

Abstract
Recent human exome-sequencing studies have implicated polymorphic Brg1-associated factor (BAF) complexes (mammalian SWI/SNF chromatin remodeling complexes) in several intellectual disabilities and cognitive disorders, including autism. However, it remains unclear how mutations in BAF complexes result in impaired cognitive function. Post-mitotic neurons express a neuron-specific assembly, nBAF, characterized by the neuron-specific subunit BAF53b. Subdomain 2 of BAF53b is essential for the differentiation of neuronal precursor cells into neurons. We generated transgenic mice lacking subdomain 2 of Baf53b (BAF53bΔSB2). Long-term synaptic potentiation (LTP) and long-term memory, both of which are associated with phosphorylation of the actin severing protein cofilin, were assessed in these animals. A phosphorylation mimic of cofilin was stereotaxically delivered into the hippocampus of BAF53bΔSB2 mice in an effort to rescue LTP and memory. BAF53bΔSB2 mutant mice show impairments in phosphorylation of synaptic cofilin, LTP, and memory. Both the synaptic plasticity and memory deficits are rescued by overexpression of a phosphorylation mimetic of cofilin. Baseline physiology and behavior were not affected by the mutation or the experimental treatment. This study suggests a potential link between nBAF function, actin cytoskeletal remodeling at the dendritic spine, and memory formation. This work shows that a targeted manipulation of synaptic function can rescue adult plasticity and memory deficits caused by manipulations of nBAF, and thereby provides potential novel avenues for therapeutic development for multiple intellectual disability disorders.
Recent human exome-sequencing data have implicated de novo mutations in genes regulating chromatin structure and synaptic function in a host of human neuropsychiatric disorders including autism, schizophrenia, depression, bipolar disorder, and intellectual disability (Vogel-Ciernia and Wood 2013). Together these large-scale sequencing efforts point to a critical role for regulation of transcription via chromatin modification (e.g., histone acetylation) and chromatin remodeling (e.g., nucleosome remodeling). Nucleosome remodeling is carried out by large, multi-subunit complexes that hydrolyze ATP to alter chromatin structure by sliding, evicting, or exchanging nucleosomes (Hargreaves and Crabtree 2011). However, the role of chromatin remodeling in regulating transcription required for cognitive function remains largely unexplored. This is a critical question considering that de novo mutations in nucleosome remodeling genes that compose the nBAF (neuron-specific Brg1-associated factor) complex are considered to be causally involved in human intellectual disability disorders (Koga et al. 2009; Loe-Mie et al. 2010; Nord et al. 2011; Hoyer et al. 2012; Neale et al. 2012; Roak et al. 2012; Santen et al. 2012; Tsurusaki et al. 2012; Van Houdt et al. 2012; Wolff et al. 2012; Chesi et al. 2013). Mutations in genes encoding nBAF subunits are associated with several developmental and intellectual disability disorders including Coffin-Siris syndrome (Santen et al. 2012; Tsurusaki et al. 2012), Nicolaides–Baraitser syndrome (Van Houdt et al. 2012), and autism spectrum disorders (ASD) (Neale et al. 2012; Roak et al. 2012). Furthermore, network analysis has implicated BAF53b in a gene coexpression module associated with decreased expression in ASD cortex samples (Voineagu et al. 2011).
How mutations in nBAF subunits lead to impaired human intellectual function is largely unknown and potentially relies on both developmental and adult functions of nBAF (Vogel-Ciernia and Wood 2013). Our prior work with BAF53b, a neuron-specific subunit of nBAF (Harata et al. 1999; Olave et al. 2002), mutant mice indicated a role for BAF53b in long-term memory and synaptic plasticity that may have been driven by deficits in actin polymerization (Vogel-Ciernia et al. 2013). In this study, we sought to investigate whether manipulation of cofilin, which ultimately regulates actin polymerization, could ameliorate BAF53b-dependent impairments in memory and synaptic plasticity.
As a critical part of the nBAF complex, BAF53b is required for nBAF's role in regulating activity-dependent gene expression and dendritic spine formation in cultured neurons (Wu et al. 2007). BAF53b is unique in that it is only expressed in post-mitotic neurons and is found only in the nBAF complex (Olave et al. 2002), making it an ideal target for examining the contribution of nBAF in neuronal function. The non-neuronal homolog of BAF53b, BAF53a is found in all other cell types and in multiple nucleosome remodeling complexes (Harata et al. 1999; Olave et al. 2002; Park et al. 2002). BAF53b and BAF53a are encoded by two separate genes that share 93% similarity with most of the divergence occurring in subdomain 2 (amino acids 39–82). Subdomain 2 of BAF53b appears to be the critical domain for regulating BAF53b's function in dendritic spine maturation. Switching subdomain 2 of BAF53a to b rescued dendritic outgrowth deficits and alterations in gene expression in Baf53b−/− knockout neuronal cultures. The reverse swap (BAF53b with subdomain 2 from BAF53a) failed to rescue the phenotype, indicating a critical role for subdomain 2 of BAF53b in synapse formation (Wu et al. 2007). These findings indicate that BAF53b, and specifically subdomain 2, may also be critically important for synaptic structure and function in the adult brain.
Consequently, to examine the unique contribution of the neuron-specific function of BAF53b, and consequently nBAF, in synaptic plasticity and memory in the adult brain we targeted subdomain 2 of BAF53b by overexpressing a mutant form of BAF53b lacking subdomain 2 in excitatory forebrain neurons (BAF53bΔSB2 transgenic mice). BAF53bΔSB2 mutant mice exhibit severe, yet specific, impairments in synaptic plasticity and memory formation. Expression of a phospho-mimetic of cofilin, which promotes actin polymerization, ameliorated both synaptic plasticity and long-term memory impairments in adult BAF53bΔSB2 mutant mice. Together, these results suggest that BAF53b may lead to a disruption in actin dynamics at the synapse, which affects synaptic plasticity and memory processes in the adult brain.
Results
Generation of BAF53bΔSB2 mutant mice
BAF53b is highly expressed throughout the adult brain and localizes to the neuronal nucleus (Fig. 1A,B). Subdomain 2 (SB2) of BAF53b (amino acids 39–82) is the region of BAF53b most divergent from BAF53a (non-neuronal homolog of BAF53b) with 84% identity (95% similarity) over the entire 429 amino acid alignment compared with amino acids 39–82 of Subdomain 2 (40% identity and 78% similarity) (Fig. 1C; Harata et al. 1999; Kuroda et al. 2002; Olave et al. 2002). To examine the neuron-specific role of BAF53b in long-term memory processes, we engineered transgenic mice that express a mutant form of BAF53b with a deletion of subdomain 2 (BAF53bΔSB2; see Fig. 1D). The BAF53bΔSB2 transgene (Fig. 1D) is expressed by the CaMKIIα promoter (Mayford et al. 1996), which allows for restricted expression to forebrain excitatory neurons during postnatal development (Kojima et al. 1997). We characterized two independently derived transgenic lines based on levels of transgene expression in dorsal hippocampus: a low-expressing line and a high expressing line (BAF53bΔSB2low and BAF53bΔSB2high; Fig. 1E). Endogenous wild-type BAF53b levels are unaltered in the BAF53bΔSB2high at baseline (Fig. 1F). Localization of BAF53bΔSB2 protein was not possible since the only antibodies that distinguish between BAF53b and BAF53a recognize the SB2 domain, which is deleted in the mutants. However, nuclear staining of other components of the nBAF complex (Brg1 and CREST, Supplemental Fig. S1A–D) indicates that expression of the transgene does not disrupt nBAF nuclear localization.
Figure 1.

Characterization of BAF53bΔSB2 transgenic mice. (A) The 4× scans of BAF53b expression in adult (12 wk) mouse brain. BAF53b expression is shown in green, NeuN expression in red, and DAPI in blue. The merged image contains all three channels. Scale bar 500 microns (B) Same panels as A but at 63× in dorsal hippocampal CA1. Scale bar 10 microns. (C) Predicted BAF53a, BAF53b, and the BAF53bΔSB2 mutant protein structure models simulated by comparison to know crystal structures for Actin using Modeller (Eswar et al. 2006). Below is shown the amino acid sequence for subdomain 2 (SB2) of BAF53b aligned to BAF53a. Note the lack of consensus across the domain. (D) BAF53bΔSB2 transgene construction. Amino acids 39–66 of the SB2 domain were deleted from wild-type BAF53b cDNA to make BAF53bΔSB2. This sequence was cloned upstream of the SV40 intron and polyadenylation signal and downstream from the 8.5 kb mouse CaMKIIα promoter. This construct was used to generate BAF53bΔSB2 transgenic mice. (E) Quantitative RT-PCR was performed with transgene specific primers to measure transgene expression in dorsal hippocampus of two independently derived lines of BAF53bΔSB2 mice (n = 9–10 animals per group; Mann–Whitney U = 4.00, P < 0.001). (F) Endogenous BAF53b expression in the dorsal hippocampus of BAF53bΔSB2high (n = 12) and wild-type (n = 12) littermates does not significantly differ (Mann–Whitney U = 56.00, P = 0.371). Mean (±SEM).
Selective disruption in hippocampal LTP within the trisynaptic circuit in slices from BAF53bΔSB2 mutant mice
To assess the potential contributions of Subdomain 2 of BAF53b in synaptic plasticity, we examined long-term potentiation (LTP) in acute hippocampal slices from adult BAF53bΔSB2low and BAF53bΔSB2high mice. We tested for differences between the two mutant groups and respective wild-type littermates at three sites in adult hippocampal slice (Fig. 2A): Schaffer-commissural projections to CA1, mossy fiber projections from the dentate gyrus to CA3, and in the lateral perforant pathway from cortex to the dentate gyrus. To compare BAF53bΔSB2low and BAF53bΔSB2high to wild types across the three regions, which differ from each other with regard to degree of stable potentiation, we normalized the mutant results to percent wild-type stable (plateau) LTP (Fig. 2 insets).
Figure 2.

Selective disruption in hippocampal LTP within the trisynaptic circuit in BAF53bΔSB2low and BAF53bΔSB2high slices. (A) Recording sites within the hippocampal slice. Schematic illustrating three synaptic recording sites within the hippocampus; R1 (blue, CA1 stratum radiatum), R2 (red, Mossy fiber projections (MFP)), and R3 (green, lateral perforant path (LPP)). The medial perforant path (MPP) runs close to the granular cell layer and can be distinguished from LPP with its characteristic paired-pulse depression. (B) LTP in CA1 stratum radiatum. Simultaneous recordings of slices receiving baseline stimulation (white circle) and five theta bursts (TBS; arrow) (gray, blue, and light blue circles). LTP gradually decayed toward baseline in slices recorded from BAF53bΔSB2low (ΔSB2low) and BAF53bΔSB2high (ΔSB2high) mice with respect to wild-type (WT) control LTP (n ≥ 4 mice per group, RM ANOVA: F(2,19) = 19.11, P < 0.0001). (Inset) fEPSP slopes from ΔSB2low and ΔSB2high slices were combined and normalized to WT controls. Plot illustrates percent potentiation in mutant (black circles) and WT (gray circles) mice beginning 10 min following TBS. (C) LTP in Mossy Fiber Projection—CA3. LTP in slices recorded from ΔSB2low (orange circle) and ΔSB2high (red circle) mice produced a slight, but significant increase in level of LTP 50 min post-HFS relative to controls WT slices (n ≥ 6 mice per group, RM ANOVA: F(2,26) = 5.36, P = 0.01). (Inset) fEPSP amplitude from ΔSB2low and ΔSB2high slices were combined and normalized to WT controls. Plot illustrates percent potentiation in mutant and WT mice beginning 5 min following HFS. When combined, there was a significantly greater potentiation in the mutants compared with WT (RM ANOVA: F(1,28) = 9.55, P = 0.005). (D) LTP in lateral perforant pathway—dentate gyrus. LTP induction and maintenance were similar in slices from ΔSB2low (light green circle), ΔSB2high (green circle), and WT mice (n ≥ 4 mice per group, RM ANOVA: F(2,30) = 0.09, P = 0.91). (Inset) fEPSP slope from ΔSB2low and ΔSB2high slices were combined and normalized to WT controls. Plot illustrates percent potentiation in mutant and WT mice beginning 10 min following HFS. Combined analysis indicate no differences between groups (RM ANOVA: F(1,31) = 0.001, P = 0.98). All inset traces represent field recordings collected during baseline stimulation (black line) and 50 min following stimulation (red line) in ΔSB2high and WT mice. Scale bars: 1 mV, 5 msec.
The Schaffer-commissural projections to the proximal apical dendrites of field CA1 had comparable input/output curves and paired-pulse facilitation (PPF) across the groups (Supplemental Fig. S2A,B), indicating that the mutations did not affect axon excitability, packing densities, or transmitter release kinetics. Five theta burst stimulation delivered to wild-type slices produced the expected rapid, marked increase in the size of fEPSPs followed by a decay over 10 min to a stable plateau at ∼50% above the pre-TBS baseline (Fig. 2B). A similar rapid increase in responses was observed for both BAF53bΔSB2low and BAF53bΔSB2high mutants but failed to stabilize with the level of potentiation falling to near baseline levels after 60 min (Fig. 2B). Comparisons between the BAF53bΔSB2low and BAF53bΔSB2high slices did not approach statistical significance (Bonferroni corrected t-tests for each time point P > 0.05 ΔSB2low versus ΔSB2high). It is evident from this analysis that BAF53bΔSB2 eliminates the stabilization of LTP in the Schaffer-commissural pathway.
A very different result was obtained in the mossy fiber connections between the dentate gyrus and field CA3 of hippocampus. Postsynaptic responses in this area are contaminated by spiking elicited by the potent (“detonator”) mossy fiber terminals, making it difficult to measure input/output curves to assess axonal excitability; accordingly, we used antidromic responses and did not detect differences between wild types and BAF53bΔSB2low or BAF53bΔSB2high slices (Supplemental Fig. S2C). Following a stable baseline recording in standard aCSF containing 100 µM D,L-APV to block associational fiber projections, two high frequency trains (HFS; 100Hz, 1 sec, 1-min interval) were applied to mossy fiber projections in the hilus. HFS of the mossy fibers in wild-type slices produced a large, short-term potentiation followed by a rapid drop to a stable plateau (∼80% above baseline) that persisted for the duration of the recording session (Fig. 2C). The BAF53bΔSB2 mutant slices also produced an elevated level of potentiation, with the BAF53bΔSB2high showing a larger potentiation compared with wild-type (Bonferroni corrected t-tests for each time point P < 0.05 WT versus ΔSB2high) (Fig. 2C). Bonferroni corrected post hoc tests did not reveal a difference between the BAF53bΔSB2low animals and wild type (last 10 min of recording P > 0.05 WT versus ΔSB2low), however BAF53bΔSB2low and BAF53bΔSB2high slices also did not significantly differ, indicating both mutant lines show mossy fiber LTP abnormalities (Bonferroni corrected t-tests for each time point P > 0.05 ΔSB2low versus ΔSB2high). To further examine the impact of the BAF53bΔSB2 transgene, we normalized the percent potentiation to the values for wild types during the plateau period, which confirmed that the mutants had a greater degree of response facilitation (Fig. 2C inset).
We next tested for the effects of impaired nBAF complex function on the lateral perforant input to the dentate gyrus, identified by a robust PPF effect (Supplemental Fig. S2D; Christie and Abraham 1994). As previously shown induction of LTP in these synapses with a single train of HFS (100 Hz, 1 sec) was facilitated by pretreating the slices with an antagonist of GABA-A receptors (10 µM picrotoxin) (Bramham and Sarvey 1996; Suzuki and Okada 2007). Under these conditions, HFS produced a marked potentiation in wild-type slices that decayed over 15 min to a stable level ∼30% above baseline. A comparable curve was found in BAF53bΔSB2low and BAF53bΔSB2high slices (Fig. 2D). Using the same normalization to stable wild-type potentiation described for the Schaffer-commissural and mossy fibers, we found no evidence for a difference between wild-type and mutant slices (Fig. 2D inset).
Phosphorylation of cofilin by LTP induction is severely impaired in BAF53bΔSB2 mutant mice
Past studies showed that theta bursts cause a rapid (<2 min) and transient phosphorylation of cofilin localized to postsynaptic densities in field CA1 (Lynch and Gall 2013). Cofilin is a constitutively active protein that severs dynamic actin filaments and thereby prevents polymerization. Phosphorylation at Ser 3 inactivates cofilin, allowing filaments to extend, elaborate, and stabilize. These latter events are critical to the consolidation of recently induced LTP (Chen et al. 2007). Disruption of cofilin inactivation is therefore a plausible explanation for the loss of CA1-LTP in BAF53bΔSB2high mutants. We tested this hypothesis using fluorescence deconvolution tomography (FDT), a technique that allows for the 3-D reconstruction of large numbers (∼100,000) of synapses in the region surrounding the pipette tip used to record fEPSPs (Rex et al. 2009; Chen et al. 2010). Multiple sections through a slice were collected and immunostained for phospho-cofilin and the postsynaptic density (PSD) marker PSD95; individual PSD95 positive structures matching the size and eccentricity constraints of synapses that were colabeled with phospho-cofilin were counted (Fig. 3A). The total number of double-labeled contacts (“contacts” refers to PSD95 labeling that both satisfies the size and eccentricity constraints of a synapse and is colocalized with phospho-cofilin) in slices collected after baseline recording was comparable in wild types (14,182 ± 332 per sampling zone; n = 13 slices) and mutants (14,826 ± 239; n = 15), indicating that baseline activity of cofilin was undisturbed in the latter (P > 0.90, t-test). This also held for the number of psds (with and without pCofilin) (P > 0.50).
Figure 3.

Theta burst induced phosphorylation of synaptic cofilin is impaired in hippocampal slices prepared from BAF53bΔSB2high mice. Slices were removed 3 min after delivery of low-frequency stimulation (LFS) or theta burst stimulation (TBS) to the Schaffer-commissural projections to the apical dendrites of field CA1b. (A) Sections were immunostained for PSD95 (green) and phospho-cofilin (red). Double-labeled structures matching the size constraints of synapses (arrows) were counted and the density of phospho-cofilin measured. Scale bars: 5 µm and 1 µm (inset). (B) Frequency distribution of the density of phospho-cofilin labeling colocalized with PSD95. The percentage of all double-labeled contacts at different phospho-cofilin intensities is described. TBS produced a highly significant (n = 13–14); (F(19,475) = 6.117; P < 0.0001) rightward skew to the curve relative to LFS. (C) Results for BAF53bΔSB2high slices; there was no statistical difference between the LFS and TBS curves (n = 15 per group; (F(19,532) = 0.5519; P = 0.9381). (D) The percent difference in pCofilin density between low-frequency stimulation and TBS slices for high-density bins (≥92.5 units) was greater in the wild-type group than in the mutants (P = 0.029, t-test, two tails). (E) Example image of Golgi staining for the first or second dendritic branch of a wild-type mouse in stratum radiatum dorsal hippocampus CA1 (left panel). Single plane image. Scale bar 1 micron. (Middle panel) Maximum intensity projection following processing in Image J. (Right panel) Analysis of the dendritic segment using NeuronStudio. The branch is first traced (green circles) and then dendritic spines are automatically detected, measured, and categorized. Stubby spines are labeled with pink dots, mushroom spines with orange dots, and thin spines with blue dots. See Materials and Methods for further details on spine categorization. (F) Same as (E) but for BAF53bΔSB2high mice. (G) Overall spine density (number of spines per micron) for wild-type and BAF53bΔSB2high mice is equivalent (Mann–Whitney U (12) = 21.00, P = 0.71) n = 7 animals per genotype, 9–11 branches per animal. (H) Overall head diameter (microns) was equivalent between the two genotypes (Mann–Whitney U (12) = 13.00, P = 0.16). (I) Overall spine length was not different between wild-type and BAF53bΔSB2high mice (Mann–Whitney U (12) = 19.00, P = 0.54). (J) Density of different spine types are equivalent between genotypes two-way ANOVA no main effect of genotype F(1,36) = 0.27, P = 0.61, significant effect of spine type F(2,36) = 569.8, P < 0.0001, no significant interaction F(2,36) = 0.39, P = 0.68. Mean (±SEM).
Next, we measured the density of phospho-cofilin labeling for each double-labeled contact and expressed the data as frequency histograms (percentage of double-labeled contacts according to density bins). Four groups of slices were measured: wild type with low-frequency stimulation; wild type removed 3 min after TBS; BAF53bΔSB2high with LFS; BAF53bΔSB2high following TBS. The distributions of phospho-cofilin densities for the LFS and TBS were then compared for the wild-type and mutant groups. As described in an earlier study (Vogel-Ciernia et al. 2013), the frequency distribution for the density of pCofilin was right-shifted in the low frequency (control) in the mutant slices relative to wild-type preparations (two-way ANOVA: F(19,520) = 4.064, P < 0.0001). TBS produced a highly significant rightward skew in the frequency curve in wild types similar to that previously described for control slices (Fig. 3B; Rex et al. 2009; Chen et al. 2010), indicating that TBS had increased phospho-cofilin density in a subpopulation of synapses. In marked contrast, the LFS and TBS curves were superimposed in the BAF53bΔSB2high slices (Fig. 3C). Similarly, the percent increase in high-density (≥92 units) pCofilin contacts produced by TBS was much greater in wild-type slices than in mutant slices (Fig. 3D).
Given that baseline counts of phospho-cofilin colocalized with PSD95 did not differ, the overall results suggest that a lack of phospho-cofilin induction in response to stimulation leads to impaired LTP in BAF53b mutant mice (Chen et al. 2010). To further evaluate any global structural changes in dendritic spine density or morphology, we examined CA1 neurons by Golgi staining. Dendritic spine density and spine morphology were examined for the first and second branches of pyramidal cells in stratum radiatum of dorsal hippocampus CA1 (Fig. 3E,F). Nine to 11 branches were analyzed for each animal (Supplemental Fig. S3A) and an average of 440–490 dendritic spines were analyzed per animal (Supplemental Fig. S3B). BAF53bΔSB2high mice were similar to wild-type littermates for overall spine density (Fig. 3G) (Mann–Whitney U (12) = 21.00, P = 0.71), head diameter (Fig. 3H) (Mann–Whitney U (12) = 13.00, P = 0.16), and spine length (Fig. 3I) (Mann–Whitney U (12) = 19.00, P = 0.54). Spines were categorized into stubby, thin or mushroom using NeuronStudio (Fig. 3J) and there were no differences in the density of each spine type between the mutants and wild-type animals (Fig. 3J) (two-way ANOVA no main effect of genotype F(1,36) = 0.27, P = 0.61, significant effect of spine type F(2,36) = 569.8, P < 0.0001, no significant interaction F(2,36) = 0.39, P = 0.68). Similarly, there were no differences in the measured head diameters or spine lengths for any of the categories of spines (Supplemental Fig. S3C–H). The lack of obvious, gross structural differences in CA1 between wild-type and mutant animals suggests specific deficits in cofilin-dependent activity-regulated spine remodeling and not large-scale, developmental abnormalities.
A mimetic of phospho-cofilin rescues LTP deficits in BAF53b mutant mice
To determine whether manipulation of phospho-cofilin could ameliorate the hippocampal synaptic plasticity deficits in BAF53b mutant mice, we directly manipulated cofilin using adeno-associated virus (AAV) to introduce a phospho-mimic cofilin protein (cofilinS3D) into the dorsal hippocampus of adult mice. In the cofilinS3D construct, serine 3 was replaced by an aspartic acid and consequently acts as a phospho-mimetic of inactivated cofilin (Shi et al. 2009; Pontrello et al. 2012; Parisiadou et al. 2014; Newell-Litwa et al. 2015; Havekes et al. 2016). CofilinS3D overexpressed in hippocampal neuronal cultures localizes to the PSD (Newell-Litwa et al. 2015) and impacts dendritic spine morphology by promoting actin polymerization (Shi et al. 2009; Pontrello et al. 2012; Parisiadou et al. 2014; Newell-Litwa et al. 2015). CofilinS3D overexpressed in vivo in the dorsal hippocampus rescues memory, synaptic plasticity, and synapse density deficits induced by sleep deprivation without impacting endogenous cofilin expression (Havekes et al. 2016).
Three weeks after viral infusion robust expression is detected throughout the dorsal hippocampus (Fig. 4A). Both paired-pulse facilitation and input/output curves were similar between genotypes and viral conditions, indicating that viral expression did not negatively affect basic neuronal function (Fig. 4B,C). To examine the impact of virally expressed, constitutively inactive cofilin (cofilinS3D) on long-term plasticity in area CA1, five theta bursts were delivered to slices from BAF53bΔSB2high mice expressing the control AAV-GFP in one hemisphere and AAV-cofilinS3D in the other. Field responses showed a significant impairment in LTP in mutants expressing AAV-GFP when compared with wild-type littermates with either AAV-GFP or AAV-cofilinS3D (Fig. 4D). The overexpression of AAV-cofilinS3D did not appear to negatively impact wild-type animals as there was no difference between animals receiving AAV-GFP or AAV-cofilinS3D. There was, however, a rescue of LTP in the BAF53bΔSB2high animals with overexpression of cofilinS3D (BAF53bΔSB2high AAV-cofilinS3D was not different than either WT group) (Fig. 4D).
Figure 4.
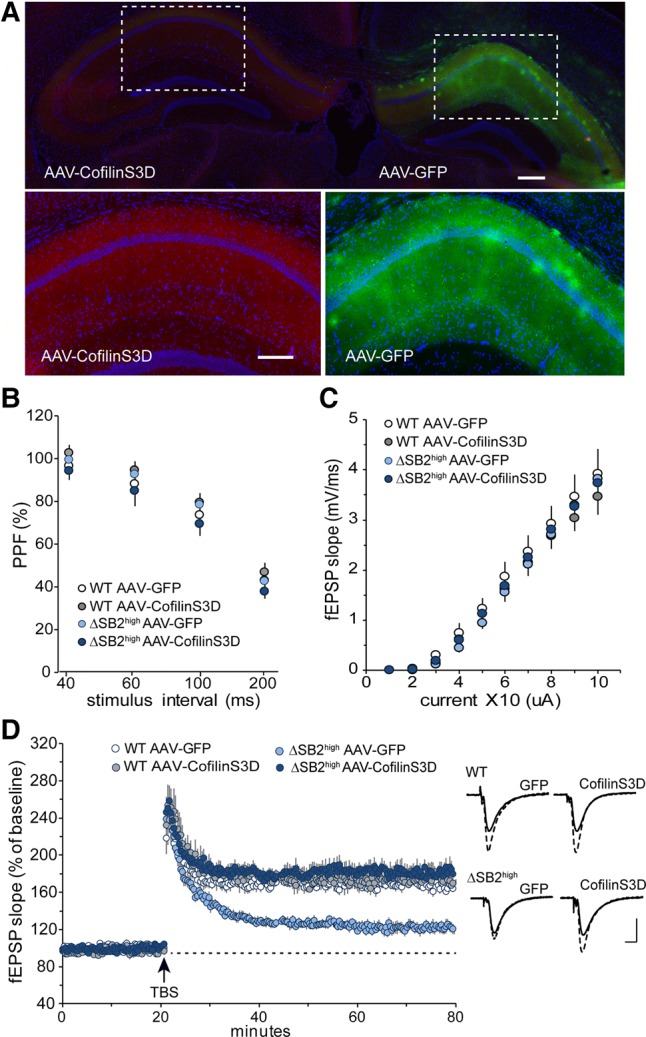
AAV-cofilinS3D expression in dorsal hippocampus rescues LTP deficits in CA1 stratum radiatum. (A) AAV-cofilinS3D and AAV-GFP are expressed throughout dorsal hippocampus (top image bilateral, 4×). Viral expression is localized by HA tag (AAV-cofilinS3D) or GFP expression (AAV-GFP) (lower images 20×). Scale bars 200 µm. (B) Paired-pulse facilitation of the initial slope of the synaptic response in hippocampal CA1 (40, 60, 100, and 200-msec inter-pulse intervals) was not significantly difference between groups (n = 5 mice/group, RM ANOVA: F(3,18) = 1.00, P = 0.417). (C) Input/output curves across a range of stimulation currents in hippocampal CA1 show no measurable different between groups (n = 5 mice/group, F(3,18) = 0.37, P = 0.776). (D). LTP in CA1 stratum radiatum following five theta bursts (TBS; arrow) in slices from wild-type or BAF53bΔSB2high mice expressing either AAV-GFP or AAV-cofilinS3D. Each animal received both viruses, one per hemisphere. LTP gradually decayed toward baseline in slices recorded from BAF53bΔSB2high mice with the control vector (BAF53bΔSB2high AAV-GFP) with respect to wild-type (WT) controls (AAV-GFP or AAV-cofilinS3D) (n = 5 mice per group, RM ANOVA: F(3,18) = 26.28, P < 0.0001). BAF53bΔSB2high AAV-cofilinS3D showed a complete rescue of the LTP deficits relative to WT controls (n = 5 mice per group, Bonferroni corrected t-tests for each minute of last 10 min of LTP: WT AAV-GFP versus WT AAV-cofilinsS3D all P > 0.05; WT AAV-GFP versus BAF53bΔSB2high AAV-GFP all P < 0.01; WT AAV-GFP versus BAF53bΔSB2high AAV-cofilinS3D all P > 0.05). Traces represent field recordings collected during baseline stimulation and 50 min following TBS for each group. Scale bar: 1 mV, 5 msec.
Phosphorylation mimetic of phospho-cofilin rescues long-term memory deficits in BAF53b mutant mice
We next examined short and long-term memory for object location (OLM; Fig. 5A) and object recognition (ORM; Fig. 5D) in the BAF53bΔSB2 mutant mice and wild-type littermates. Both anxiety and motor function were normal in BAF53bΔSB2 mice when compared with wild-type controls for open field, distance traveled during habituation to a novel context, and elevated plus maze (Supplemental Fig. S4A–F). Wild-type, BAF53bΔSB2low and BAF53bΔSB2high mice all demonstrated robust short-term OLM and ORM (tested at 90 min) (Fig. 5B,E; as measured by preference for the novel location over the familiar location and calculated with a discrimination index (DI)). There were no significant differences in total exploration time between the groups during testing (OLM: ANOVA F(2,42) = 0.10, P = 0.91; ORM: ANOVA F(2,35) = 1.60, P = 0.22) or training (OLM: ANOVA F(2,42) = 0.78, P = 0.47; ORM: ANOVA F(2,35) = 0.12, P = 0.89). In a different cohort of animals, the wild-type mice showed significant long-term OLM and ORM. However, both the high- and low-expressing lines of BAF53bΔSB2 mice exhibited significantly reduced DIs compared with wild-type littermates (Fig. 5C,F). There were no significant differences in total exploration time between the groups during testing (OLM: ANOVA F(2,39) = 0.29, P = 0.75; ORM: ANOVA F(2,35) = 0.78, P = 0.46) or training (OLM: ANOVA F(2,39) = 0.67, P = 0.52; ORM: ANOVA F(2,35) = 1.61, P = 0.21). Together, results shown in Figure 5A–F indicate that the subdomain 2 of BAF53b plays a critical role in long-term memory formation.
Figure 5.

BAF53bΔSB2 transgenic mice have intact short-term and impaired long-term memory. (A) Mice received 10 min training in an environment with two identical objects and received a retention test either 90 min (short-term) or 24 h (long-term) later in which one object is moved to a new location (OLM). (B) Wild-type (n = 22), BAF53bΔSB2low (n = 10), and BAF53bΔSB2high mutant mice (n = 13) all exhibit similar 90-min short-term hippocampus-dependent OLM (ANOVA F(2,42) = 0.60, P = 0.55). All three genotypes showed a DI significantly different from zero (wild-type t-test t(21) = 11.54, P < 0.0001; BAF53bΔSB2low t-test t(9) = 8.31, P < 0.0001 and BAF53bΔSB2high t-test t(12) = 9.80, P < 0.0001) (C) BAF53bΔSB2low (n = 12) and BAF53bΔSB2high mutant mice (n = 9) show significant deficits in 24-h long-term OLM compared with wild-type littermates (n = 21) (ANOVA F(2,39) = 30.37, P < 0.0001, Bonferroni corrected t-test: BAF53bΔSB2low versus wild type t(31) = 5.81, P < 0.05; BAF53bΔSB2high versus wild type t(28) = 6.82, P < 0.05). (D) Mice received 10-min training in an environment with two identical objects and received a retention test 90 min (short-term) or 24 h (long-term) later in which one object is replaced with a novel one. (E) In a hippocampus-independent object recognition task BAF53bΔSB2low (n = 9) and BAF53bΔSB2high (n = 8) exhibit 90-min short-term ORM similar to wild-type littermates (n = 21) (Kruskal–Wallis H(2,35) = 2.19, P = 0.33). All three genotypes showed a DI significantly different from zero (wild-type t-test t(20) = 13.14, P < 0.0001; BAF53bΔSB2low t-test t(8) = 10.53, P < 0.0001 and BAF53bΔSB2high t-test t(7) = 9.01, P < 0.0001). (F) BAF53bΔSB2low (n = 10) and BAF53bΔSB2high mutant mice (n = 11) show significant deficits in 24 h long-term ORM compared with wild-type littermates (n = 17) (ANOVA F(2,35) = 15.93, P < 0.0001, Bonferroni corrected t-test: BAF53bΔSB2low versus wild type t(25) = 4.26, P < 0.05; BAF53bΔSB2high versus wild type t(26) = 5.05, P < 0.05). (G) AAV-cofilinS3D is expressed throughout dorsal hippocampus of wild-type and BAF53bΔSB2high mutant mice. Viral expression is localized by HA tag. Scale bar 200 µm. (H) BAF53bΔSB2high mice with AAV-GFP viral expression in dorsal hippocampus (n = 13) show impaired OLM compared with wild-type littermates with hippocampal AAV-GFP expression (n = 14) (ANOVA no main effect for genotype F(1,57) = 2.01, P = 0.16, no effect of virus F(1,57) = 4.00, P = 0.05 but a significant interaction F(1,57) = 4.57, P = 0.04). Overexpression of AAV-cofilinS3D in dorsal hippocampus of BAF53bΔSB2high mice (n = 18) completely restored the long-term memory formation compared with wild-type littermates with AAV-cofilinS3D expression (n = 16) (Bonferroni corrected t-test: AAV-GFP and BAF53bΔSB2high mice with AAV-GFP t(25) = 2.38, P < 0.05; wild-type AAV-cofilinS3D and BAF53bΔSB2high mice with AAV-cofilinS3D t(32) = 0.54, P > 0.05). Mean (±SEM).
To further test the ability of the phospho-mimetic cofilin to rescue impairments produced by the loss of BAF53b function, we examined whether introduction of phospho-mimetic cofilin (AAV-cofilinS3D) into the dorsal hippocampus of adult BAF53bΔSB2high mice could reverse deficits in long-term memory formation (Fig. 5G). Three weeks following viral infusion, BAF53bΔSB2high mice and wild-type littermates were tested for alterations in anxiety with elevated plus maze and then examined for long-term OLM. All four groups (BAF53bΔSB2high mice and wild-type littermates with either AAV-cofilinS3D or AAV-GFP control virus) showed comparable levels of anxiety and habituation to the OLM context (Supplemental Fig. S4G,H). When tested for long-term OLM, BAF53bΔSB2high mice with the AAV-GFP virus showed long-term memory impairments relative to wild-type littermates with the control virus. Overexpression of cofilinS3D in BAF53bΔSB2high mice rescued the long-term memory impairments (Fig. 5H). There were no differences in overall exploration at test (ANOVA no effect of genotype F(1,57) = 0.83, P = 0.34, no effect of virus F(1,57) = 0.85, P = 0.36 and no interaction F(1,57) = 1.06, P = 0.31) or during training (ANOVA no effect of genotype F(1,57) = 0.30, P = 0.58, no effect of virus F(1,57) = 1.26, P = 0.27, and no interaction F(1,57) = 1.84, P = 0.18). Previous work demonstrated that AAV-cofilinS3D does not negatively impact transcriptionally independent short-term object location memory (Havekes et al. 2016). Consequently, the rescue of long-term OLM with AAV-cofilinS3D suggests a potential link between BAF53b disruption of long-term memory formation and synaptic function at the level of actin cytoskeleton signaling.
Discussion
Epigenetic regulation of gene expression and disruption of synaptic function are emerging as key contributors to human neuropsychiatric disorders. Large-scale, exome-sequencing studies of both intellectual disability and autism spectrum disorders have identified mutations in chromatin remodelers (Ben-David and Shifman 2012). Specifically, de novo mutations in multiple different subunits of the BAF complex have been found in patients with intellectual disability disorders (Santen et al. 2012; Tsurusaki et al. 2012; Van Houdt et al. 2012) and ASD (Neale et al. 2012; Roak et al. 2012). Our prior work on BAF53b, a key subunit of the nBAF chromatin remodeling complex, suggested a critical role for long-term memory formation and deficits in synaptic plasticity. To further explore a link between BAF53b's specific function in neurons and synaptic plasticity we targeted subdomain 2 of BAF53b, the domain of BAF53b that confers it's neuron-specific function (Wu et al. 2007; Tea and Luo 2011). Disruption of nBAF by targeted overexpression of BAF53b lacking subdomain 2 significantly disrupted LTP in the hippocampal trisynaptic circuit, phosphorylation of cofilin, and long-term memory formation. These deficits were rescued by introducing a constitutively inactive phosphorylation mimetic of cofilin, providing a connection between BAF53b-induced memory and plasticity deficits and actin remodeling at the dendritic spine.
The most profound LTP deficits were observed in CA1 stratum radiatum, a region of the hippocampus where phosphorylation of cofilin is the final signaling step in the actin remodeling pathway that underlies LTP maintenance (Chen et al. 2007). In response to neuronal activity the transient phosphorylation of cofilin at Ser3 blocks cofilin's interaction with F-actin and inhibits actin severing. The consequent shift in actin dynamics results in both dendritic spine remodeling and regulation of AMPA receptor dynamics (Gu et al. 2010; Rust et al. 2010). Cofilin phosphorylation has been linked to spine enlargement and stabilization following LTP induction (Chen et al. 2007), destabilization and shrinkage following long-term depression (Zhou et al. 2004), and the transformation of mature mushroom spines to immature thin spines (Shi et al. 2009). A lack of phosphorylation of cofilin in multiple models of BAF53b mutant mice (Vogel-Ciernia et al. 2013) indicates excessively active cofilin (lack of activity-induced phosphorylation) that may prevent activity-dependent dendritic spine remodeling through disruption of actin dynamics. These deficits appear to be linked specifically to long-term, stable changes in synaptic connectivity as only long-term memory and LTP maintenance are impaired in multiple types of BAF53b mutant mice (Vogel-Ciernia et al. 2013) while overall synapse morphology and structure remain intact (Fig. 3).
Overexpression of a phospho-mimetic cofilin (cofilinS3D) has been used previously to examine the role of cofilin phosphorylation on dendritic spine morphology (Shi et al. 2009; Pontrello et al. 2012; Parisiadou et al. 2014; Newell-Litwa et al. 2015) and to rescue memory and LTP deficits induced by sleep deprivation (Havekes et al. 2016). Similar to endogenous phosphorylation of cofilin, cofilinS3D fails to bind F or G actin in vitro (Moriyama et al. 1996) and consequently may act as a competitive inhibitor by binding to Slingshot phosphatases and increasing endogenous p-cofilin levels at the synapse (Havekes et al. 2016). A similar type of mechanism was shown for a synthetic cell-permeable peptide of cofilin that enhanced the level of nonphosphorylated cofilin by effectively competing with endogenous cofilin as a substrate for LIM kinase (Aizawa et al. 2001). Overexpressing the cofilinS3D mutant may restore the p-cofilin/cofilin equilibrium by inhibiting dephosphorylation of endogenous p-cofilin. In support of this conclusion, cofilinS3D overexpression can reverse plasticity-induced changes in spine morphology observed with inhibition of other components of the actin signaling cascade including EphB2, FAK (Shi et al. 2009), and β-arrestin (Pontrello et al. 2012). Future work will be needed to carefully dissect which components of the actin signaling pathway are disrupted in BAF53b mutant animals, and whether BAF53b and the nBAF complex directly regulate expression of actin signaling pathway genes including cofilin.
The analysis of mossy fiber potentiation further supports the conclusion that disruption of conventional LTP in BAF53b mutants reflects disturbances to spine dynamics. Unlike CA1 where LTP has been firmly linked to reorganization of the postsynaptic cytoskeleton, activity-driven increases in transmission in the mossy fibers are expressed by presynaptic modifications (Staubli and Lynch 1990; Zalutsky and Nicoll 1990). This latter effect does not depend on spine changes and was not impaired in the BAF53b mutants, but instead was measurably greater than in wild types. Whether small but reliable enhancement reflects dysregulation of cofilin responses in mossy terminals to HFS is an open question, but there is evidence relating the actin severing protein and cytoskeletal remodeling to vesicle mobilization and exocytosis (Wolf et al. 2015). The discreteness of the plasticity defects in the BAF53b transgenics was further emphasized by the apparent normalcy of LTP in the lateral perforant path. Recent studies demonstrated that activity-driven enhancement of responses in these contacts is induced postsynaptically but expressed and stabilized presynaptically with the endocannabinoid 2-archidonoylglycerol serving as the requisite retrograde (spine to terminal) messenger. In marked contrast to CA1, actin polymerization in spines is not required for the production of LTP in the dentate gyrus (Wang et al. 2016). Collectively, the results indicate that disruptions of BAF53b selectively affect memory-related synaptic modifications that depend on reorganization of the spine cytoskeleton.
The findings from this study and others (Vogel-Ciernia et al. 2013) have demonstrated a role for BAF53b and the nBAF complex in learning and memory. In all of the different manipulations of BAF53b (BAF53bΔSB2 mice, Baf53b+/− heterozygous knockout mice, and dominant-negative BAF53bΔHD mice (Vogel-Ciernia et al. 2013)) induction of LTP was normal and long-term maintenance of LTP was impaired, indicating a critical role for BAF53b and nBAF in regulating processes required for long-term, stable changes in neuronal circuits. BAF53b appears to play a critical role in the adult brain, independent from the potential role of BAF53b in neuronal differentiation and development, as reintroduction of BAF53b into the adult Baf53b+/− heterozygous knockout mice rescued long-term memory (Vogel-Ciernia et al. 2013). These findings are similar to those in the BAF53bΔSB2 mutant mice in that delivery of cofilinS3D in the adult hippocampus rescued long-term OLM and synaptic plasticity deficits. The reversibility of the memory phenotype in the adult animal suggests that BAF53b may serve as a novel therapeutic target for reversing memory and cognitive symptoms associated with intellectual disability and ASD.
Together these findings indicate a critical role for subdomain 2 of BAF53b in regulating synaptic plasticity in the adult brain. The ability of cofilinS3D to rescue both long-term memory and synaptic plasticity in the BAF53bΔSB2 transgenic mice suggests that a failure in the activity-dependent induction of p-cofilin may be a key deficit in these animals. This study shows that a direct, targeted manipulation of synaptic activity-dependent cytoskeletal remodeling can rescue functional impairments driven by manipulations of neuron-specific chromatin remodeling machinery. These findings offer novel insight into the potential mechanism by which a neuron-specific chromatin remodeling complex for nervous system functions and helps explain why mutations impairing complex function in human neurodevelopmental disorders result in severe intellectual impairment (Koga et al. 2009; Loe-Mie et al. 2010; Nord et al. 2011; Hoyer et al. 2012; Neale et al. 2012; Roak et al. 2012; Santen et al. 2012; Tsurusaki et al. 2012; Van Houdt et al. 2012; Wolff et al. 2012; Chesi et al. 2013). Elucidating how chromatin remodeling complexes like nBAF shape the chromatin landscape and ultimately regulate gene expression and synaptic function is critical for further development of novel, activity-dependent synaptic therapeutics for ID and ASD.
Materials and Methods
Animals
All animals were between 8–15 wk old at the time of behavioral testing. Mice had free access to food and water and lights were maintained on a 12:12 h light–dark cycle, with all behavioral testing performed during the light portion of the cycle. All animals were group housed and backcrossed at least five generations to C57BL/6 (Jackson Labs). All experiments were conducted according to National Institutes of Health guidelines for animal care and use and were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
AAV Production and infusion
The AAV2.9-CamKII-cofilinS3D-HA virus (AAV-cofilinS3D) expresses a modified cofilin protein where serine 3 was replaced by an aspartic acid to serve as a phospho-mimetic of inactivated cofilin (Ser3-P). The construct was HA tagged and expression was driven by the neuron-specific CamKII promoter. The control virus AAV2.9-CMV-eGFP expresses green fluorescent protein under the CMV promoter. Both viruses were purchased from the University of Pennsylvania Vector Core.
Three weeks prior to the behavioral experiments 1.0 µL of virus was infused bilaterally into the dorsal hippocampus (AP 2.0 mm; ML ±1.5 mm; 1.5 mm DV from bregma) at a rate of 6 µL/h (Vogel-Ciernia et al. 2013). All infusions were confirmed by immunohistochemistry for the HA tag.
Immunohistochemistry and Western blot analysis
Immunohistochemistry and Western blot analysis were performed as previously described (Vogel-Ciernia et al. 2013). Primary and secondary antibodies: Mouse anti BAF53b (1:500, N332B/15, NeuroMab), Rabbit anti NeuN-Cy3 (1:500 Millipore, N78C3), Mouse anti CREST (1:200, 12439-1-AP, Proteintech), Rabbit anti Brg1 (1:1000, ab70588, Abcam), and Rat anti HA tag 3F10 (1:1,000, 11867423001, Sigma). Secondary antibodies: Alexa Fluor 555-conjugated anti-rabbit IgG (1:500, 4413S, Cell Signaling), Alexa Fluor 488 conjugated anti-mouse IgG (1:500, A11017, Invitrogen), Alexa Fluor 555-conjugated anti-rat IgG (1:1,000, A-21434, Invitrogen), donkey anti-mouse-HRP (1:10,000; 715-036-150, Jackson ImmunoResearch) rabbit anti-GAPDH (1:10,000; SC25778, Santa Cruz Biotechnology) and donkey anti-rabbit-HRP (1:10,000; 711-036-152, Jackson ImmunoResearch).
Hippocampal slice physiology
Hippocampal slices were prepared from BAF53bΔSB2low, BAF53bΔSB2high, and wild-type mice (∼2 mo of age). For details in slice preparation see Vogel-Ciernia et al. (2013).
Field excitatory postsynaptic potentials (fEPSPs) were recorded from three regions within the hippocampus in three separate experiments using a single glass pipette (2–3 MΩ) filled with 2 M NaCl positioned in; (1) area CA1b stratum radiatum, (2) lateral perforant path (LPP) in the dentate molecular layer, and (3) area CA3b stratum lucidum (Fig. 2A). Placement of electrodes for area CA1 and LPP were positioned as previously described (Vogel-Ciernia et al. 2013; Trieu et al. 2015).
For area CA3 recordings, the stimulating electrode was positioned on the hilar side of the dentate gyrus granular cell layer to activate and record mossy fiber (MF) synaptic responses in area CA3b stratum lucidum. Identification of MF fEPSPs was based on several criteria; (1) negative waveform restricted to stratum lucidum, (2) short latency (<5 msec), (3) fast time course (<10 msec), and (4) strong paired-pulse facilitation (PPF) (>100% at 40-msec interstimulus interval; PPF was expressed as the amplitude ratio between the second and the first fEPSP of the pair). Stimulation intensity was adjusted to elicit a fEPSP amplitude that was 50%–60% of maximum amplitude. Because mossy fiber LTP is NMDAR-independent (Harris and Cotman 1986; Jaffe and Johnston 1990), aCSF contained 100 µM D-APV to eliminate the possibility of signal contamination from recurrent collateral fibers expressing NMDAR-dependent LTP in area CA3. MF potentiation was induced by applying two trains of HFS, each consisting of 100 pulses at 100 Hz, separated by 1 min. Data were collected and digitized by NAC 2.0 Neurodata Acquisition System (Theta Burst Corp.) and stored on a disk.
Data in the text are presented as means ± SEM, and the fEPSP slope was measured at 10%–90% fall of the slope. Data in figures on LTP were normalized to the last 10 min of baseline. Data were collected from control slices in parallel with experimental tissue. At the end of the study, measurements from control slices were pooled and used as the comparison to slices from experimental groups.
Fluorescence deconvolution tomography
Hippocampal slices used for fluorescence deconvolution tomography were prepared as described above. Following at least 1.5 h incubation period, stimulation electrodes were positioned at two sites (CA1a and CA1c) in the apical Schaffer collateral-commissural projection to provide convergent activation of CA1b pyramidal cells. During low-frequency stimulation (control), baseline pulses were administered in an alternating manner to the two electrodes at a frequency of 0.05 Hz using a current that elicited 50% maximum response. Synaptic potentiation was induced by delivering TBS (10 bursts of 4 pulses at 100 Hz separated by 200 msec) to the two stimulation electrodes within 5 sec of each other, the slice was then collected 3 min after the second theta train and fixed in 4% PFA in 0.1 M of sodium phosphate buffer; control slices that did not receive TBS continued to receive baseline stimulation for an equivalent period before being collected and submerged in fixative. Following fixation (1.5–2 h), slices were sectioned at 20 µm and then processed for immunolabeling (Seese et al. 2013) using primary antisera cocktails including rabbit anti-phospho-cofilin S3 (1:500, Abcam, ab12866), in combination with goat anti-PSD95 (1:1000, Abcam, ab12093). Secondary antisera included AlexaFluor 594 anti-rabbit IgG and AlexaFluor 488 anti-goat IgG (1:1000, Life Technologies A21207, A11055). Epifluorescence image z-stacks were collected at 63× from CA1b stratum radiatum of 5–8 sections through each hippocampal slice and then processed through restorative deconvolution (99% confidence, Volocity 4.0, Improvision). Using in house software, image z-stacks were normalized for background intensity and iteratively binarized at multiple intensity thresholds. The z-stack images were then used to construct a 3D montage of each sample field (136 × 105 × 2 µm) and elements meeting size constraints of synapses, and detected across multiple intensity thresholds, were quantified. By this technique approximately 15,000 synapses were reconstructed per z-stack (Rex et al. 2009; Seese et al. 2013). Elements were considered double-labeled if there was overlap in fields labeled with the two fluorophores as assessed in 3D. The density of the label colocalized with PSD95 was measured for each synapse with results expressed as a percent of all cases belonging to each member of a series of increasing density bins. Two-way ANOVAs were used to test for differences between the frequency distributions of the different groups. Three cohorts of mutant and wild-type slices were tested. Percent increases in synapses associated with high-density phospho-cofilin produced by theta burst stimulation relative to low-frequency stimulation were calculated within cohorts for each group.
Golgi staining and dendritic spine analysis
Golgi staining was performed as described for the superGolgi Kit (Bioenno Tech, LLC cat# 003010). Eight-week-old wild-type and BAF53bΔSB2high mice were sacrificed directly from their homecage. All animals were perfused with 0.9% saline for 3 min and then placed directly in impregnation buffer (solution A; 10 mL per hemisphere). All brains were incubated in impregnation solution for 11 d (solution refreshed after the first 24 h). Brains were then rinsed in water and transferred to post-impregnation buffer (solution B) for 48 h (renewed after the first 24 h). Sections were then collected using a vibratome at 150 µm and mounted onto gelatin-coated slides (FD NeuroTechnology, cat# PO101). Slides were covered in the staining jar and allowed to try overnight. The next day slides were washed with 0.01M PBS-Tween (30 min), washed in water, and incubated in staining solution C for 20 min. The slides were then rinsed in water and placed in post-staining buffer for 20 min followed by three 10 min washes in PBS-Tween. Slides were dehydrated in 100% ethanol for three 10 min washes followed by coverslipping in DPX (Sigma, cat# 06522).
Bright-field images were taken with a 100× oil objective (NA 1.4) using a stereology workstation (StereoInvestigator, MicroBrightField) for the first or second dendritic branch within stratum radiatum of CA1 of dorsal hippocampus. Images from 9–10 neurons were collected per animal by an investigator blind to genotype. All images were then converted to 8 bit, inverted and background subtracted using a sliding paraboloid (4 pixels) in Fiji (ImageJ) (Schindelin et al. 2012). Dendritic spine analysis was then conducted using NeuronStudio (Rodriguez et al. 2008). Briefly, for each image dendritic spines were automatically and quantified for spine size, shape, and volume using the rayburst algorithm (Rodríguez et al. 2006) in NeuronStudio. Voxel size was set to 0.064 × 0.064 by 0.1 µm (x, y, z) and the default spine classification settings were used with the following exceptions: minimum spine height 0.3 µm, maximum spine height 3.0 µm, and maximum spine width 3.0 µm. In order to account for the lower limit of optical resolution any spines with head diameters less than 0.3 µm were removed from analysis. NeuronStudio classifies spines based on the following default parameters: neck ratio 1.1 µm, thin ratio 2.5 µm, and mushroom size 0.35 µm. Spines lacking a neck are classified as stubby. Spines with a neck are classified as either mushroom or thin based on head diameter (<0.35 µm is thin). Spine classification has been verified using manual counting (Rodriguez et al. 2008).
Object location and novel object recognition paradigms
Object location and object recognition were performed as previously described (Vogel-Ciernia et al. 2013). Prior to training, mice are handled for 2 min per day for 5 d and then habituated to the experimental apparatus for 5 min a day for 6 d in the absence of objects. During the training period, mice were placed into the experimental apparatus with two identical objects (100 mL beakers or tins or vases) and allowed to explore for 10 min. During the retention test, (24 h for long-term memory or 90 min for short-term memory), mice were allowed to explore the experimental apparatus for 5 min. Exploration was scored when a mouse's head was oriented toward the object within a distance of 1 cm or when the nose was touching the object. The relative exploration time (t) was recorded and expressed as a discrimination index (DI = (tnovel − tfamiliar)/(tnovel + tfamiliar) × 100%). Mean exploration times were then calculated and the discrimination indexes between treatment groups compared. Animals that explored <3 sec total for both objects during either training or testing were removed from further analysis. Animals that demonstrated an object preference during training (DI > ±20) were also removed. Distance traveled during habituation was tracked using ANY-maze (Stoelting).
Open field
Animals were placed in a novel environment (46 × 61 × 27 cm tall made of white acrylic) and allowed to freely explore for 5 min. Illumination was set at 45 Lux and held constant across the environment. Total distance traveled and speed of travel (m/sec) were recorded throughout the session. A 5 cm perimeter was defined around the walls of the box (edge) and compared with the remaining interior of the chamber (center).
Elevated plus maze
The plus maze consisted of two open arms, 30 × 5 cm, and two enclosed arms, 30 (1) × 5 (w) × 15 (h) cm. The arms extended from a central platform 5 × 5 cm, and the apparatus was raised to a height of 40 cm above the floor. The light level was adjusted to 15 Lux. Testing (5 min) consisted of placing an animal onto the central platform of the maze facing an open arm. Between subjects the maze was cleaned with 70% ethanol. The percentage of time spent in the closed and open arms was scored using EthoVision 3.1 (Noldus Information Technology).
BAF53a, BAF53b wild-type and BAF53bΔSB2-predicted 3D structures
The protein structures for wild-type BAF53a, BAF53b and the BAF53bΔSB2 transgene were predicted using the comparative protein structure modeling software Modeller (Fig. 1C; Eswar et al. 2006). Model templates with at least 90% of the amino acids aligning to BAF53a or BAF53b wild-type sequence and with at least 30% sequence identity were selected from the Protein Data Bank (Supplemental Table S1; Berman et al. 2000).
Statistical analysis
Statistical analysis was conducted as indicated in the text and figure legends using Prism (GraphPad). Parametric tests (t-test or ANOVA) were used where assumptions of normality (Shapiro–Wilk) and equal variance (F test or Bartlett's test) were met and were replaced by nonparametric equivalents where appropriate (Mann–Whitney U or Kruskal–Wallis). Main effects and interactions for all ANOVAs or Kruskal–Wallis tests are described in the text. Repeated-measures ANOVA is abbreviated as RM ANOVA. The appropriate post hoc tests (Bonferroni or Dunn's) are listed for each condition examined. All t-tests were two-tailed using a P value of 0.05. Sex was considered as a biological variable in all experiments conducted. In no experiment was there a main effect of sex or a significant interaction with sex.
Supplementary Material
Acknowledgments
We thank J. Guzowski for use of the BX61 microscope and XC10 camera. This work was supported by grants from NIMH (MH101491) to M.A.W. and G.L.; NIDA (DA036984) to M.A.W.; NIMH (F31-MH098565) to A.V.C.; Hewitt Foundation to T.J.H. The work of C.N.M. and P.B. was supported by grants NSF IIS-1321053 and NIH LM010235 to P.B. The work of T.A. was supported by NIMH (MH087463).
Author contributions: A.V.C. and M.A.W. designed the experiments and wrote the manuscript. A.V.C. conducted the experiments, oversaw data analysis, and managed the project. D.P.M. conducted experiments and designed and created the transgenic mice. E.A.K. and B.T. conducted the electrophysiology experiments. G.L., E.A.K., and W.W. designed the electrophysiology experiments, analyzed, and interpreted the data. R.H. and T.A. designed, tested, and contributed the AAV-cofilinS3D virus. T.J.H., K.S., A.T., S.A., A.L., R.D., J.T., R.M.B., and R.J.P. ran behavioral experiments and assisted with data analysis. C.N.M. and P.B. created the predicted BAF53b structural models.
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.044602.116.
References
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, et al. 2001. Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci 4: 367–373. [DOI] [PubMed] [Google Scholar]
- Ben-David E, Shifman S. 2012. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry 18: 1054–1056. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Sarvey JM. 1996. Endogenous activation of mu and delta-1 opioid receptors is required for long-term potentiation induction in the lateral perforant path: dependence on GABAergic inhibition. J Neurosci 16: 8123–8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. 2007. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci 27: 5363–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramár EA, Lynch G, Gall CM, Lauterborn JC. 2010. Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci 30: 10977–10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi A, Staahl B, Jovičić A, Couthouis J, Fasolino M, Raphael A, Yamazaki T, Elias L, Polak M, Kelly C, et al. 2013. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci 16: 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Abraham WC. 1994. Differential regulation of paired-pulse plasticity following LTP in the dentate gyrus. Neuroreport 5: 385–388. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2006. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics 10.1002/0471250953.bi0506s15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, et al. 2010. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci 13: 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harata M, Mochizuki R, Mizuno S. 1999. Two isoforms of a human actin-related protein show nuclear localization and mutually selective expression between brain and other tissues. Biosci Biotechnol Biochem 63: 917–923. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. 2011. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21: 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E, Cotman C. 1986. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl d-aspartate antagonists. Neurosci Lett 70: 132–137. [DOI] [PubMed] [Google Scholar]
- Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, Bruinenberg VM, Poplawski SG, Day JP, Aton SJ, et al. 2016. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife 5: R774–R788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, et al. 2012. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 90: 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe D, Johnston D. 1990. Induction of long-term potentiation at hippocampal mossy-fiber synapses follows a Hebbian rule. J Neurophysiol 64: 948–960. [DOI] [PubMed] [Google Scholar]
- Koga M, Ishiguro H, Yazaki S, Horiuchi Y, Arai M, Iritani S, Itokawa M, Inada T, Iwata N, Ozaki N. 2009. Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum Mol Genet 18: 2483–2494. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER. 1997. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Neurobiology 94: 4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda Y, Oma Y, Nishimori K, Ohta T, Harata M. 2002. Brain-specific expression of the nuclear actin-related protein ArpN a and its involvement in mammalian SWI/SNF chromatin remodeling complex. Biochem Biophys Res Commun 299: 328–334. [DOI] [PubMed] [Google Scholar]
- Loe-Mie Y, Maussion G, Doron-Faigenboim A, Aggerbeck L, Pupko T, Gorwood P, Imbeaud S, Simonneau M, Moalic J, Lepagnol-Bestel A, et al. 2010. SMARCA2 and other genome-wide supported schizophrenia-associated genes: regulation by REST/NRSF, network organization and primate-specific evolution. Hum Mol Genet 19: 2841–2857. [DOI] [PubMed] [Google Scholar]
- Lynch G, Gall CM. 2013. Mechanism based approaches for rescuing and enhancing cognition. Front Neurosci 7: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. 1996. Control of memory formation through regulated expression of a CaMKII transgene. Science 274: 1678–1683. [DOI] [PubMed] [Google Scholar]
- Moriyama K, Iida K, Yahara I. 1996. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1: 73–86. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin C-F, Stevens C, Wang L-S, Makarov V, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa KA, Badoual M, Asmussen H, Patel H, Whitmore L, Horwitz AR. 2015. ROCK1 and 2 differentially regulate actomyosin organization to drive cell and synaptic polarity. J Cell Biol 210: 225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Roeb W, Dickel D, Walsh T, Kusenda M, O'Connor KL, Malhotra D, McCarthy SE, Stray SM, Taylor SM, et al. 2011. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur J Hum Genet 19: 727–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olave I, Wang W, Xue Y, Kuo A, Crabtree GR. 2002. Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev 16: 2509–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu X-L, Lin X, Crowley NA, Lovinger DM, et al. 2014. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci 17: 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Wood MA, Cole MD. 2002. BAF53 forms distinct nuclear complexes and functions as a critical c-Myc-interacting nuclear cofactor for oncogenic transformation. Mol Cell Biol 22: 1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontrello CG, Sun M, Lin A, Fiacco TA, Defea KA, Ethell IM. 2012. Cofilin under control of β-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci 109: E422–E451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. 2009. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol 186: 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roak BJO, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. 2012. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez A, Ehlenberger DB, Hof PR, Wearne SL. 2006. Rayburst sampling, an algorithm for automated three-dimensional shape analysis from laser scanning microscopy images. Nat Protoc 1: 2152–2161. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger D, Dickstein D, Hof P, Wearne S. 2008. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3: e1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust M, Gurniak C, Renner M, Vara H, Morando L, Rlich A, Pognetto M, Banchaabouchi M. 2010. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J 29: 1889–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GWE, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EAJ, Hilhorst-hofstee Y, et al. 2012. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet 44: 379–380. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seese RR, Chen LY, Cox CD, Schulz D, Babayan AH, Bunney WE, Henn FA, Gall CM, Lynch G. 2013. Synaptic abnormalities in the infralimbic cortex of a model of congenital depression. J Neurosci 33: 13441–13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. 2009. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci 29: 8129–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Lynch G. 1990. Stable depression of potentiated synaptic responses in the hippocampus with 1–5 Hz stimulation. Brain Res 513: 113–118. [DOI] [PubMed] [Google Scholar]
- Suzuki E, Okada T. 2007. Regional differences in GABAergic modulation for TEA-induced synaptic plasticity in rat hippocampal CA1, CA3 and dentate gyrus. Neurosci Res 59: 183–190. [DOI] [PubMed] [Google Scholar]
- Tea JS, Luo L. 2011. The chromatin remodeling factor Bap55 functions through the TIP60 complex to regulate olfactory projection neuron dendrite targeting. Neural Dev 6: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu BH, Kramár EA, Cox CD, Jia Y, Wang W, Gall CM, Lynch G. 2015. Pronounced differences in signal processing and synaptic plasticity between piriform-hippocampal network stages: a prominent role for adenosine. J Physiol 593: 2889–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Naritomi K, Kawame H, Wakui K, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, et al. 2012. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44: 376–378. [DOI] [PubMed] [Google Scholar]
- Van Houdt JKJ, Nowakowska BA, Sousa SB, van Schaik BDC, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard M-JH, Bottani A, et al. 2012. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 44: 445–449. [DOI] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Wood MA. 2013. Neuron-specific chromatin remodeling: a missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 80: 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Matheos DP, Barrett RM, Kramár EA, Azzawi S, Chen Y, Magnan CN, Zeller M, Sylvain A, Haettig J, et al. 2013. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci 16: 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. 2011. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Trieu BH, Palmer LC, Jia Y, Pham DT, Jung K-M, Karsten CA, Merrill CB, Mackie K, Gall CM, et al. 2016. A primary cortical input to hippocampus expresses a pathway-specific and endocannabinoid-dependent form of long-term potentiation. eNeuro 3 10.1523/ENEURO.0160-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff D, Endele S, Azzarello-Burri S, Hoyer J, Zweier M, Schanze I, Schmitt B, Rauch A, Reis A, Zweier C. 2012. In-frame deletion and missense mutations of the C-terminal helicase domain of SMARCA2 in three patients with Nicolaides-Baraitser syndrome. Mol Syndromol 2: 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M, Zimmermann A-M, Görlich A, Gurniak CB, Sassoè-Pognetto M, Friauf E, Witke W, Rust MB. 2015. ADF/cofilin controls synaptic actin dynamics and regulates synaptic vesicle mobilization and exocytosis. Cereb Cortex 25: 2863–2875. [DOI] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR. 2007. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56: 94–108. [DOI] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. 1990. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science 248: 1619–1624. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. 2004. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44: 749–757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.