

Abstract
Our understanding of the pathophysiological basis of chronic thromboembolic pulmonary hypertension (CTEPH) will be accelerated by an animal model that replicates the phenotype of human CTEPH. Sprague-Dawley rats were administered a combination of a single dose each of plastic microspheres and vascular endothelial growth factor receptor antagonist in polystyrene microspheres (PE) + tyrosine kinase inhibitor SU5416 (SU) group. Shams received volume-matched saline; PE and SU groups received only microspheres or SU5416, respectively. PE + SU rats exhibited sustained pulmonary hypertension (62 ± 13 and 53 ± 14 mmHg at 3 and 6 weeks, respectively) with reduction of the ventriculoarterial coupling in vivo coincident with a large decrement in peak rate of oxygen consumption during aerobic exercise, respectively. PE + SU produced right ventricular hypokinesis, dilation, and hypertrophy observed on echocardiography, and 40% reduction in right ventricular contractile function in isolated perfused hearts. High-resolution computed tomographic pulmonary angiography and Ki-67 immunohistochemistry revealed abundant lung neovascularization and cellular proliferation in PE that was distinctly absent in the PE + SU group. We present a novel rodent model to reproduce much of the known phenotype of CTEPH, including the pivotal pathophysiological role of impaired vascular endothelial growth factor–dependent vascular remodeling. This model may reveal a better pathophysiological understanding of how PE transitions to CTEPH in human treatments.
The field of pulmonary hypertension research lacks a reproducible, economically feasible, and pathologically accurate animal model of chronic thromboembolic pulmonary hypertension (CTEPH). Categorized as group 4 in the World Health Organization's classification of pulmonary hypertension (PH), CTEPH uniquely occurs as a direct result of pulmonary embolism.1, 2 Between 0.4% and 9.1% of patients presenting a single or multiple episodes of acute pulmonary embolism progress to CTEPH.3, 4, 5
CTEPH develops as a consequence of persistent thrombotic obstruction and scarring of pulmonary arteries, causing progressive PH and eventual right ventricular (RV) failure.1 One factor heavily associated with CTEPH is multiple episodes of pulmonary embolism without resolution of perfusion defects on scintillation lung scanning.6 The pathophysiological mechanisms that cause CTEPH are not fully elucidated, but probably represent the intersection of predisposing patient phenotype and genotype that leads to resistance to pulmonary intravascular thrombotic clearance, together with persistent hypercoagulability, platelet activation,7 pulmonary vascular inflammation, vasospasm, inflammatory reaction, and vascular cell mitogenesis.8 No specific plasmatic biomarker profile has emerged that predicts or characterizes CTEPH, which hinders the development of much-needed prophylactic treatments after first pulmonary embolism diagnosis.9, 10, 11 Although CTEPH can be treated with pulmonary thromboendarterectomy, patient eligibility and accessibility limit the use of this complicated surgery, which sometimes fails to cure the disease.12 Treatment with the guanylate cyclase stimulator riociguat may reverse vasospasm and bring partial symptom relief, but no medical treatment reverses or even inhibits the progression of CTEPH.13 Therefore, it is imperative to generate a generalizable animal model to accelerate the study of CTEPH pathogenic characteristics and develop new therapeutic strategies that go beyond treating vasospasm. Many CTEPH animal models have been attempted with varying success, but no rodent model has reproduced even the main features of the human disease, which at minimum requires embolic occlusion followed by progressive PH with RV remodeling.14 Therefore, our goal was to develop a rodent model of CTEPH that recapitulates the major phenotypic characteristics of the disease but, most important, exhibits a sustained and progressive PH. We hypothesized that two distinct insults to the pulmonary vascular bed would be required, comprising the injection of polystyrene microspheres, together with an inhibitor of the vascular endothelial growth factor (VEGF) receptor tyrosine kinase (SU5416) to disrupt VEGF signaling and block the cognate endothelial response to the pulmonary vascular occlusion.
Materials and Methods
All experimental procedures were approved by the Animal Care and Use Committee of Indiana University School of Medicine (Indianapolis, IN), and were conducted in accordance with NIH guidelines on animal care and use.15
Induction of Chronic Thromboembolic Pulmonary Hypertension
Male Sprague-Dawley rats weighing initially 400 to 420 g were used (39 in total; Harlan Laboratories, Indianapolis, IN). Animals were assigned into four different groups: i) PE + SU group rats received a combination of a single i.v. (tail vein) dose of polystyrene microspheres (97.000/100 g; 85 μm; Thermo Scientific, Fremont, CA), dissolved in 0.01% Tween 20,16, 17 followed by a single s.c. injection of the tyrosine kinase inhibitor [SU5416; Tocris Bioscience, Bristol, UK; 20 mg/kg in dimethyl sulfoxide (DMSO)] to disrupt post-receptor signaling for the VEGF receptor.18 Injection of the 85-μm polystyrene microspheres was performed to produce vascular occlusion of the fourth-order branches of the rat pulmonary vasculature, approximately corresponding to segmental branching in humans.19, 20 In previous work, we found that autologous clot would not produce persistent occlusion, even in the Copenhagen rat treated with the plasmin inhibitor tranexamic acid.21 ii) PE group received the same dose of polystyrene microspheres described above and volume-matched DMSO as vehicle. iii) SU group received vehicle (0.01% Tween 20) and SU5416 (20 mg/kg, s.c. dissolved in DMSO). iv) Sham group received volume-matched vehicles only (0.001% Tween 20 and DMSO). To assess timing of disease progression, animals were euthanized at 3 weeks (early stage, n = 16) and another subset at 6 weeks (late stage, n = 22) after PE + SU induction or vehicle injection. The dose of SU5416 was chosen on the basis of previous studies with Sugen/hypoxia-induced pulmonary hypertension model.22, 23
Aerobic Capacity Testing
Aerobic capacity was assessed with exercise testing for maximal rate of oxygen consumption (VO2max), as previously described,24 using a motorized rodent treadmill (Columbus Instruments, Columbus, OH). All rats were familiarized with the treadmill for 8 days before exercise testing. Familiarization sessions were performed 4 days per week using gradually increasing treadmill speeds (6 to 24 m/minute) and inclines (0 to 20 degrees) and a mild electric stimulus at the back of the treadmill belt to promote learning of running behavior. The duration of each familiarization session was kept to 5 minutes to minimize chronic training effects. Exercise testing was conducted using an enclosed treadmill chamber that permits metabolic measurements via analysis of expired gases (Oxymax; Columbus Instruments). Gas analyzer calibrations were conducted before testing using standardized gas mixtures (Praxair, Indianapolis, IN). For determination of VO2max, a modified incremental treadmill protocol was used25 in 3-minute stages as follows: 10 m/minute at 0 degrees, 15 m/minute at 5 degrees, 18 m/minute at 10 degrees, 20 m/minute at 15 degrees, 22 m/minute at 20 degrees, and 25 m/minute at 20 degrees. The test was terminated when VO2 plateaued despite increasing workload25 or when the rat was unable to maintain position on the belt and received three consecutive electrical stimuli without recovery to the front of the treadmill. The highest VO2 achieved was recorded as VO2max (as mL/kg per hour) and is expressed relative to body weight.
Echocardiography
Rats were lightly anesthetized with 2% isoflurane in 100% O2 via nose cone. Images were obtained in the parasternal two-dimensional short axis and long axis with a 13- to 24-MHz scan probe (Vevo 2100; Visual Sonics, Toronto, ON, Canada). Chamber sizes, wall thicknesses, pressures, and other derived values were assessed in accordance with published methods.26
In Vivo Hemodynamics
Rats were anesthetized (isoflurane 2% in 100% O2), intubated, and ventilated via tracheostomy (80 breaths/minute; inspiratory/expiratory ratio, 1:1; Micro-Ventilator; Kent Scientific, Torrington, CT). Systemic pressures were evaluated in the right carotid artery, and instantaneous RV pressure-volume measurements were obtained with 1.9 F admittance catheters (Transonic Systems Inc., Ithaca, NY) positioned in the right ventricle via right thoracotomy and myocardial puncture. To obtain load-independent measures of function, the inferior vena cava was transiently occluded with manual tension held on a silk thread for several seconds. Heparinized arterial blood was collected and then centrifuged at 956 × g, 15 minutes at 4°C and, frozen in liquid nitrogen, then stored at −80°C for biochemical analysis.
Ex Vivo Isolated Perfused Hearts
After euthanasia with ketamine and xylazine overdose, hearts were excised and perfused via the aorta at a constant pressure (60 mmHg) in the Langendorff apparatus with Krebs-Henseleit bicarbonate buffer, as previously described.16, 17 A fluid-filled balloon connected to a pressure transducer was inserted into the RV chamber and the Frank-Starling relationship of RV function (RV-developed pressure and maximum rate of pressure development) was explored by filling the balloon with saline to increase the RV end diastolic pressure from 0 to 40 mmHg with electrical pacing at 350 beats/minute (S44 stimulator; Grass Instruments, Quincy, MA).
After ex vivo perfusion, the RV was carefully separated from the left ventricle and septum (LV + S). After determination of RV and LV + S masses, RV/LV + S ratio was calculated to determine RV hypertrophy.
High-Resolution CT Pulmonary Vascular Imaging
In euthanized rats obtained from the above listed groups, heparinized saline (99 parts 0.9% NaCl and 1 part heparin) was infused through the right jugular vein to flush the pulmonary circulation free of blood. A barium sulfate contrast medium (BriteVu; Scarlett Imaging, LLC, Murray, UT) was infused through the jugular vein catheter with the lungs inflated with 20 mmHg air, and the trachea was ligated to maintain pressure. Heart and lung blocks were excised and placed on a carbon-fiber bed and scanned with small-animal high-resolution micro–computed tomography (CT) scanner (Skyscan 1176; Bruker Biospin Corp., Kontich, Belgium). The micro-CT images were acquired at 33.63 μm using three continuous bed positions to cover the entirety of the lungs, where the tube voltage, current, step-size filter, and exposure times were 80 kV, 313 μA, 0.9 degrees/step, 0.5 mm Al filter, and 20 milliseconds, respectively. On completion of the micro-CT scans, images were reconstructed using filtered back projection via SkyScan NRecon (Kontich, Belgium) software version 1.6.9.4, where a Hamming filter of 0.25 (α) and a Nyquist frequency cutoff of 100 Hz yielded isotropic voxels with a 33.63-μm resolution. For analysis, DICOM image volumes were imported into Analyze 12.0 (AnalyzeDirect, Overland Park, KS), signal intensity was normalized (ie, 0.0, 1.0), and tissue segmentation was accomplished via segmented threshold for background plus soft tissue (ie, 0.0 to 0.1) and lung vasculature (ie, 0.1 to 1.0). Segmented object maps were then quantified for total vascular volumes using AnalyzePro's ROITool (AnalyzeDirect).
Telemetric Pressure Monitoring
To assess hemodynamics over model development and during exercise, we instrumented an additional rat with a dual-pressure telemetry transmitter (model HD-S21; Data Sciences International, New Brighton, MN) for simultaneous RV and aortic pressure measures, as previously described by our group.24 Briefly, via laparotomy, one catheter was placed into the abdominal aorta and secured with methacrylate; a second catheter was inserted into the RV via thoracotomy and myocardial puncture and secured with a purse-string. The animal was allowed to recover for 2 weeks before induction of PE + SU. In addition to daily resting measures, exercise pressures were recorded during VO2max testing at baseline, and at 3 and 6 weeks after PE + SU induction.
Histology of Heart and Lungs
Formalin-fixed organs were divided into sections (6 μm thick) and stained with Masson's trichrome for collagen and Verhoeff–Van Giesen for collagen in elastic fibers. Lung cellular proliferation was assessed with Ki-67 immunohistochemistry: slides were blocked for 5 minutes using hydrogen peroxide to quench endogenous peroxidase, and anti–Ki-67 rabbit monoclonal antibody (RM-9106-S; Thermo Scientific, Fremont, CA) was applied at a 1:200 dilution for 30 minutes. Envision+ anti-rabbit kit (Dako Agilent Pathology Solutions, Santa Clara, CA) was used as secondary antibody. Sections were developed with diaminobenzidine and counterstained with hematoxylin. Two observers blinded to study group examined digitized photomicrographs (Leica Microsystems, Wetzlar, Germany) to count the number of nuclei staining Ki-67 positive normalized by the total number of nuclei per moderate power field. The average of the two observers is reported as percentage of Ki-67–positive cells. Right ventricular fibrosis was assessed on formalin-fixed, paraffin-embedded RV sections as percentage positively stained area with Masson's trichrome staining.
Intact Platelet Mitochondrial Oxygen Consumption
Platelets were obtained by serial centrifugation of EDTA-treated blood (300 × g for 15 minutes and 4800 × g for 5 minutes subsequently) and were resuspended in platelet-poor plasma. Approximately 400 × 106 cells/mL were placed in each chamber of the Oxygraph (Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria) at 37°C in MIR05 buffer containing 5 mmol/L glucose and 0.4 mmol/L octanoate. Oxygen consumption was measured in the absence (routine or state 4) and presence of oligomycin (leak or state 3), with the protonophore, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (uncoupled) for electron transport system and extramitochondrial oxygen consumption was measured by adding rotenone (complex I inhibitor) and antimycin-A (complex III inhibitor).
Plasma Biomarkers of Endothelial Injury and Inflammation and Arterial Blood Gas Measurements
Tissue inhibitor of metalloproteinases 1, total plasminogen activator inhibitor-1, caveolin-1, cytokine-induced neutrophil chemoattractant-1 (CINC-1/CXCL1, a homolog to human IL-8), monocyte chemoattractant protein-1, tumor necrosis factor-α, VEGF, adiponectin, soluble intercellular adhesion molecule-1, and soluble E-selectin were determined using a commercially available assay (Milliplex mean arterial pressure rat vascular injury panels 1 and 2, RV1MAG-26K and RV1MAG-26K; EMD Millipore Corp., Billerica, MA). P-selectin (CD62P) concentrations were measured on plasma samples using a rat sandwich enzyme-linked immunosorbent assay kit (LS-F5274; LifeSpan Biosciences, Inc., Seattle, WA). Arterial blood was examined for lactate, sodium, potassium, hematocrit, and partial pressures of oxygen and carbon dioxide (PaO2 and PaCO2, respectively) using an i-STAT device (Abbott Laboratories, Abbott Park, IL). PaO2 and PaCO2 were acquired under spontaneous breathing 21% O2 (room air) or 100% O2 (delivered by nose cone), and during mechanical ventilation 100% O2. Alveolar-arterial oxygenation gradient, A-a O2 = [(barometric pressure − water vapor pressure) × FiO2 − PaCO2] − PaO2, during mechanical ventilation of 100% O2.
Statistical Analysis
Data are presented as means ± SEM. Means were compared with a one-way analysis of variance, followed by the Student-Newman-Keuls multiple comparison post hoc test. VO2max and PaO2 means were compared with two-way analysis of variance, followed by Bonferroni post hoc test. The null hypothesis was rejected at P < 0.05.
Results
Progressive PH Caused by PE + SU
The combination of a single dose of microspheres and SU5416 (PE + SU) produced increases in RV systolic pressure (RVSP) (Figure 1A) and pulmonary resistance (Figure 1B) at 3 and 6 weeks (P < 0.05), without change in the mean arterial pressure (P > 0.05) (Figure 1C). Neither the injection of microspheres nor SU5416 alone (PE and SU groups, respectively) was associated with changes in RVSP, pulmonary resistance, or mean arterial pressure compared to the sham group (P > 0.05) (Figure 1, A–C). To better understand the time-severity course of PH development, we induced PE + SU in a rat implanted with telemetric monitoring for serial measures of simultaneous RV and aortic pressures at rest and during maximal exercise testing. Figure 1D shows that the linear increase in resting RV systolic pressure occurred between weeks 1 and 3 and plateaued thereafter at an elevated 80 to 100 mmHg. Throughout this time, the heart rate and aortic pressure remained stable (Figure 1E). RV pressure increase relative to increasing exercise workload during VO2max testing (Figure 1F) was steeper at 3 weeks after PE + SU induction (Figure 1F) compared to predisease (Figure 1F), but became severely blunted at 6 weeks (Figure 1F). In addition to a failure to generate greater RV pressure with exercise, later disease was also accompanied by a reduction in resting RV pressure where, at approximately week 11, the previously stable but elevated RV values of 80 to 100 mmHg declined to the mid-60s. The animal then developed gross peripheral edema and overt respiratory distress, prompting euthanasia. Necropsy revealed hepatomegaly and ascites. These data indicate that neither pulmonary vascular occlusion nor VEGF antagonism alone produce persistent pulmonary hypertension, but the two insults combined produce progressive, severe pulmonary hypertension, culminating in RV failure, a prerequisite to CTEPH.
Figure 1.

In vivo hemodynamics assessment data on early stage (3 weeks) and late stage (6 weeks). Measurements from admittance catheter placed in the right ventricle (RV) of anesthetized rats (A–C), and a conscious, freely moving rat implanted with telemetry system transmitting real-time data from indwelling pressure-sensing catheters (D and F). A: RV systolic pressure (RVSP). B: Index of pulmonary vascular resistance [RVSP normalized by cardiac output (CO)]. C: Mean arterial pressure (MAP). D: RV pressures (systolic and diastolic). E: Systemic pressures (systolic and diastolic) and heart rate. F: Real-time RVSP tracings recorded in the telemetrically instrumented animal during VO2max testing performed at baseline (light gray line) and at 3 weeks (dark gray line) and 6 weeks (black line) after PE + SU induction. Five minutes of resting collection on a stationary treadmill belt (rest), and a 2-minute warm-up period (warm up) preceded the start (arrow) of the incremental treadmill protocol in 3-minute stages. Maximal oxygen uptake (VO2max, in mL/kg/hour), and maximal heart rate (HRmax, in bpm) are indicated in call-out boxes at each test's termination point (time to exhaustion) in addition to maximal RVSP. Values are expressed as means ± SEM (A–C). n = 4 (3 weeks; A–C); n = 4 to 6 (6 weeks; A–C). ∗P < 0.05 versus PE + SU group; †P < 0.05 versus PE group. CTEPH, chronic thromboembolic pulmonary hypertension; dSAP, diastolic–systemic arterial pressure; HR, heart rate; RVDP, RV diastolic pressure; sSAP, systolic–systemic arterial pressure.
RV Dysfunction and Hypertrophy Caused by PE + SU
Echocardiography revealed that PE + SU rats had increased RV wall thickness at 3 and 6 weeks (Figure 2A), coincident with a profound decrease in RV fractional systolic shortening (Figure 2B) and decreased cardiac output (Figure 2C). With PE alone, the changes in RV wall thickness, fractional systolic shortening, and cardiac output were clearly less severe than with PE + SU (Figure 2, A–C). Neither PE nor PE + SU changed the LV wall thickness (Figure 2D) or diameter (Figure 2E), but as might be predicted, PE + SU increased the mean ratio of the RV/LV diameter (Figure 2F). The static two-dimensional echocardiography images displayed in Figure 2G capture the physiological consequence of this increase in RV diameter and illustrate that PE + SU results mimic the echocardiographic pattern seen in humans with CTEPH (namely, that the RV displaces the septum to the left and compresses the LV). Rats with PE + SU also demonstrated a significant reduction in the pulmonary artery acceleration time/RV ejection time and 100% of PE + SU rats had the midsystolic notch, as observed in representative images of pulmonary artery velocities on Doppler echocardiogram (Supplemental Figure S1, A and B), whereas no rat in any other group had the midsystolic notch.
Figure 2.

Assessment of cardiac function and morphology by echocardiography at 3 (white bars) and 6 (black bars) weeks after chronic thromboembolic pulmonary hypertension (CTEPH) induction. A: Right ventricular (RV) wall thickness. B: RV fractional shortening. C: Cardiac output. D: Left ventricular (LV) wall thickness. E: LV dimension in diastole (LVIDd). F: RV/LV diameter ratio (as a parameter of enlargement). G: Representative images from short-axis view echocardiography at early and late stages. Note the strong RV dysfunction and hypertrophy phenotype in PE + SU rats after CTEPH induction. Values are expressed as means ± SEM (A–F). n = 4 per group (3 weeks; A–F); n = 4 to 6 per group (6 weeks; A–F). ∗P < 0.05 versus PE + SU group; †P < 0.05 versus PE group; ‡P < 0.05 for PE versus PE + SU group.
Figure 3 shows results of load-independent measurements of right ventricular function. End-systolic elastance (P < 0.05) (Figure 3A) and arterial elastance (P < 0.05) (Figure 3B) were elevated in PE + SU group, resulting in reduction of the ventriculoarterial coupling (end-systolic elastance/arterial elastance, P < 0.05) (Figure 3C). No changes in LV function were found.
Figure 3.

Pressure-volume analyses of the right ventricle at 3 (white bars) and 6 (black bars) weeks. End-systolic elastance (Ees; A) and arterial elastance (Ea; B) increase significantly by PE + SU treatment. C: Because of insufficient enhancement in right ventricular contractility to counterpoise elevated afterload (Ea), right ventricular-pulmonary arterial coupling (Ees/Ea) reduces in PE + SU group. Bar graphs represent means ± SEM. n = 4 per group (3 weeks); n = 4 to 6 per group (6 weeks). ∗P < 0.05.
We then measured RV intrinsic contractile function in isolated perfused hearts. Hearts isolated from PE + SU rats at 3 and 6 weeks had significantly worsened RV-developed pressure and maximum rate of pressure development normalized by RV mass, compared with sham and SU groups (Table 1). After perfusions, the heart and each ventricle were weighed to assess the Fulton index, which revealed a significant increase in RV mass (greater than twofold, P < 0.05), but no change in LV + Septum mass leading to an increase in the RV/LV + S ratio (RV mass normalized by LV + S). Thus, PE + SU produced RV contractile dysfunction, both in vivo and ex vivo, coincident with marked RV hypertrophy.
Table 1.
Ex Vivo RV Intrinsic Contractile Function Parameters Evaluated by Isolated Perfused Hearts and Cardiac Mass (Heart, RV, and LV + Septum) at 3 and 6 Weeks after CTEPH
| Parameters | Sham | SU | PE | PE + SU | Comparison |
|---|---|---|---|---|---|
| 3 Weeks | |||||
| Developed pressure/RV weight | |||||
| RVEDP at 0 mmHg | 145 ± 14 | 151 ± 27 | 95 ± 41 | 51 ± 9 | P > 0.05 |
| RVEDP at 40 mmHg | 307 ± 12∗ | 299 ± 30∗ | 214 ± 37 | 128 ± 12† | P < 0.05 |
| dP/dtmax/RV weight | |||||
| RVEDP at 0 mmHg | 4231 ± 287 | 5087 ± 926∗ | 3101 ± 1313 | 1524 ± 260 | P < 0.05 |
| RVEDP at 40 mmHg | 5895 ± 170∗ | 5822 ± 746∗ | 3493 ± 1320 | 2282 ± 149 | P < 0.05 |
| RV weight (g) | 0.29 ± 0.02∗ | 0.30 ± 0.02∗ | 0.34 ± 0.01 | 0.62 ± 0.04† | P < 0.05 |
| LV + septum weight (g) | 1.23 ± 0.04 | 1.22 ± 0.01 | 1.27 ± 0.04 | 1.12 ± 0.08 | P > 0.05 |
| Heart weight (g) | 1.53 ± 0.04 | 1.53 ± 0.02 | 1.61 ± 0.04 | 1.75 ± 0.11 | P > 0.05 |
| RV/(LV + septum) ratio | 0.24 ± 0.01∗ | 0.24 ± 0.02∗ | 0.27 ± 0.01 | 0.55 ± 0.03† | P < 0.05 |
| 6 Weeks | |||||
| Developed pressure/RV weight | |||||
| RVEDP at 0 mmHg | 149 ± 12∗ | 127 ± 12∗ | 93 ± 46 | 57 ± 3 | P < 0.05 |
| RVEDP at 40 mmHg | 280 ± 11∗ | 258 ± 12∗ | 227 ± 52 | 126 ± 4† | P < 0.05 |
| dP/dtmax/RV weight | |||||
| RVEDP at 0 mmHg | 4699 ± 802∗ | 4093 ± 300∗ | 2593 ± 1160 | 1797 ± 135 | P < 0.05 |
| RVEDP at 40 mmHg | 3267 ± 1282∗ | 4836 ± 209∗ | 3267 ± 1282 | 2269 ± 119 | P < 0.05 |
| RV weight (g) | 0.30 ± 0.01∗ | 0.34 ± 0.01∗ | 0.33 ± 0.02 | 0.71 ± 0.03† | P < 0.05 |
| LV + septum weight (g) | 1.18 ± 0.04 | 1.26 ± 0.04 | 1.18 ± 0.04 | 1.17 ± 0.07 | P > 0.05 |
| Heart weight (g) | 1.49 ± 0.05∗ | 1.61 ± 0.06∗ | 1.52 ± 0.05 | 1.88 ± 0.09† | P < 0.05 |
| RV/(LV + septum) ratio | 0.25 ± 0.01∗ | 0.27 ± 0.02∗ | 0.28 ± 0.01 | 0.65 ± 0.06† | P < 0.05 |
RV-developed pressure and the maximum rate of increase in RV pressure during systole (dP/dtmax) were corrected for RV weights. Developed pressure/RVweight (mmHg/g) and dP/dtmax/RVweight (mmHg/s/g) were acquired at different RV end diastolic pressures (0 and 40 mmHg). RV/LV + septum ratio was used as a marker of RV hypertrophy. Values are means ± SEM. n = 4 to 6 per group.
CTEPH, chronic thromboembolic pulmonary hypertension; dP/dtmax, RV-developed pressure and maximum rate of pressure development; LV, left ventricular; PE, polystyrene microsphere; RV, right ventricular; RVEDP, RV end diastolic pressure; SU, tyrosine kinase inhibitor SU5416.
P < 0.05 versus PE + SU group.
P < 0.05 for PE versus PE + SU group.
PE + SU Worsens Exercise Capacity of Rats and Causes Hypoxemia
PE + SU produced a 40% and 32% reduction in VO2max at 3 and 6 weeks compared to sham rats at the same time points (P < 0.05) (Figure 4, A and B). In parallel, we observed decrements in exercise time, whereas the respiratory exchange ratio increased above unity, suggesting dominant lactate oxidation (P < 0.05) (Figure 4, C–F). PE alone produced 6% reduction in mean VO2max at 3 weeks and 11% at 6 weeks (P > 0.05) (Figure 4, A and B). Arterial blood samples demonstrated mild reduction in the PaO2 in rats of PE and PE + DMSO groups compared to sham rats during spontaneous breathing of 100% O2 (P < 0.05) (Figure 5A). However, during mechanical ventilation, 100% O2 only PE group showed lower PaO2 compared to sham rats (P < 0.05) (Figure 5A). Increased alveolar-arterial oxygenation gradient (A-a O2) was found elevated in PE and PE + SU groups compared to sham rats during mechanical ventilation of 100% O2 (P < 0.05) (Figure 5B). Although A-a O2 gradient tended to be higher in PE group rather than PE + SU group, it was not significantly different at the α = 0.05 level (Figure 5B). In addition, arterial lactate concentrations were increased in rats with PE + SU (8.9 ± 2.4 mmol/L) versus sham rats (2.5 ± 0.4 mmol/L) at 6 weeks (P < 0.05) (Figure 5C). Although sodium plasma concentrations were similar between all groups (P > 0.05) (Supplemental Figure S2A), PE + SU was associated with mild increase in serum potassium concentrations and lower hematocrit compared to sham and PE groups (P < 0.05) (Supplemental Figure S2, B and C).
Figure 4.

Maximal aerobic capacity expressed as VO2max (A and B), total exercise time (C and D), and respiratory exchange ratio (E and F) at 3 and 6 weeks after PE + SU induction. Values are means ± SEM (A–F). ∗P < 0.05 for sham versus PE + SU group; †P < 0.05 for sham versus PE group; ‡P < 0.05 for PE versus PE + SU group.
Figure 5.

A: Partial pressure of arterial oxygen (PaO2) was assessed during spontaneous breathing of 21% O2 (room air), spontaneous breathing of 100% O2, and mechanical ventilation of 100% O2 after 6 weeks of PE + SU induction. B: Alveolar-arterial oxygenation gradient (A-a O2) was calculated during mechanical ventilation of 100% O2. C: Simultaneously, arterial blood samples were used to measure lactate concentrations in sham, PE, and PE + SU rats at 6 weeks. ∗P < 0.05 versus PE + SU group; †P < 0.05 for sham versus PE group.
Association of a CTEPH-Like State with Circulating Markers of Endothelial Injury, Inflammation, and Platelet Activation
Taken together, the hemodynamic, echocardiographic, morphological, intrinsic contractility, and histological data suggest that PE + SU in rats produces many of the same consequences of CTEPH in humans. The next question was to determine whether PE + SU produces changes in plasma biomarkers of hypofibrinolysis—reduced collagen proteolysis and platelet activation, suggestive of a phenotype of clot persistence, RV and vascular hypertrophy, and small-vessel occlusion, respectively. Indeed, the plasma concentrations of tissue inhibitor of metalloproteinases 1, total plasminogen activator inhibitor-1, and P-selectin were elevated in the PE + SU group compared to the others groups (Figure 6, A–C). Intact platelets isolated from PE + SU rats also had significantly elevated oxygen consumption in two states of respiration, including unstimulated (routine, 55 ± 8 versus 31 ± 2 pmol × s−1/106 cells) and uncoupled (electron transport system, 86 ± 13 versus 44 ± 4 pmol × s−1/106 cells) conditions with greater wasted oxygen use (leak, 10 ± 1 versus 5 ± 1 pmol × s−1/106 cells), compared to sham rats (P < 0.05) (Figure 6D). These results indicate marked elevated rates of mitochondrial oxidative phosphorylation with mild uncoupling in platelets of PE + SU rats. No extramitochondrial oxygen consumption was detected. Additional biomarkers at 5 days, 3 weeks, and 6 weeks after CTEPH are shown in Supplemental Figure S2 (SU group was not included because no difference was found between sham group). PE + SU produced elevations on caveolin-1, CINC-1, and tumor necrosis factor-α, and reductions on adiponectin at 5 days after CTEPH induction in PE + SU group (P < 0.05) (Supplemental Figure S3, A, B, D, and F). In addition, tumor necrosis factor-α and adiponectin decreased in PE group at 5 days (P < 0.05) (Supplemental Figure S3, D and F). At 3 weeks, CINC-1, VEGF, soluble E-selectin, and soluble intercellular adhesion molecule-1 increased in plasma of PE + SU rats, whereas CINC-1 decreased in PE group (P < 0.05) (Supplemental Figure S3, B, E, G and H). At 6 weeks, only monocyte chemoattractant protein-1 and VEGF were elevated on PE + SU group, and CINC-1 decreased in PE and PE + SU groups (P < 0.05) (Supplemental Figure S3, B, C, and E).
Figure 6.

Plasma concentrations of tissue inhibitor of metalloproteinases 1 (TIMP-1; A), total plasminogen activator inhibitor-1 (tPAI-1; B), and P-selectin (C) in sham, SU, PE, and PE + SU groups at 3 weeks and 6 weeks. D: Platelet mitochondrial respiration in sham, PE, and PE + SU groups at 6 weeks. Values are means ± SEM. n = 4 (A–C, 3 weeks); n = 4 to 6 (A–C, 6 weeks); n = 8 (D, sham group); n = 5 (D, PE group); n = 7 (D, PE + SU group). ∗P < 0.05 for PE + SU group versus other groups; †P < 0.05 for sham versus PE + SU group; ‡P < 0.05 for PE versus PE + SU group.
High-Resolution CT Angiography Demonstrates Pulmonary Vascular Rarefaction with PE + SU
Figure 7, A–C, shows three-dimensional images from micro-CT scan that demonstrate robust increase in the number and distension of small (greater than the fourth order branch) pulmonary arteries and arterioles in PE group compared to sham group (Figure 7, A and B). However, Figure 7C shows that lungs from rats treated 6 weeks before with PE + SU demonstrate vascular pruning, and frank loss of peripheral vasculature beyond fourth-order branching. The pulmonary vascular volume was found to be reduced in PE + SU group (789 ± 114 mm3) compared to sham group (1369 ± 175 mm3, P < 0.05) (Figure 7D). However, in PE group, pulmonary vascular volume tended to be elevated (2312 ± 816 mm3) (Figure 7D).
Figure 7.

Lung computed tomography imaging at 6 weeks after chronic thromboembolic pulmonary hypertension. Representative three-dimensional micro–computed tomographic images and magnifications (left and right sides) of sham rats injected with vehicle only (A), PE rats injected with microspheres only (B), and PE + SU rats injected with a combination of microspheres and SU5416 (20 mg/kg; C). D: Quantification of the pulmonary vascular volume in sham, PE, and PE + SU groups. n = 4 per group (D). ∗P < 0.05 for sham versus PE + SU group.
Increased RV Fibrosis and Lung Vascular Hyperplasia in Parallel with Impaired Lung Growth
PE + SU rats had marked decrease in cellular proliferation, expressed as Ki-67– positive cells in percentage of total cells nuclei per field, compared with rats in the sham and PE groups (P < 0.05) (Figure 8, A and B). Lung sections stained with Verhoeff–Van Giesen (Figure 8C) demonstrated marked medial hypertrophy in small arterioles, occasional intimal proliferation with obliteration of the lumen, but without plexiform lesions. In addition, trichrome-stained sections (P < 0.05) (Figure 8E and Supplemental Figure S4) demonstrate moderate RV-specific fibrosis with PE alone, and showed more severe RV-specific fibrosis with PE + SU. No significant increase in fibrosis was observed in the LV with any group or in the RV with SU alone (Figure 8D).
Figure 8.

A: Cellular proliferation was assessed by quantification of the Ki-67–positive cells (expressed as Ki-67–positive cells in percentage of total cells nuclei per field) in the sham, PE, and PE + SU rats. B: Representative images of immunohistochemistry for Ki-67 stain (positive cells indicated by arrows) on rat lung sections from animals in the sham, PE, and PE + SU groups at 6 weeks. C: Verhoeff–van Gieson staining reveals medial hypertrophy and lumen obliteration in small pulmonary arterioles in PE + SU rats. Histological sections of Masson's trichrome staining [left ventricular (LV; (D) and right ventricular (RV; E)] on sham, PE, PE + SU, and SU groups at 6 weeks after chronic thromboembolic pulmonary hypertension. Masson's trichrome stain showing RV fibrosis (blue color indicates collagen accumulation) in PE group but more extensive in the PE + SU group. No LV fibrosis is observed. n = 6 per group (A). ∗P < 0.05 for sham and PE versus PE + SU group. Scale bars: 50 μm (B, D, and E); 200 μm (C). Original magnification, ×200 (B–E).
Discussion
In a novel model, rats with pulmonary vascular occlusion only with impervious microspheres failed to manifest CTEPH-like states, but when subjected to inhibition of the endothelial proliferative response mediated by VEGF with SU5416 in PE + SU group, the animals show phenotypic profiles suggestive of CTEPH. Without variability, 100% of rats treated with PE + SU manifested progressive elevation of pulmonary pressure and resistance, concomitant with RV dysfunction, enlargement and hypertrophy, exercise intolerance,27 platelet activation,7 and pulmonary vascular atrophy.28 Only approximately 10% of rats with PE + DMSO developed PH, and in data not shown, we repeated weekly microsphere embolization (up to five times over 6 weeks but without SU5416) in dozens of rats and were unable to produce consistent PH.16, 17 Similarly, repeated injections of fibrinogen/collagen-covered polystyrene microspheres together with thrombin produced minor elevations of the RVSP (<33 mmHg) in a rat model of CTEPH.29 These findings reaffirm the requirement of a combined insult of pulmonary vascular occlusion and inhibition of endothelial proliferation (accomplished herein via blocking tyrosine kinase, required for VEGF signaling) to produce persistent PH after PE.30 We observe that >90% of rats with PE but without VEGF antagonism have systolic pulmonary arterial pressures <35 mmHg, and together with obviously increased vascularity on high-resolution CT pulmonary angiography and lung cellular proliferation on Ki-67 staining. Taken together, these findings increased pulmonary vascular cross-sectional area by either pulmonary arterial dilation, or more likely, growing new vessels to increase the net pulmonary vascular cross-sectional area. With PE alone, the lung cellular proliferation estimated with Ki-67 staining did not differ from the sham group, whereas with PE + SU, the lung cellular proliferation decreased, coincident with marked vascular pruning on high-resolution CT angiography (Figures 7 and 8). Loss of peripheral vascularity is considered a radiological hallmark of CTEPH in humans.31, 32, 33 However, on histological examination, humans with CTEPH demonstrate a range of vascular morphometric abnormalities, from complete loss of vessels to intimal and muscular hyperplasia, and occasional plexiform lesion formation.31, 34, 35 Indirect evidence has suggested that pulmonary arteriolar/capillary remodeling and vasospasm contribute to persistent PH after PE, given that the increased pulmonary vascular resistance correlates poorly with the degree of angiographic pulmonary vascular bed obstruction in humans (with pressures most often disproportionately high).36, 37 Kasahara et al38 found that SU5416 alone can cause emphysema by endothelial cell apoptosis. However, in that work, the animals were injected s.c. three times per week for 3 weeks with 20 mg/kg SU5416. In our model, we only used one injection of 20 mg/kg SU5416. Although we did not measure emphysema, no enlarged airspaces were found in rats injected with SU5416, as was seen by Kasahara et al.38 In addition, we believe that vasoconstriction will not play an important role on our imaging because the CT scans were performed post-mortem. Therefore, the pulmonary vascular rarefaction may be the most important effect for intravascular barium exclusion in lungs of rats in PE + SU group.
If future work confirms that healthy rats with PE alone forestall PH by VEGF-dependent pulmonary vascular expansion, our model may help advance a treatment strategy to increase lung angiogenesis to prevent or possibly reverse CTEPH. Precedent literature has described cytotherapy with mesenchymal stem cells to partially reverse the phenotype of pulmonary arterial hypertension produced by monocrotaline.39 This would be especially important in the so-called type 4 CTEPH, caused by widespread distal emboli, rendering it less amenable to surgical treatment or pulmonary balloon angioplasty of proximal obstruction.40, 41 The lack of a markedly widened A-a O2 tension difference gradient in animals with PE alone argues against a large physiological shunt from bronchiolar dilation or collateral vessel formation, which has been suggested to occur in CTEPH.42, 43 Our A-a O2 tension difference gradients (35 ± 11 mmHg) are consistent with those found by McCabe et al44 in 15 humans with CTEPH (46 ± 12 mmHg). Therefore, the vascular compensation in rats with PE alone appears to be either salutary or at least not harmful, in terms of pulmonary ventilation perfusion and gas exchange. The age of the rats may affect this vascular expansion as a compensatory response. Herein, we studied male rats, all <6 months old. Given that a Sprague-Dawley rat's mean lifespan is approximately 3 years in captivity, it must be considered that our model represents the juvenile response to acute PE. Whether aged rats can compensate equally well remains unknown.
We found an elevation of biomarkers of hypofibrinolysis and reduced collagen proteolysis and platelet activation (PAI-1, tissue inhibitor of metalloproteinases 1, and p-Selectin, with marked increase in intact platelet oxygen consumption) (Figure 6) in the CETPH group (PE + SU). Increased levels of PAI-1, in the thrombi, and highly activated platelets (P-selectin–positive cells) have been reported in CTEPH patients.7, 45 In addition, PAI-1 and tissue inhibitor of metalloproteinases 1 have been reported to inhibit angiogenesis, through VEGF pathway disruption46 and protease inhibition,47 respectively. Furthermore, low expression of proangiogenic factors in white CTEPH thrombi has been proposed to impair angiogenesis-induced thrombi resolution.48
Limitations include use of polystyrene microspheres instead of native thrombi21 and male juvenile rats. Only the animal instrumented with implantable telemetry was studied out to the point of frank RV failure, incident at 10 weeks. The exercise hemodynamics recorded for this animal reflected a failing RV in later disease, where increase in pulmonary pressure relative to workload became distinctly plateaued, similar to a pattern that has been reported to be prognostic for patients with pulmonary arterial hypertension in RV exercise catheterization studies.49 We do not know if this time frame to RV failure will be reproducible, but the mean lactate values of 9 mmol/L at 6 weeks suggests animals had circulatory insufficiency.
In conclusion, in the present model, we found that the interaction of pulmonary arterial mechanical obstruction and VEGF receptor inhibition recapitulate main features of clinical CTEPH, consistent with the dual pulmonary injury concept. This novel model provides experimental evidence for the relation between progressive pulmonary pressure elevation and deficient lung vascularization in the CTEPH pathophysiology and opens the way to better understanding of the role of pulmonary vascular expansion as a therapeutic target to prevent CTEPH in the setting of unresolved PE.
Acknowledgments
We thank Dr. Jonathan Tune, who shared equipment and expertise for histology imaging; the Immunohistochemistry Research core at the University of Indiana School of Medicine, which provided immunohistochemistry service; and Gary Long, M.S., who performed serial exercise testing for the telemetrically instrumented animal.
Footnotes
Supported by a Lilly Endowment Foundation Physician Scientist Initiative (J.A.K.); an American Heart Association postdoctoral fellowship (E.M.N.-M.); an American Heart Association Midwest Affiliates Scientist Development grant (M.B.B.); and the Indiana Clinical and Translational Sciences Institute, funded in part by the NIH, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award, Collaboration in Translational Research Pilot Program grant UL1TR001108.
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.12.004.
Supplemental Data
Supplemental Figure S1.

A: Pulmonary arterial acceleration time/right ventricular ejection time (PAAT/RVET) reduction in chronic thromboembolic pulmonary hypertension–induced rats (PE + SU) as compared to sham and SU groups at 3 and 6 weeks. B: Representative images of pulmonary arterial velocities on Doppler echocardiogram showing a characteristic notch indicating pulmonary hypertension (arrows) in PE + SU group. Values are means ± SEM or dot plot (A and B). n = 4 (3 weeks; A); n = 4 to 6 (6 weeks; A). ∗P < 0.05 versus PE + SU group.
Supplemental Figure S2.
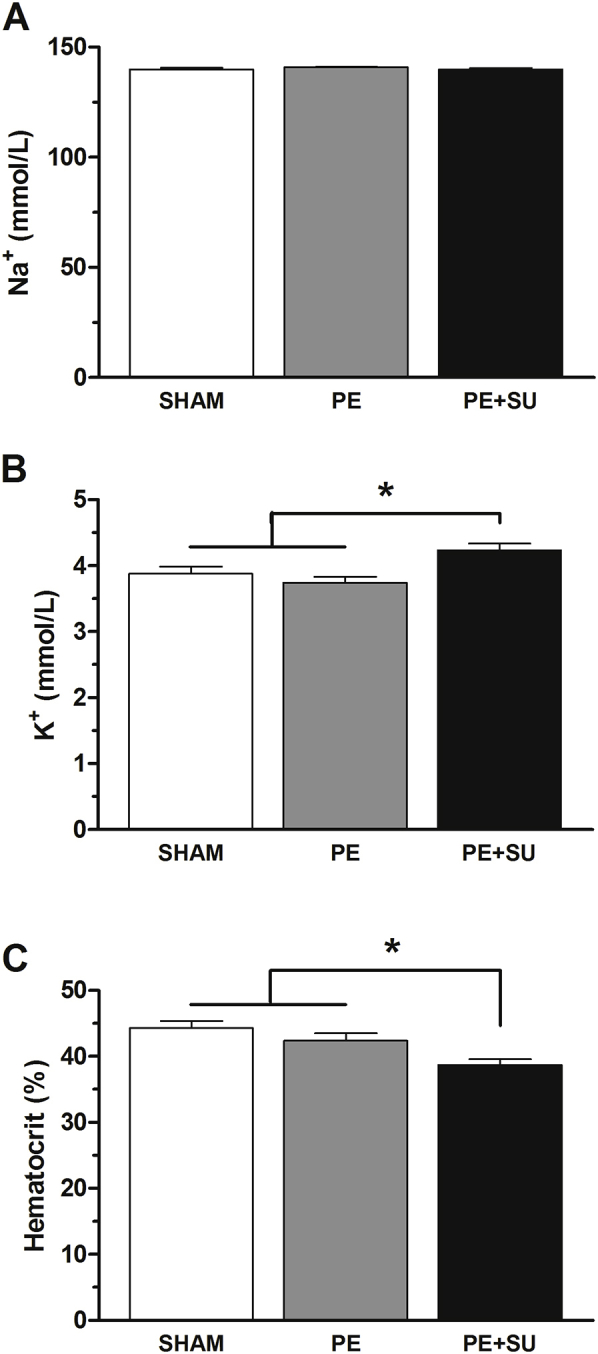
Arterial blood samples were used to measure sodium (Na+; A) and potassium (K+; B) concentrations, and hematocrit (C) in sham, PE, and PE + SU rats at 6 weeks. n = 4 to 6 per group. ∗P < 0.05 versus PE + SU group.
Supplemental Figure S3.

Markers of endothelial injury and inflammation in the plasma of sham, PE, and PE + SU groups at 5 days, 3 weeks, and 6 weeks of chronic thromboembolic pulmonary hypertension induction. A: Caveolin-1. B: Cytokine-induced neutrophil chemoattractant 1 (CINC-1). C: Monocyte chemoattractant protein-1 (MCP-1). D: Tumor necrosis factor (TNF)-α. E: Vascular endothelial growth factor (VEGF). F: Adiponectin. G: Soluble E-selectin (sE-selectin). H: Soluble intercellular adhesion molecule 1 (sICAM-1). Values are means ± SEM or dot plot (A–H). ∗P < 0.05 versus PE + SU group; †P < 0.05 for PE + SU versus PE group; ‡P < 0.05 versus sham group.
Supplemental Figure S4.
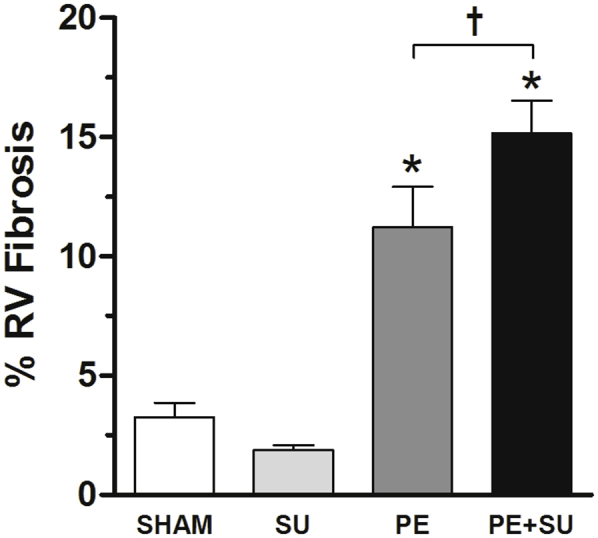
Measurement of the right ventricular (RV) fibrosis in sham, SU, PE, and PE + SU groups at 6 weeks. n = 5 to 9 per group. ∗P < 0.05 versus sham group; †P < 0.05 for PE versus PE + SU group.
References
- 1.Simonneau G., Galie N., Rubin L.J., Langleben D., Seeger W., Domenighetti G., Gibbs S., Lebrec D., Speich R., Beghetti M., Rich S., Fishman A. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G., Gatzoulis M.A., Adatia I., Celermajer D., Denton C., Ghofrani A., Gomez Sanchez M.A., Krishna Kumar R., Landzberg M., Machado R.F., Olschewski H., Robbins I.M., Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 3.Lang I. Chronic thromboembolic pulmonary hypertension: a distinct disease entity. Eur Respir Rev. 2015;24:246–252. doi: 10.1183/16000617.00001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lang I.M., Madani M. Update on chronic thromboembolic pulmonary hypertension. Circulation. 2014;130:508–518. doi: 10.1161/CIRCULATIONAHA.114.009309. [DOI] [PubMed] [Google Scholar]
- 5.Pengo V., Lensing A.W., Prins M.H., Marchiori A., Davidson B.L., Tiozzo F., Albanese P., Biasiolo A., Pegoraro C., Iliceto S., Prandoni P., Thromboembolic Pulmonary Hypertension Study Group Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257–2264. doi: 10.1056/NEJMoa032274. [DOI] [PubMed] [Google Scholar]
- 6.Pepke-Zaba J., Delcroix M., Lang I., Mayer E., Jansa P., Ambroz D., Treacy C., D'Armini A.M., Morsolini M., Snijder R., Bresser P., Torbicki A., Kristensen B., Lewczuk J., Simkova I., Barbera J.A., de Perrot M., Hoeper M.M., Gaine S., Speich R., Gomez-Sanchez M.A., Kovacs G., Hamid A.M., Jais X., Simonneau G. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation. 2011;124:1973–1981. doi: 10.1161/CIRCULATIONAHA.110.015008. [DOI] [PubMed] [Google Scholar]
- 7.Yaoita N., Shirakawa R., Fukumoto Y., Sugimura K., Miyata S., Miura Y., Nochioka K., Miura M., Tatebe S., Aoki T., Yamamoto S., Satoh K., Kimura T., Shimokawa H., Horiuchi H. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014;34:2486–2494. doi: 10.1161/ATVBAHA.114.304404. [DOI] [PubMed] [Google Scholar]
- 8.Lang I.M., Pesavento R., Bonderman D., Yuan J.X. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J. 2013;41:462–468. doi: 10.1183/09031936.00049312. [DOI] [PubMed] [Google Scholar]
- 9.Tiede S.L., Gall H., Dorr O., Troidl C., Liebetrau C., Voss S., Voswinckel R., Schermuly R.T., Seeger W., Grimminger F., Zeiher A.M., Dimmeler S., Mollmann H., Hamm C.W., Ghofrani H.A., Nef H.M. New potential diagnostic biomarkers for pulmonary hypertension. Eur Respir J. 2015;46:1390–1396. doi: 10.1183/13993003.00187-2015. [DOI] [PubMed] [Google Scholar]
- 10.Quarck R., Wynants M., Verbeken E., Meyns B., Delcroix M. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur Respir J. 2015;46:431–443. doi: 10.1183/09031936.00009914. [DOI] [PubMed] [Google Scholar]
- 11.Zabini D., Heinemann A., Foris V., Nagaraj C., Nierlich P., Balint Z., Kwapiszewska G., Lang I.M., Klepetko W., Olschewski H., Olschewski A. Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur Respir J. 2014;44:951–962. doi: 10.1183/09031936.00145013. [DOI] [PubMed] [Google Scholar]
- 12.Mayer E. Surgical and post-operative treatment of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2010;19:64–67. doi: 10.1183/09059180.00007409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghofrani H.A., D'Armini A.M., Grimminger F., Hoeper M.M., Jansa P., Kim N.H., Mayer E., Simonneau G., Wilkins M.R., Fritsch A., Neuser D., Weimann G., Wang C., CHEST-1 Study Group Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369:319–329. doi: 10.1056/NEJMoa1209657. [DOI] [PubMed] [Google Scholar]
- 14.Mercier O., Fadel E. Chronic thromboembolic pulmonary hypertension: animal models. Eur Respir J. 2013;41:1200–1206. doi: 10.1183/09031936.00101612. [DOI] [PubMed] [Google Scholar]
- 15.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . ed 8. National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 16.Watts J.A., Gellar M.A., Obraztsova M., Kline J.A., Zagorski J. Role of inflammation in right ventricular damage and repair following experimental pulmonary embolism in rats. Int J Exp Pathol. 2008;89:389–399. doi: 10.1111/j.1365-2613.2008.00610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watts J.A., Zagorski J., Gellar M.A., Stevinson B.G., Kline J.A. Cardiac inflammation contributes to right ventricular dysfunction following experimental pulmonary embolism in rats. J Mol Cell Cardiol. 2006;41:296–307. doi: 10.1016/j.yjmcc.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 18.Spiekermann K., Faber F., Voswinckel R., Hiddemann W. The protein tyrosine kinase inhibitor SU5614 inhibits VEGF-induced endothelial cell sprouting and induces growth arrest and apoptosis by inhibition of c-kit in AML cells. Exp Hematol. 2002;30:767–773. doi: 10.1016/s0301-472x(02)00837-8. [DOI] [PubMed] [Google Scholar]
- 19.Hislop A., Reid L. Normal structure and dimensions of the pulmonary arteries in the rat. J Anat. 1978;125:71–83. [PMC free article] [PubMed] [Google Scholar]
- 20.Molthen R.C., Karau K.L., Dawson C.A. Quantitative models of the rat pulmonary arterial tree morphometry applied to hypoxia-induced arterial remodeling. J Appl Physiol (1985) 2004;97:2372–2384. doi: 10.1152/japplphysiol.00454.2004. discussion 54. [DOI] [PubMed] [Google Scholar]
- 21.Runyon M.S., Gellar M.A., Sanapareddy N., Kline J.A., Watts J.A. Development and comparison of a minimally-invasive model of autologous clot pulmonary embolism in Sprague-Dawley and Copenhagen rats. Thromb J. 2010;8:3. doi: 10.1186/1477-9560-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frump A.L., Goss K.N., Vayl A., Albrecht M., Fisher A., Tursunova R., Fierst J., Whitson J., Cucci A.R., Brown M.B., Lahm T. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol. 2015;308:L873–L890. doi: 10.1152/ajplung.00006.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bogaard H.J., Natarajan R., Henderson S.C., Long C.S., Kraskauskas D., Smithson L., Ockaili R., McCord J.M., Voelkel N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 24.Brown M.B., Chingombe T.J., Zinn A.B., Reddy J.G., Novack R.A., Cooney S.A., Fisher A.J., Presson R.G., Lahm T., Petrache I. Novel assessment of haemodynamic kinetics with acute exercise in a rat model of pulmonary arterial hypertension. Exp Physiol. 2015;100:742–754. doi: 10.1113/EP085182. [DOI] [PubMed] [Google Scholar]
- 25.Bedford T.G., Tipton C.M., Wilson N.C., Oppliger R.A., Gisolfi C.V. Maximum oxygen consumption of rats and its changes with various experimental procedures. J Appl Physiol Respir Environ Exerc Physiol. 1979;47:1278–1283. doi: 10.1152/jappl.1979.47.6.1278. [DOI] [PubMed] [Google Scholar]
- 26.Liu J., Rigel D.F. Echocardiographic examination in rats and mice. Methods Mol Biol. 2009;573:139–155. doi: 10.1007/978-1-60761-247-6_8. [DOI] [PubMed] [Google Scholar]
- 27.Richter M.J., Sommer N., Gall H., Voswinckel R., Seeger W., Mayer E., Wiedenroth C.B., Rieth A., Grimminger F., Guth S., Ghofrani H.A. Pulmonary hemodynamic response to exercise in chronic thromboembolic pulmonary hypertension before and after pulmonary endarterectomy. Respiration. 2015;90:63–73. doi: 10.1159/000398815. [DOI] [PubMed] [Google Scholar]
- 28.Hirsch A.M., Moser K.M., Auger W.R., Channick R.N., Fedullo P.F. Unilateral pulmonary artery thrombotic occlusion: is distal arteriopathy a consequence? Am J Respir Crit Care Med. 1996;154:491–496. doi: 10.1164/ajrccm.154.2.8756827. [DOI] [PubMed] [Google Scholar]
- 29.Arias-Loza P.A., Jung P., Abeßer M., Umbenhauer S., Williams T., Frantz S., Schuh K., Pelzer T. Development and characterization of an inducible rat model of chronic thromboembolic pulmonary hypertension. Hypertension. 2016;67:1000–1005. doi: 10.1161/HYPERTENSIONAHA.116.07247. [DOI] [PubMed] [Google Scholar]
- 30.Nor J.E., Christensen J., Mooney D.J., Polverini P.J. Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol. 1999;154:375–384. doi: 10.1016/S0002-9440(10)65284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoeper M.M., Mayer E., Simonneau G., Rubin L.J. Chronic thromboembolic pulmonary hypertension. Circulation. 2006;113:2011–2020. doi: 10.1161/CIRCULATIONAHA.105.602565. [DOI] [PubMed] [Google Scholar]
- 32.Dong C., Zhou M., Liu D., Long X., Guo T., Kong X. Diagnostic accuracy of computed tomography for chronic thromboembolic pulmonary hypertension: a systematic review and meta-analysis. PLoS One. 2015;10:e0126985. doi: 10.1371/journal.pone.0126985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M., Ma Z., Guo X., Zhang H., Yang Y., Wang C. Computed tomographic pulmonary angiography in the assessment of severity of chronic thromboembolic pulmonary hypertension and right ventricular dysfunction. Eur J Radiol. 2011;80:e462–e469. doi: 10.1016/j.ejrad.2010.08.035. [DOI] [PubMed] [Google Scholar]
- 34.Yi E.S., Kim H., Ahn H., Strother J., Morris T., Masliah E., Hansen L.A., Park K., Friedman P.J. Distribution of obstructive intimal lesions and their cellular phenotypes in chronic pulmonary hypertension: a morphometric and immunohistochemical study. Am J Respir Crit Care Med. 2000;162:1577–1586. doi: 10.1164/ajrccm.162.4.9912131. [DOI] [PubMed] [Google Scholar]
- 35.Jamieson S.W., Kapelanski D.P. Pulmonary endarterectomy. Curr Probl Surg. 2000;37:165–252. doi: 10.1016/s0011-3840(00)80005-2. [DOI] [PubMed] [Google Scholar]
- 36.Azarian R., Wartski M., Collignon M.A., Parent F., Herve P., Sors H., Simonneau G. Lung perfusion scans and hemodynamics in acute and chronic pulmonary embolism. J Nucl Med. 1997;38:980–983. [PubMed] [Google Scholar]
- 37.Tuder R.M., Abman S.H., Braun T., Capron F., Stevens T., Thistlethwaite P.A., Haworth S.G. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–S9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Kasahara Y., Tuder R.M., Taraseviciene-Stewart L., Le Cras T.D., Abman S., Hirth P.K., Waltenberger J., Voelkel N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106:1311–1319. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Laarse A., Cobbaert C.M., Umar S. Stem and progenitor cell therapy for pulmonary arterial hypertension: effects on the right ventricle (2013 Grover Conference Series) Pulm Circ. 2015;5:73–80. doi: 10.1086/679701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thistlethwaite P.A., Mo M., Madani M.M., Deutsch R., Blanchard D., Kapelanski D.P., Jamieson S.W. Operative classification of thromboembolic disease determines outcome after pulmonary endarterectomy. J Thorac Cardiovasc Surg. 2002;124:1203–1211. doi: 10.1067/mtc.2002.127313. [DOI] [PubMed] [Google Scholar]
- 41.Ogawa A., Matsubara H. Balloon pulmonary angioplasty: a treatment option for inoperable patients with chronic thromboembolic pulmonary hypertension. Front Cardiovasc Med. 2015;2:4. doi: 10.3389/fcvm.2015.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimizu H., Tanabe N., Terada J., Masuda M., Sakao S., Kasahara Y., Takiguchi Y., Tatsumi K., Kuriyama T. Dilatation of bronchial arteries correlates with extent of central disease in patients with chronic thromboembolic pulmonary hypertension. Circ J. 2008;72:1136–1141. doi: 10.1253/circj.72.1136. [DOI] [PubMed] [Google Scholar]
- 43.Jandik J., Endrys J., Rehulova E., Mraz J., Sedlacek J., De Geest H. Bronchial arteries in experimental pulmonary infarction: angiographic and morphometric study. Cardiovasc Res. 1993;27:1076–1083. doi: 10.1093/cvr/27.6.1076. [DOI] [PubMed] [Google Scholar]
- 44.McCabe C., Deboeck G., Harvey I., Ross R.M., Gopalan D., Screaton N., Pepke-Zaba J. Inefficient exercise gas exchange identifies pulmonary hypertension in chronic thromboembolic obstruction following pulmonary embolism. Thromb Res. 2013;132:659–665. doi: 10.1016/j.thromres.2013.09.032. [DOI] [PubMed] [Google Scholar]
- 45.Lang I.M., Marsh J.J., Olman M.A., Moser K.M., Loskutoff D.J., Schleef R.R. Expression of type 1 plasminogen activator inhibitor in chronic pulmonary thromboemboli. Circulation. 1994;89:2715–2721. doi: 10.1161/01.cir.89.6.2715. [DOI] [PubMed] [Google Scholar]
- 46.Wu J., Strawn T.L., Luo M., Wang L., Li R., Ren M., Xia J., Zhang Z., Ma W., Luo T., Lawrence D.A., Fay W.P. Plasminogen activator inhibitor-1 inhibits angiogenic signaling by uncoupling vascular endothelial growth factor receptor-2-alphaVbeta3 integrin cross talk. Arterioscler Thromb Vasc Biol. 2015;35:111–120. doi: 10.1161/ATVBAHA.114.304554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson M.D., Kim H.R., Chesler L., Tsao-Wu G., Bouck N., Polverini P.J. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase. J Cell Physiol. 1994;160:194–202. doi: 10.1002/jcp.1041600122. [DOI] [PubMed] [Google Scholar]
- 48.Alias S., Redwan B., Panzenbock A., Winter M.P., Schubert U., Voswinckel R., Frey M.K., Jakowitsch J., Alimohammadi A., Hobohm L., Mangold A., Bergmeister H., Sibilia M., Wagner E.F., Mayer E., Klepetko W., Holzenbein T.J., Preissner K.T., Lang I.M. Defective angiogenesis delays thrombus resolution: a potential pathogenetic mechanism underlying chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014;34:810–819. doi: 10.1161/ATVBAHA.113.302991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tolle J.J., Waxman A.B., Van Horn T.L., Pappagianopoulos P.P., Systrom D.M. Exercise-induced pulmonary arterial hypertension. Circulation. 2008;118:2183–2189. doi: 10.1161/CIRCULATIONAHA.108.787101. [DOI] [PMC free article] [PubMed] [Google Scholar]