965) under the accession number SRP077633. The draft assembly is deposited in the NCBI whole-genome shotgun database (BioProject PRJNA326965) under the submission number SUB1661288 (released upon article acceptance).
Supplementary Material
Acknowledgements
The authors thank Travis Glenn, Troy Kieren, Swarnali Louha, the Georgia Genomics Facility, and Brant Faircloth for generating and sequencing five DNA libraries that were used in this study. We additionally thank Nolan Kane for allowing us access to the DupPipe software and Mathew Sharples for assembling and annotating the Ruellia breedlovei chloroplast genome, which facilitated our cp assembly of Ruellia speciosa.
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by National Science Foundation’s Division of Environmental Biology (Award #1354963 and #1355138) to E.A.T. and Lucinda McDade.
References
- 1. Stevens P. 2001, Angiosperm phylogeny website. Version 12, July 2012 [and more or less continuously updated since].
- 2. Funk V., Susanna A., Stuessy T., Bayer R. (Editors). 2009, Systematics, evolution and biogeography of the Compositae, International Association of Plant Taxonomy, Vienna, Austria.
- 3. Duvick J., Fu A., Muppirala U., et al. 2008, PlantGDB: a resource for comparative plant genomics, Nucleic Acids Res., 36, D959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aversano R., Contaldi F., Ercolano M.R., et al. 2015, The Solanum commersonii genome sequence provides insights into adaptation to stress conditions and genome evolution of wild potato relatives, Plant Cell, 27, 954–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hirakawa H., Shirasawa K., Miyatake K., et al. 2014, Draft genome sequence of eggplant (Solanum melongena L.): the representative Solanum species indigenous to the old world, DNA Res., 21, 649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tomato Genome Consortium. 2012, The tomato genome sequence provides insights into fleshy fruit evolution, Nature, 485, 635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Potato Genome Sequencing Consortium. 2011, Genome sequence and analysis of the tuber crop potato, Nature, 475, 189–95. [DOI] [PubMed] [Google Scholar]
- 8. Sierro N., Battey J.N., Ouadi S., et al. 2013, Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis, Genome Biol., 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bombarely A., Rosli H.G., Vrebalov J., Moffett P., Mueller L.A., Martin G.B.. 2012, A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research, Mol. Plant Microbe Interact., 25, 1523–30. [DOI] [PubMed] [Google Scholar]
- 10. Kim S., Park M., Yeom S.-I., et al. 2014, Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species, Nat. Genet., 46, 270–8. [DOI] [PubMed] [Google Scholar]
- 11. Qin C., Yu C., Shen Y., et al. 2014, Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization, Proc. Natl. Acad. Sci., 111, 5135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bombarely A., Moser M., Amrad A., et al. 2016, Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida, Nat. Plants, 2, 16074. [DOI] [PubMed] [Google Scholar]
- 13. Hellsten U., Wright K.M., Jenkins J., et al. 2013, Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing, Proc. Natl. Acad. Sci., 110, 19478–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ibarra-Laclette E., Lyons E., Hernández-Guzmán G., et al. 2013, Architecture and evolution of a minute plant genome, Nature, 498, 94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tripp E.A. 2007, Evolutionary relationships within the species-rich genus Ruellia (Acanthaceae), Syst. Botany, 32, 628–49. [Google Scholar]
- 16. Tripp E.A. 2010, Taxonomic revision of Ruellia section Chiropterophila (Acanthaceae): a lineage of rare and endemic species from Mexico, Syst. Botany, 35, 629–61. [Google Scholar]
- 17. Tripp E.A., Manos P.S.. 2008, Is floral specialization an evolutionary dead‐end? Pollination system transitions in Ruellia (Acanthaceae), Evolution, 62, 1712–37. [DOI] [PubMed] [Google Scholar]
- 18. Tripp E.A., McDade L.A.. 2013, Time-calibrated phylogenies of hummingbirds and hummingbird-pollinated plants reject a hypothesis of diffuse co-evolution, Aliso. Discipline · Botany, 31, 89–103. [Google Scholar]
- 19. Verwer B. 2002, BD DACSDiVA Option, BD Biosciences, Pharmingen; Becton, Dickson and Company, White Paper, 1–22. [Google Scholar]
- 20. Zimin A.V., Marçais G., Puiu D., Roberts M., Salzberg S.L., Yorke J.A.. 2013, The MaSuRCA genome assembler, Bioinformatics, 29, 2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo R., Liu B., Xie Y., et al. 2012, SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler, Gigascience, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Simpson J.T., Wong K., Jackman S.D., Schein J.E., Jones S.J., Birol I.. 2009, ABySS: a parallel assembler for short read sequence data, Genome Res., 19, 1117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bolger A.M., Lohse M., Usadel B.. 2014, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boetzer M., Henkel C.V., Jansen H.J., Butler D., Pirovano W.. 2011, Scaffolding pre-assembled contigs using SSPACE, Bioinformatics, 27, 578–9. [DOI] [PubMed] [Google Scholar]
- 25. Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M.. 2015, BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs, Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- 26. Yoshida K., Schuenemann V.J., Cano L.M., et al. 2013, The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine, Elife, 2, e00731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H., Durbin R.. 2009, Fast and accurate short read alignment with Burrows–Wheeler transform, Bioinformatics, 25, 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H., Handsaker B., Wysoker A., et al. 2009, The sequence alignment/map format and SAMtools, Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barker M.S., Dlugosch K.M., Dinh L., et al. 2010, EvoPipes. net: bioinformatic tools for ecological and evolutionary genomics, Evol. Bioinform., 6, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang Z. 2007, PAML 4: phylogenetic analysis by maximum likelihood, Mol. Biol. Evol., 24, 1586–91. [DOI] [PubMed] [Google Scholar]
- 31. Chaudhuri P., Marron J.S.. 1999, SiZer for exploration of structures in curves, J. Am. Stat. Assoc., 94, 807–23. [Google Scholar]
- 32. Marçais G., Kingsford C.. 2011, A fast, lock-free approach for efficient parallel counting of occurrences of k-mers, Bioinformatics, 27, 764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Campbell M.S., Law M., Holt C., et al. 2014, MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations, Plant Physiol., 164, 513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J.. 2005, Repbase Update, a database of eukaryotic repetitive elements, Cytogenet. Genome Res., 110, 462–7. [DOI] [PubMed] [Google Scholar]
- 35. Smit A.F., Hubley R., Green P.. 1996, RepeatMasker. http://www.repeatmasker.org. (06 Decemebr 2016, date last accessed).
- 36. Korf I. 2004, Gene finding in novel genomes, BMC Bioinformatics, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stanke M., Waack S.. 2003, Gene prediction with a hidden Markov model and a new intron submodel, Bioinformatics, 19, ii215–25. [DOI] [PubMed] [Google Scholar]
- 38. Proost S., Van Bel M., Sterck L., et al. 2009, PLAZA: a comparative genomics resource to study gene and genome evolution in plants, Plant Cell, 21, 3718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Denton J.F., Lugo-Martinez J., Tucker A.E., Schrider D.R., Warren W.C., Hahn M.W.. 2014, Extensive error in the number of genes inferred from draft genome assemblies, PLoS Comput. Biol., 10, e1003998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Upadhyay A.K., Chacko A.R., Gandhimathi A., et al. 2015, Genome sequencing of herb Tulsi (Ocimum tenuiflorum) unravels key genes behind its strong medicinal properties, BMC Plant Biol., 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang X., Lu P., Luo Z.. 2013, GMATo: a novel tool for the identification and analysis of microsatellites in large genomes, Bioinformation, 9, 541–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng J., Zhao Z., Li B., et al. 2016, A comprehensive characterization of simple sequence repeats in pepper genomes provides valuable resources for marker development in Capsicum, Sci. Rep., 6, 18919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Camacho C., Coulouris G., Avagyan V., et al. 2009, BLAST+: architecture and applications, BMC Bioinformatics, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boeckmann B., Bairoch A., Apweiler R., et al. 2003, The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003, Nucleic Acids Res., 31, 365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. UniProt Consortium. 2014, UniProt: a hub for protein information, Nucleic Acids Res., 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gene Ontology Consortium. 2004, The Gene Ontology (GO) database and informatics resource, Nucleic Acids Res., 32, D258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bairoch A.. 2007, UniProtKB/Swiss-Prot: The manually annotated section of the UniProt KnowledgeBase. Methods Mol. Biol, 406, 89–112.18287689 [Google Scholar]
- 48. Du Z., Zhou X., Ling Y., Zhang Z., Su Z.. 2010, AgriGO: a GO analysis toolkit for the agricultural community, Nucleic Acids Res., 38, W64–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ye J., Fang L., Zheng H., et al. 2006, WEGO: a web tool for plotting GO annotations, Nucleic Acids Res., 34, W293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waterhouse R.M., Tegenfeldt F., Li J., Zdobnov E.M., Kriventseva E.V.. 2013, OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs, Nucleic Acids Res., 41, D358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wegrzyn J.L., Liechty J.D., Stevens K.A., et al. 2014, Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation, Genetics, 196, 891–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li W., Godzik A.. 2006, CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences, Bioinformatics, 22, 1658–9. [DOI] [PubMed] [Google Scholar]
- 53. Lechner M., Findeiß S., Steiner L., Marz M., Stadler P.F., Prohaska S.J.. 2011, Proteinortho: detection of (co-) orthologs in large-scale analysis, BMC Bioinformatics, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Katoh K., Misawa K., Kuma K.I., Miyata T.. 2002, MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform, Nucleic Acids Res., 30, 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huelsenbeck J.P., Hillis D.M., Jones R.. 1996, Parametric bootstrapping in molecular phylogenetics: applications and performance In: Molecular Zoology: Advances, Strategies, and Protocols. Wiley-Liss, New York, pp. 19–45. [Google Scholar]
- 56. Le S.Q., Gascuel O.. 2008, An improved general amino acid replacement matrix, Mol. Biol. Evol., 25, 1307–20. [DOI] [PubMed] [Google Scholar]
- 57. Tamura K., Stecher G., Peterson D., Filipski A., Kumar S.. 2013, MEGA6: molecular evolutionary genetics analysis version 6.0, Mol. Biol. Evol., 30, 2725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Siltberg J., Liberles D.. 2002, A simple covarion‐based approach to analyse nucleotide substitution rates, J. Evol. Biol., 15, 588–94. [Google Scholar]
- 59. Rhee S.Y., Beavis W., Berardini T.Z., et al. 2003, The Arabidopsis information resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community, Nucleic Acids Res., 31, 224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Finn R.D., Mistry J., Schuster-Böckler B., et al. 2006, Pfam: clans, web tools and services, Nucleic Acids Res., 34, D247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cominelli E., Galbiati M., Vavasseur A., et al. 2005, A guard-cell-specific MYB transcription factor regulates stomatal movements and plant drought tolerance, Curr. Biol., 15, 1196–200. [DOI] [PubMed] [Google Scholar]
- 62. Chen Y., Zhang X., Wu W., Chen Z., Gu H., Qu L.J.. 2006, Overexpression of the wounding‐responsive gene AtMYB15 activates the shikimate pathway in Arabidopsis, J. Integr. Plant Biol., 48, 1084–95. [Google Scholar]
- 63. Gigolashvili T., Berger B., Mock H.P., Müller C., Weisshaar B., Flügge U.I.. 2007, The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana, Plant J., 50, 886–901. [DOI] [PubMed] [Google Scholar]
- 64. Hirai M.Y., Sugiyama K., Sawada Y., et al. 2007, Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis, Proc. Natl. Acad. Sci., 104, 6478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gigolashvili T., Engqvist M., Yatusevich R., Müller C., Flügge U.I.. 2008, HAG2/MYB76 and HAG3/MYB29 exert a specific and coordinated control on the regulation of aliphatic glucosinolate biosynthesis in Arabidopsis thaliana, New Phytol., 177, 627–42. [DOI] [PubMed] [Google Scholar]
- 66. Matus J.T., Aquea F., Arce-Johnson P.. 2008, Analysis of the grape MYB R2R3 subfamily reveals expanded wine quality-related clades and conserved gene structure organization across Vitis and Arabidopsis genomes, BMC Plant Biol., 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Van der Ent S., Verhagen B.W., Van Doorn R., et al. 2008, MYB72 is required in early signaling steps of rhizobacteria-induced systemic resistance in Arabidopsis, Plant Physiol., 146, 1293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li L., Yu X., Thompson A., et al. 2009, Arabidopsis MYB30 is a direct target of BES1 and cooperates with BES1 to regulate brassinosteroid‐induced gene expression, Plant J., 58, 275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Segarra G., Van der Ent S., Trillas I., Pieterse C.. 2009, MYB72, a node of convergence in induced systemic resistance triggered by a fungal and a bacterial beneficial microbe, Plant Biol., 11, 90–6. [DOI] [PubMed] [Google Scholar]
- 70. Seo P.J., Park C.-M.. 2009, Auxin homeostasis during lateral root development under drought condition, Plant Signal. Behav., 4, 1002–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang Y., Cao G., Qu L.-J., Gu H.. 2009, Characterization of Arabidopsis MYB transcription factor gene AtMYB17 and its possible regulation by LEAFY and AGL15, J. Genet. Genomics, 36, 99–107. [DOI] [PubMed] [Google Scholar]
- 72. Dubos C., Stracke R., Grotewold E., Weisshaar B., Martin C., Lepiniec L.. 2010, MYB transcription factors in Arabidopsis, Trends Plant Sci., 15, 573–81. [DOI] [PubMed] [Google Scholar]
- 73. Ambawat S., Sharma P., Yadav N.R., Yadav R.C.. 2013, MYB transcription factor genes as regulators for plant responses: an overview, Physiol. Mol. Biol. Plants, 19, 307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Araki R., Hasumi A., Nishizawa O.I., et al. 2013, Novel bioresources for studies of Brassica oleracea: identification of a kale MYB transcription factor responsible for glucosinolate production, Plant Biotech. J., 11, 1017–27. [DOI] [PubMed] [Google Scholar]
- 75. Chen Y., Chen Z., Kang J., Kang D., Gu H., Qin G.. 2013, AtMYB14 regulates cold tolerance in Arabidopsis, Plant Mol. Biol. Rep., 31, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pourcel L., Irani N.G., Koo A.J., Bohorquez‐Restrepo A., Howe G.A., Grotewold E.. 2013, A chemical complementation approach reveals genes and interactions of flavonoids with other pathways, Plant J., 74, 383–97. [DOI] [PubMed] [Google Scholar]
- 77. Gibbs D.J., Voß U., Harding S.A., et al. 2014, AtMYB93 is a novel negative regulator of lateral root development in Arabidopsis, New Phytol., 203, 1194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xie R., Zheng L., Deng L., et al. 2014, The role of R2R3MYB transcription factors in plant stress tolerance, J. Anim. Plant Sci., 24, 1821–33. [Google Scholar]
- 79. Roberts A., Feng H., Pachter L.. 2013, Fragment assignment in the cloud with eXpress-D, BMC Bioinformatics, 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Warnes G.R., Bolker B., Bonebakker L., et al. 2009, gplots: various R programming tools for plotting data. R package version, 2. [Google Scholar]
- 81. Kent W.J. 2002, BLAT—the BLAST-like alignment tool, Genome Res., 12, 656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Altschul S.F., Madden T.L., Schäffer A.A., et al. 1997, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25, 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Langmead B., Salzberg S.L.. 2012, Fast gapped-read alignment with Bowtie 2, Nat. Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bushnell B. 2016, BBMap short read aligner. http://sourceforge.net/projects/bbmap (17 June 2016, date last accessed).
- 85. Bankevich A., Nurk S., Antipov D., et al. 2012, SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing, J. Comp. Biol., 19, 455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu C., Shi L., Zhu Y., et al. 2012, CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences, BMC Genomics, 13, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nowak M.D., Russo G., Schlapbach R., Huu C.N., Lenhard M., Conti E.. 2015, The draft genome of Primula veris yields insights into the molecular basis of heterostyly, Genome Biol., 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Clouse J., Adhikary D., Page J., et al. 2016, The amaranth genome: genome, transcriptome, and physical map assembly, Plant Genome, 9. [DOI] [PubMed] [Google Scholar]
- 89. Claros M.G., Bautista R., Guerrero-Fernández D., Benzerki H., Seoane P., Fernández-Pozo N.. 2012, Why assembling plant genome sequences is so challenging, Biology, 1, 439–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Michael T.P., Jackson S.. 2013, The first 50 plant genomes, Plant Genome, 6. [Google Scholar]
- 91. Van Bakel H., Stout J.M., Cote A.G., et al. 2011, The draft genome and transcriptome of Cannabis sativa, Genome Biol., 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Peng Y., Lai Z., Lane T., et al. 2014, De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms, Plant Physiol., 166, 1241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Van Hoeck A., Horemans N., Monsieurs P., Cao H.X., Vandenhove H., Blust R.. 2015, The first draft genome of the aquatic model plant Lemna minor opens the route for future stress physiology research and biotechnological applications, Biotechnol. Biofuels, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rahman A.Y.A., Usharraj A.O., Misra B.B., et al. 2013, Draft genome sequence of the rubber tree Hevea brasiliensis, BMC Genomics, 14, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jia J., Zhao S., Kong X., et al. 2013, Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation, Nature, 496, 91–5. [DOI] [PubMed] [Google Scholar]
- 96. Ling H.-Q., Zhao S., Liu D., et al. 2013, Draft genome of the wheat A-genome progenitor Triticum urartu, Nature, 496, 87-90. [DOI] [PubMed] [Google Scholar]
- 97. Otto S.P. 2007, The evolutionary consequences of polyploidy, Cell, 131, 452–62. [DOI] [PubMed] [Google Scholar]
- 98. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. 2013, TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions, Genome Biol., 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Conesa A., Madrigal P., Tarazona S., et al. 2016, A survey of best practices for RNA-seq data analysis, Genome Biol., 17, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Glass K., Quackenbush J., Silverman E.K., et al. 2014, Sexually-dimorphic targeting of functionally-related genes in COPD, BMC Syst. Biol., 8, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cai Z., Mao X., Li S., Wei L.. 2006, Genome comparison using Gene Ontology (GO) with statistical testing, BMC Bioinformatics, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kubis S., Schmidt T., Heslop-Harrison J.S.P.. 1998, Repetitive DNA elements as a major component of plant genomes, Ann. Bot., 82, 45–55. [Google Scholar]
- 103. Brenchley R., Spannagl M., Pfeifer M., et al. 2012, Analysis of the bread wheat genome using whole-genome shotgun sequencing, Nature, 491, 705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mehrotra S., Goyal V.. 2014, Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function, Genomics Proteomics Bioinformatics, 12, 164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hoshino A.A., Bravo J.P., Morelli K.A., Nobile P.M.. 2012, Microsatellites as tools for genetic diversity analysis, Genet Divers Microorganisms., 6, 149–170. [Google Scholar]
- 106. Miah G., Rafii M.Y., Ismail M.R., et al. 2013, A review of microsatellite markers and their applications in rice breeding programs to improve blast disease resistance, Int. J. Mol. Sci., 14, 22499–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rastogi S., Kalra A., Gupta V., et al. 2015, Unravelling the genome of Holy basil: an “incomparable” “elixir of life” of traditional Indian medicine, BMC Genomics, 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Raven P.H. 1972, Why are bird-visited flowers predominantly red?, Evolution, 26, 674. [DOI] [PubMed] [Google Scholar]
- 109. Melendez-Ackerman E., Campbell D.R.. 1998, Adaptive significance of flower color and inter-trait correlations in an Ipomopsis hybrid zone, Evolution, 1293–303. [DOI] [PubMed] [Google Scholar]
- 110. Fulton M., Hodges S.A.. 1999, Floral isolation between Aquilegia formosa and Aquilegia pubescens, Proc. R. Soc. Lond., 266, 2247–52. [Google Scholar]
- 111. Bradshaw H., Schemske D.W.. 2003, Allele substitution at a flower colour locus produces a pollinator shift in monkeyflowers, Nature, 426, 176–8. [DOI] [PubMed] [Google Scholar]
- 112. Irwin R.E., Strauss S.Y.. 2005, Flower color microevolution in wild radish: evolutionary response to pollinator‐mediated selection, Am. Nat., 165, 225–37. [DOI] [PubMed] [Google Scholar]
- 113. de Pascual-Teresa S., Moreno D.A., García-Viguera C.. 2010, Flavanols and anthocyanins in cardiovascular health: a review of current evidence, Int. J. Mol. Sci., 11, 1679–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Falginella L., Castellarin S.D., Testolin R., Gambetta G.A., Morgante M., Di Gaspero G.. 2010, Expansion and subfunctionalisation of flavonoid 3′, 5′-hydroxylases in the grapevine lineage, BMC Genomics, 11, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wessinger C.A., Rausher M.D.. 2015, Ecological transition predictably associated with gene degeneration, Mol. Biol. Evol., 32, 347–54. [DOI] [PubMed] [Google Scholar]
- 116. Campanella J.J., Smalley J.V., Dempsey M.E.. 2014, A phylogenetic examination of the primary anthocyanin production pathway of the Plantae, Bot. Stud., 55, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Cone K.C., Cocciolone S.M., Burr F.A., Burr B.. 1993, Maize anthocyanin regulatory gene pl is a duplicate of c1 that functions in the plant, Plant Cell, 5, 1795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Seitz C., Ameres S., Schlangen K., Forkmann G., Halbwirth H.. 2015, Multiple evolution of flavonoid 3′, 5′-hydroxylase, Planta, 242, 561–73. [DOI] [PubMed] [Google Scholar]
- 119. Winkel B.S. 2006, The biosynthesis of flavonoids In: The Science of Flavonoids. Springer, New York, pp. 71–95. [Google Scholar]
- 120. Johnson E.T., Ryu S., Yi H., Shin B., Cheong H., Choi G.. 2001, Alteration of a single amino acid changes the substrate specificity of dihydroflavonol 4‐reductase, Plant J., 25, 325–33. [DOI] [PubMed] [Google Scholar]
- 121. Smith S.D., Wang S., Rausher M.D.. 2013, Functional evolution of an anthocyanin pathway enzyme during a flower color transition, Mol. Biol. Evol., 30, 602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Miosic S., Thill J., Milosevic M., et al. 2014, Dihydroflavonol 4-reductase genes encode enzymes with contrasting substrate specificity and show divergent gene expression profiles in Fragaria Species, PLoS One, 9, e112707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Quattrocchio F., Wing J.F., Va K., Mol J.N., Koes R.. 1998, Analysis of bHLH and MYB domain proteins: species‐specific regulatory differences are caused by divergent evolution of target anthocyanin genes, Plant J., 13, 475–88. [DOI] [PubMed] [Google Scholar]
- 124. Quattrocchio F., Wing J., van der Woude K., et al. 1999, Molecular analysis of the anthocyanin2 gene of Petunia and its role in the evolution of flower color, Plant Cell, 11, 1433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Albert N.W., Lewis D.H., Zhang H., Schwinn K.E., Jameson P.E., Davies K.M.. 2011, Members of an R2R3‐MYB transcription factor family in Petunia are developmentally and environmentally regulated to control complex floral and vegetative pigmentation patterning, Plant J., 65, 771–84. [DOI] [PubMed] [Google Scholar]
- 126. Zhang Y., Li L., Yan T.L., Liu Q.. 2014, Complete chloroplast genome sequences of Praxelis (Eupatorium catarium Veldkamp), an important invasive species, Gene, 549, 58-69. [DOI] [PubMed] [Google Scholar]
- 127. Clegg M.T., Gaut B.S., Learn G.H., Morton B.R.. 1994, Rates and patterns of chloroplast DNA evolution, Proc. Natl. Acad. Sci., 91, 6795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Martin G., Baurens F.-C., Cardi C., Aury J.-M., D’Hont A.. 2013, The complete chloroplast genome of banana (Musa acuminata, Zingiberales): insight into plastid monocotyledon evolution, PLoS One, 8, e67350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hansen D.R., Dastidar S.G., Cai Z., et al. 2007, Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae), Mol. Phylogenet. Evol., 45, 547–63. [DOI] [PubMed] [Google Scholar]
- 130. Stebbins G.L. 1970, Adaptive radiation of reproductive characteristics in angiosperms, I: pollination mechanisms, Annu. Rev. Ecol. Evol. Syst., 1, 307–26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.