

Abstract
Background
The aim of this study was to characterize the molecular relationship, origin and historical biogeography of the species in important brown rot fungal genus Laetiporus from East Asia, Europe, Pan-America, Hawaii and South Africa. We used six genetic markers to estimate a genus-level phylogeny including (1) the internal transcribed spacer (ITS), (2) nuclear large subunit rDNA (nrLSU), (3) nuclear small subunit rDNA (nrSSU), (4) translation elongation factor 1-α (EF-1α), (5) DNA-directed RNA polymerase II subunit 2 (RPB2), and (6) mitochondrial small subunit rDNA (mtSSU).
Results
Results of multi-locus phylogenetic analyses show clade support for at least seventeen species-level lineages including two new Laetiporus in China. Molecular dating using BEAST estimated the present crown group diverged approximately 20.16 million years ago (Mya) in the early Miocene. Biogeographic analyses using RASP indicated that Laetiporus most likely originated in temperate zones with East Asia and North America having the highest probability (48%) of being the ancestral area.
Conclusions
Four intercontinental dispersal routes and a possible concealed dispersal route were established for the first time.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-017-0948-5) contains supplementary material, which is available to authorized users.
Keywords: Laetiporus, Wood rot fungi, Phylogeny, Biogeography, Molecular clock
Background
Since the late Tertiary period, severe climatic change and major geological events have played important roles in driving species diversity and in shaping the biogeographic distribution of extant organisms. Benefiting from the development of DNA technology and molecular analysis methods, studies of fungal molecular phylogeny and biogeography have been conducted in recent decades [1–3]. Based on molecular dating, many phylogenetic studies have revealed striking chronological and geographical correlations between evolutionary divergence and geological events [3–8].
Laetiporus Murrill (Fomitopsidaceae, Polyporales) is a cosmopolitan genus, typified by L. sulphureus (Bull.) Murrill [9]. Species in this genus grow from cold temperate to tropical zones and are associated with Betulaceae, Burseraceae, Elaeocarpaceae, Fabaceae, Fagaceae, Meliaceae, Myrtaceae, Oleaceae, Pinaceae, Salicaceae, Sapindaceae and Taxaceae [10–15]. Laetiporus spp. have been considered to be forest pathogens and to cause brown cubical heart rot [16, 17], which is implicated in the cycle of the forest ecosystem [13, 15]. Chemical composition research determined that this cultivable mushroom is a potential food due to its rich digestible bioactive substances and lack of detectable levels of poisonous microelements [18]. Some taxa of Laetiporus are also valuable sources of medicine, such as ergosterol and acetyl eburicoic acid [19, 20].
Recently, several studies were carried out to clarify the species diversity and phylogeny of Laetiporus [11–15]. In these studies, six new species were described, and four new lineages were identified: Clade I, Clade H, Clade L and Clade M. In addition, L. sulphureus and L. versisporus (Lloyd) Imazeki were shown to each be divisible into three different lineages [11–15], which are represented here as Clade C, Clade E1/E2 and Clade G1/G2/G3, respectively.
To date, eleven species and four undescribed taxa of Laetiporus have been accepted as belonging to this genus [15]: L. ailaoshanensis B.K. Cui & J. Song, L. cremeiporus Y. Ota & T. Hatt., L. versisporus and L. zonatus B.K. Cui & J. Song from East Asia; L. cincinnatus (Morgan) Burds., Banik & T.J. Volk, L. conifericola Burds. & Banik and L. huroniensis Burds. & Banik from North America; L. caribensis Banik & D.L. Lindner from Central America; L. montanus Černý ex Tomšovský & Jankovský from East Asia and Europe; L. sulphureus from North America, South America and Europe; L. gilbertsonii Burds. from Pan-America; L. sp. 1 from Hawaii; L. sp. 2 from South America; L. sp. 3 and L. sp. 4 from Central America [11–15]. However, the interspecies relationships within Laetiporus, as well as the origin and biogeography of the genus, remain unclear.
Here, we present multi-locus phylogenetic analyses using sequences from the internal transcribed spacer (ITS), nuclear large subunit rDNA (nrLSU), nuclear small subunit rDNA (nrSSU), translation elongation factor 1-α (EF-1α), DNA-directed RNA polymerase II subunit 2 (RPB2), and mitochondrial small subunit rDNA (mtSSU) to gain insight into the evolution of species in Laetiporus. Our study sought to (1) explore the evolutionary relationships between Laetiporus species, and (2) estimate the divergence time and examine hypotheses about the origin and biogeography of Laetiporus species.
Results
Phylogenetic analyses
The combined dataset (ITS + nrLSU + nrSSU + mtSSU + EF-1α + RPB2) has an aligned length of 3850 characters, of which 3086 are constant, 247 are variable and parsimony uninformative, and 517 are parsimony informative. The tree obtained from the Maximum likelihood (ML) analysis and the maximum parsimony (MP), maximum likelihood (ML) and Bayesian posterior probability (BPP) values based on the dataset are shown in Fig. 1. The aligned ITS matrix comprises 514 positions, of which 378 are constant, 11 are variable and parsimony uninformative, and 125 are parsimony informative. The tree inferred from the ML analysis and the MP, ML and BPP values are shown in Fig. 2.
Fig. 1.
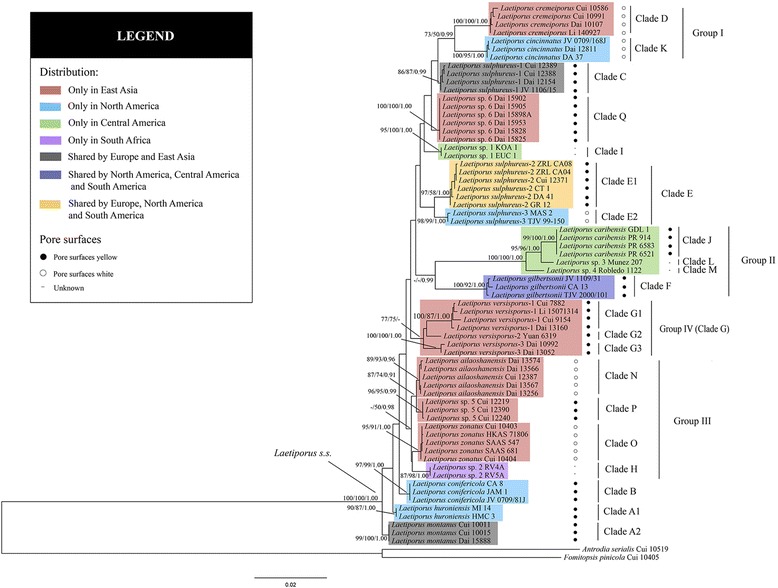
Phylogenetic consensus tree inferred from the maximum likelihood (ML) analysis based on a concatenated, multi-locus dataset (ITS + nrLSU + nrSSU + mtSSU + EF-1α + RPB2). Branches are labeled where MP/BS support is greater than 75% and collapsed below that support threshold. BPP is labeled where greater than 0.90
Fig. 2.
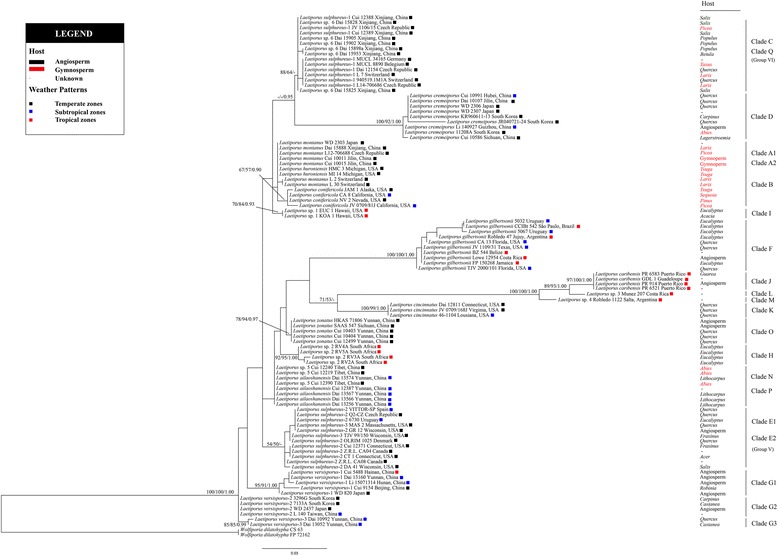
Phylogenetic tree inferred from the maximum likelihood (ML) analysis based on the ITS sequences. Branches are labeled with MP/BS values if greater than 50% and with BPP values if greater than 0.90
The combined dataset and ITS dataset inferred similar topologies (Figs. 1 and 2). The genus Laetiporus was supported with low levels of support on the stem branches. Moreover, 21 different phylogenetic lineages were inferred and significantly supported by both datasets.
In the combined dataset topology (Fig. 1), L. sulphureus was divided into three different well-supported clades: Clade C (86% MP, 87% ML, 0.99 BPP) and Clade E1 (97% MP, 58% ML, 1.00 BPP) with yellow pore surfaces and Clade E2 (98% MP, 99% ML, 1.00 BPP) with a white pore surface. L. versisporus was also divided into three different clades: Clade G1 (100% MP, 87% ML, 1.00 BPP), Clade G2 and Clade G3 (100% MP, 100% ML, 1.00 BPP). Three sister lineages (L. montanus, L. huroniensis and L. conifericola) that grow on coniferous trees were well supported (Fig. 1). Two novel phylogenetic species from western China formed two significantly supported terminal lineages and were named Clade P (96% MP, 95% ML, 0.99 BPP) and Clade Q (100% MP, 100% ML, 1.00 BPP). Moreover, four groups were recognized (Fig. 1). Group I is well supported by Bayesian inference (BI) (0.99 BPP) and moderately supported by MP and ML analyses (73% MP, 50% ML), it is composed of two cold-temperate to subtropical Laetiporus species with white pore surfaces. Group II is well supported by BI (0.99 BPP) but weakly supported by MP and ML analyses, and it contains four North American, Central American and South American Laetiporus species. Group III was well supported by BI (0.98 BPP), moderately supported by ML analysis (50% ML) and includes four Laetiporus species with a disjunct distribution from Hengduan-Himalayan zones to South Africa. Group IV was supported by MP and ML analyses (77% MP, 75% ML) and only includes the East Asian species L. versisporus with a yellow pore surface.
The ITS dataset (Fig. 2) inferred a similar topology despite some existing differences. Clade E1 and Clade E2 clustered together and formed a novel group (Group V) with moderate support from MP and ML analyses (54% MP; 50% ML) and weak support from BI. Notably, this group was weakly supported by BI, MP and ML in the analyses using the combined dataset. The novel phylogenetic species Clade Q clustered together with Clade C and formed a novel group (Group VI) supported by MP and ML analyses (86% MP; 64% ML) but only weakly supported by BI. Moreover, of the 21 lineages identified in the phylogeny, 14 lineages (67%) have temperate distribution, 9 lineages (43%) have subtropical distribution and 9 lineages (43%) have tropical distribution (Fig. 2).
Bayesian estimation of divergence time and the historical biogeography of Laetiporus
The alignment of the two concatenated datasets (ITS + nrLSU + nrSSU and EF-1α + RPB2), which were 2172 and 1137 bp in length, respectively, consisted of 44 taxa. The aligned ITS dataset was 514 bp in length and was established to estimate the divergence time and biogeographical history of Laetiporus.
Analyses were calibrated using two methods. First, based on the divergence between Ascomycota and Basidiomycota, at 582 million years ago (Mya), Paleopyrenomycites devonicus Taylor, Hass, Kerp, M. Krings & Hanlin (Fig. 3) was used to estimate the divergence time of Polyporales at 194.56 ± 0.89 Mya (141.93–247.52 Mya, 95% higher posterior density (HPD)), which is consistent with a previous inference [21]. The initial diversification of Laetiporus occurred during the early Miocene, 20.17 ± 0.12 Mya (12.66–29.09 Mya, 95% HPD), similar to the date of the diversification of the main Laetiporus host plants, such as Quercus, Salix, Populus, Abies, Picea and Pinus [1, 4, 22–25]. Based on the second calibration point, Quatsinoporites cranhamii S.Y. Sm., Currah & Stockey, the divergence between Ascomycota and Basidiomycota was estimated to have occurred at 332.93 ± 3.03 Mya (232.23–447.89 Mya, 95% HPD), which was much more recent than the minimal divergence age of the Ascomycota/Basidiomycota (400 Mya). Meanwhile, the crown age of Laetiporus estimated based on the calibration point was approximately 12.26 ± 0.13 Mya (7.04–18.48 Mya, 95% HPD), which was also significantly more recent than is required for the estimated divergence time of the main host plants. Thus, the second calibration point seemed to vastly underestimate the divergence time of Laetiporus. Therefore, the first calibration point was used for subsequent analyses, and the divergence times of the main nodes are showed in Fig. 3 and summarized in Additional file 1: Table S1.
Fig. 3.
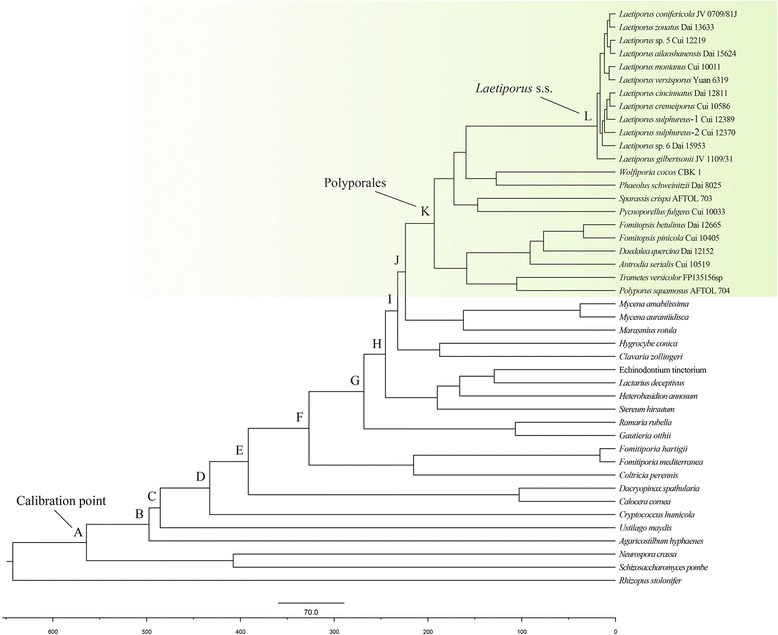
Chronogram and estimated divergence times of Laetiporus generated from molecular clock analysis using the ITS + nrLSU + nrSSU and EF-1α + RPB2 datasets. A chronogram obtained using the Ascomycota–Basidiomycota divergence time of 582 Mya as the calibration point is shown. The calibration point and objects of this study are marked in the chronogram. The lineages in the Polyporales are highlighted in green. The geological time scale is in millions of years ago (Mya)
The inferred historical biogeographic scenarios from analyses using RASP are shown in Fig. 4. The divergence times of the main groups based on the ITS dating analysis are also showed in Fig. 4 and summarized in Additional file 1: Table S3. The results of the Dispersal-Extinction-Cladogenesis (DEC) analysis suggest a complex biogeographic history for Laetiporus. Fifteen dispersal events and six vicariance events were necessary to explain the current distribution of the genus. The ancestral area of Laetiporus was ambiguous. In the reconstruction of their ancestral geographic range, several areas contribute to the geography in different proportions: the probability for East Asia and North America was 48%, that for Europe and North America was 42%, and that for North America was 10%. Thus, the geographic range of East Asia and North America had the highest probability (48%) of being the ancestral area. The most probable (100%) ancestral area for Group I was East Asia and North America. The most probable (43%) ancestral area for Group II was North America and Central America. East Asia was the most probable ancestral area for Group III and Group IV, at 72% and 85%, respectively. The most probable (100%) ancestral area for Group V was North America. The most probable (74%) ancestral area for Group VI was East Asia. Furthermore, four dispersal routes and a possible concealed dispersal route were inferred: East Asia–eastern North America, North America–Central America–South America, East Asia–South Africa, East Asia–Europe and East Asia–Malay Archipelago–Australia–Hawaii (Fig. 5).
Fig. 4.
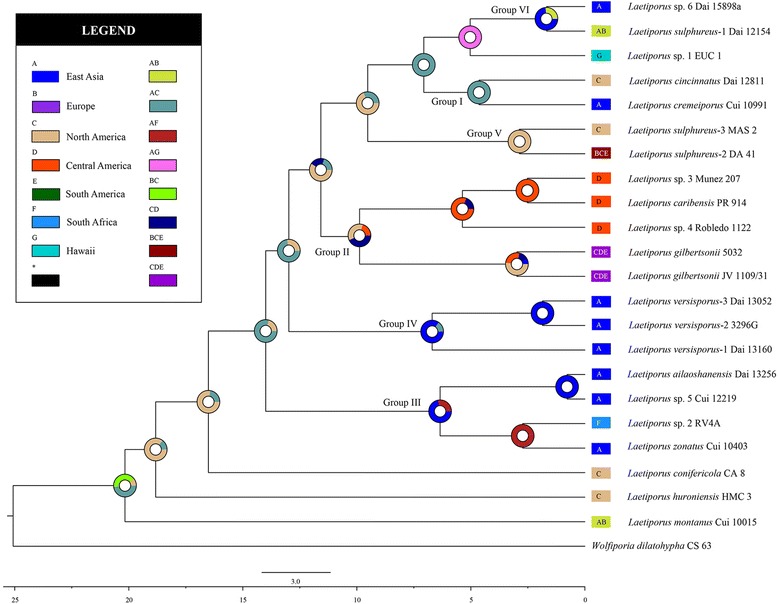
Divergence time estimation and ancestral area reconstruction of Laetiporus using the ITS dataset. The chronogram was obtained via molecular clock analysis using BEAST. A pie chart at each node indicates the possible ancestral distributions inferred from dispersal-extinction-cladogenesis (DEC) analysis implemented in RASP. A black asterisk represents other ancestral ranges
Fig. 5.
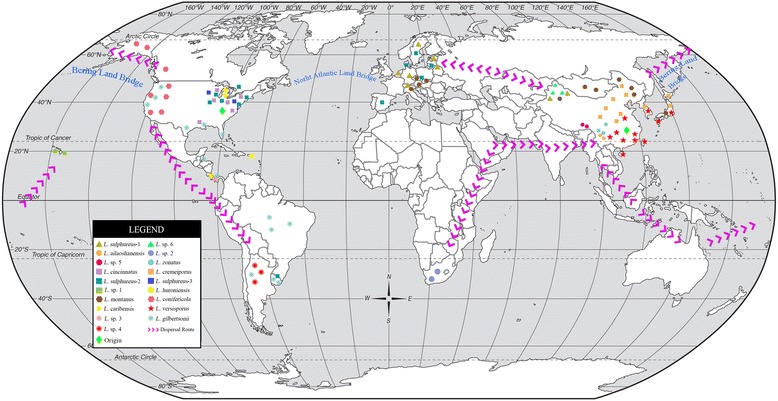
Map of the geographic distribution of Laetiporus and possible dispersal routes generated by ArcGIS v10.1. A hypothetical schematic depiction of the original locations, the migration routes, the rapid radiation and the speciation of Laetiporus
Discussion
Laetiporus has been shown to be a monophyletic group [11–15, 26]. Unexpectedly, despite conducting multi-locus phylogenetic analyses, our study is still unable to entirely resolve the stem relationships within Laetiporus. Nevertheless, novel phylogenetic species and certain clustering tendencies are described. Findings regarding the origin, ancestral area and diversification are also inferred.
Group I contains two sister clades, Clade D and Clade K, with disjunct distribution (Fig. 1). Phylogenetically, this group is supported by the combined dataset analyses (73% MP, 50% ML, 0.99 BPP). However, Clade D and Clade K are distant in the ITS topology. Previous studies showed that both species grow on hardwood with the common cool temperate to subtropical habitat, producing an orange pileal surface and cream pore surface (11, 13, 15). The complete gene information, as well as a similar growth habit and morphology between Clade D and Clade K, suggests that the phylogeny inferred from the analyses of the combined dataset is more reliable.
Group II consists of four North/Central/South American Laetiporus clades (Fig. 1). Within this group, Clade F is known to reside in temperate to tropical areas with a Pan-American distribution [10, 12, 27]. This distribution indicates a strong adaptive ability. The other three members of Laetiporus that behave as sister species are known to reside only in Central America [14], which is part of the Mesoamerican biodiversity hotspot [28]. Species in this group are found on hardwood and share an orange pileal surface and yellow pore surface, although the characters of Clade L and Clade M are uncertain [15]. Notably, their host plants are usually Fagaceae in North America, tropical plants such as Guarea and Dacryodes in Central America and mainly Eucalyptus in South America [11, 14, 27].
Group III contains four Laetiporus clades from East Asia and South Africa, including the novel phylogenetic species Clade P (Fig. 1). Clade P is found on Abies in cool temperate areas in the Himalayan region. It acts as a sister species with L. ailaoshanensis (Fig. 1), which has been found on Lithocarpus and Castanopsis in subtropical areas in the Hengduan Mountains [15]. Clade O is the other species collected from the Hengduan Mountains, and it grows on Quercus in temperate areas [15]. Clade H is found on Eucalyptus from South Africa, but its characters remain unclear [12]. The relationships between Clade H and the other three species are uncertain due to the low support in the topology of the combined dataset (Fig. 1). Further studies using samples from South Africa are necessary.
Group IV consists of only L. versisporus (Clade G), which has a yellow pore surface (Fig. 1). Previous studies have shown that this species is usually divided into two or three clades [13, 15]. In the current study, L. versisporus specimens grouped together with significant support from MP and ML analyses. L. versisporus covers most parts of East Asia from the Yunnan-Guizhou Plateau, Hainan to Japan and South Korea, and associate with Robinia, Castanea, Quercus, Elaeocarpus and Castanopsis [13, 15]. Infraspecific variation and infraspecific hybridization are considered to occur simultaneously [15].
Group V consists of Clade E1 and Clade E2 (Fig. 1). It is obvious that they are closely related and share similar morphology except for the pore surface [11]. Clade E1 is associated with Quercus, Eucalyptus, Salix, Acer and Fraxinus and has a disjunct temperate to subtropical areas distribution in North America, South America and Europe. Besides, it produces a yellow pore surface [10, 11]. Clade E2 is distributed in temperate areas of North America, is associated with Quercus and Fraxinus, and produces a white pore surface [10, 11].
Group VI consists of Clade C and the novel phylogenetic species Clade Q (Fig. 2). This group is only supported by the ITS phylogeny, and the phylogeny analyses do not indicate an obvious species boundary. This suggests a close relationship between Clade C and Clade Q. Laetiporus Clade C has previously been reported only from Europe [11, 13]. Our study presents the first report of Clade C in Xinjiang, China. This species usually grows on hardwoods and conifers such as Quercus, Sorbus, Populus, Castanea, Prunus, Taxus, Larix and Picea in temperate areas, producing a yellow pore surface. Clade Q is also found in temperate areas in Xinjiang, China, where it is associated with hardwoods such as Salix, Betula and Populus and produces a yellow pore surface.
The maximum crown age of Laetiporus is estimated at the early Miocene (20.17 ± 0.12 Mya) and East Asia and North America are inferred to be the most probable ancestral areas (Figs. 3 and 4). The notable finding is that three coniferous species (L. montanus, L. huroniensis and L. conifericola) in temperate areas behave as sister species in the analyses of the combined dataset (Fig. 1). Moreover, the temperate host plants are diverse, including Quercus, Salix, Populus, Picea, Larix, Abies, Tsuga, Lithocarpus, Fraxinus and Acer; in contrast, the tropical host plants are limited in variety, including Eucalyptus and Guarea [10–15]. Based on these findings, an origin in temperate East Asia and North America is proposed.
The independent sister species in Group I indicate an East Asian–eastern North American dispersal route before the estimated divergence time (4.64 Mya) in the early Pliocene (Fig. 4). This divergence time is close to the break time of the Bering Land Bridge (BLB) at approximately 5.4–5.5 Mya [5]. We speculate that their ancestor covered East Asia and North America via the BLB route and that regional speciation after the vicariance emerged due to the disconnection of the BLB and the severe climate change at that time [29–31]. This route is also present in the dispersal of other organisms, especially the common host plant Quercus [1]. There may be a strong dispersal and vicariance correlation between Laetiporus spp. and their host plants.
Four Laetiporus species in Group II with Pan-American distribution exhibit a North American–Central American–South American dispersal route. This group first diverged at approximately 9.88 Mya. North and Central America are inferred to be the most probable ancestral areas. Clade J, Clade L and Clade M are from Central America and the estimated crown age is approximately 5.38 Mya, which coincides with the paleo-elevations that occurred during the late Miocene and early Pliocene [32]. The second intercontinental distribution between North America and South America is exhibited in Group V (Fig. 4). This route has been confirmed by biogeographical research on plants and animals [1, 33–36]. We speculate that the severe climate change that has occurred since 15 Mya [29] drove the migration from North and Central America to South America and the adaptation to tropical host plants such as Eucalyptus, Guarea and Dacryodes. The vicariance due to tectonic activity is thought to be responsible for the endemism of Laetiporus in Central America.
In Group III, four Laetiporus species from East Asia and South Africa are closely related (Fig. 1). The estimated divergence time of this group is 6.35 Mya. The DEC analysis inferred East Asia as the most probable ancestral area. However, it is notable that Clade H does not form a robust sister relationship with Clade O (Fig. 1). We speculate that there is incomplete sampling from the Indian Subcontinent to Africa because suitable host plants, such as Eucalyptus, are abundant in these areas [12, 37]. Although the estimated divergence time is potentially inaccurate, the dispersal route between East Asia and South Africa is proposed.
The species in Group V also exhibit a continuous distribution in Europe and eastern North America (Fig. 4). The DEC analysis inferred a North American origin for this group, with an estimated divergence time of 2.89 Mya. Clade E1 is found in the eastern North America and Europe with low host-plant specificity. The short-lived North Atlantic Land Bridge acted as a dispersal route until the low Oligocene [6, 36]. Migration to Europe seems unlikely, so the reasonable interpretation is that the human activity introduced this species into new habitats as proposed by Feng et al. [3]. The wind and ocean current could be another driving force and reasonable explanation for the dispersal of fungal basidiospores between Europe and eastern North America [8].
The species in Group VI and Clade A2 have an East Asian-European dispersal route. This route is probable because an exchange of species occurs for Laetiporus and its most common host plants such as Quercus, Salix, Populus, Picea, Abies and Larix [1, 22–25, 38]. It is reasonable to accept this route because the Eurasian Plate is continuous.
Group IV consists of three different types of L. versisporus that are endemic in East Asia (Figs. 1 and 4). The infraspecific variation is obvious in these three types, but gene exchange and recombination still exist according to the clonal research of Ota et al. [13]. This finding indicates that vicariance is important for regional speciation.
In addition, the migration of Clade I to Hawaii is surprising and worth exploring. We speculate that this example results from an incomplete sampling of molecular data. However, there are many standalone islands in the South Pacific indirectly connecting Hawaii, Australia and Malay Archipelago. The frequent strong winds and continuous ocean currents are potentially responsible for the dispersal of basidiospores between islands. The humid climate and abundant host plants such as Quercus, Castanea and Eucalyptus from the Malay Archipelago to Australia [39, 40] are suitable for Laetiporus. A dispersal route of East Asia–Malay Archipelago–Australia–Hawaii seems unlikely. Interestingly, Eucalyptus, the host plant of Clade I has been proven to colonize Hawaii via this route [2].
In our study, the samples of Laetiporus are scanty in some areas around the world, such as South America, Indian Subcontinent, South Africa and Australia. The taxonomic situation is still unclear, and the evolutionary history of Laetiporus remains incompletely understood. A wider range of sampling and further morphological studies, incompatibility tests, and more information of host range and distribution are needed.
Conclusion
The evolutionary history of Laetiporus remains incompletely understood. However, this study presents some progress on this topic. (1) Two novel phylogenetic species in East Asia were identified. (2) Our reconstruction and analysis of ancestral areas suggest that Laetiporus originated during the early Miocene (20.16 ± 0.13 Mya) in temperate zones and that the combination of East Asia and North America has the highest probability (48%) of being the ancestral area. (3) We also predict that Laetiporus may be present in the Indian Subcontinent, in Australia and in the Malay Archipelago. (4) Four intercontinental dispersal routes and a possible concealed dispersal route are proposed. (5) Vicariance is suggested to play an important role in regional speciation, and recent human activity may render some geographical distribution inexplicable. Further sampling and more molecular data are needed to further clarify the species affinity.
Methods
Taxon sampling
This study included 105 samples of Laetiporus from East Asia, Europe, North America, Central America, South America, Hawaii and South Africa. Basidiomata of several Laetiporus species were shown in Fig. 6. The sequences of the samples obtained for this study were deposited in the herbaria of the Institute of Microbiology, Beijing Forestry University (BJFC), Institute of Microbiology, Chinese Academy of Sciences (HMAS), and Institute of Applied Ecology, Chinese Academy of Sciences (IFP). Each specimen’s scientific name, GenBank accession numbers and other relevant information are listed in Additional file 1: Table S2.
Fig. 6.

Basidiomata of Laetiporus species. a–L. ailaoshanensis. b–L. cremeiporus. c–L. montanus. d–L. sulphurous. e–L. zonatus. f–L. versisporus
DNA extraction, PCR, and DNA sequencing
Genomic DNA was extracted from dried fruiting bodies using a cetyltrimethylammonium bromide rapid plant genome extraction kit (Aidlab Biotechnologies Co., Ltd., Beijing) according to the manufacturer’s instructions with some modifications [26]. Six DNA gene fragments were analyzed, including those coding for RPB2 and EF-1α, along with four non-protein coding regions: ITS, nrLSU, nrSSU and mtSSU. These fragments were actually appropriate in determining the taxonomic status of Laetiporus. The primer pairs ITS5/4 [29], LR0R/LR7 [41], PNS1/NS41 [42], MS1/MS2 [29], and 983F/1567R [43] were used to amplify ITS, nrLSU, nrSSU, mtSSU and EF-1α, respectively. Initial attempts to amplify RPB2 using previously published primers that were designed for fungi [44] resulted in weak or non-specific amplification. To improve the success rate of RPB2 amplification, a new primer pair, 6F-1 (CCTCGTCAACTGCACAACA) and 7R-1 (TCTTCCTCGGCATCCAA), was designed based on eleven obtained sequences using Primer-Premier 5 (Premier Biosoft International, Palo Alto, CA, USA).
PCR was performed in a reaction mixture containing 25 μl of 2 × EasyTaq® PCR SuperMix, 2 μl of Forward Primer (10 μM), 2 μl of Reverse Primer (10 μM), and 2 μl of Template DNA. The total volume was adjusted to 50 μl with sterile deionized H2O. The PCR amplifications were conducted using an Eppendorf Master Cycler (Eppendorf, Netheler-Hinz, Hamburg, Germany), and the cycling conditions were follows: pre-denaturation at 95 °C for 4 min; 35 cycles of denaturation at 94 °C for 40 s, annealing at 50 °C–54 °C for 45 s (ITS, mtSSU, EF-1α and RPB2) or for 60 s (nrLSU and nrSSU), and elongation at 72 °C for 60 s (ITS, mtSSU, EF-1α and RPB2) or for 90 s (nrLSU and nrSSU); and a final elongation at 72 °C for 10 min. The PCR products were visualized by agarose gel electrophoresis and stored at −20 °C after visualization. The PCR products were purified and sequenced at the Beijing Genomics Institute (China) using the same primers as those used for amplification. Of the 370 sequences of Laetiporus used in this paper, 226 sequences of Laetiporus were newly generated, including 28 ITS (27% new), 27 nrLSU (40% new), 41 nrSSU (100% new), 47 mtSSU (78% new), 46 EF-1α (85% new), and 37 RPB2 (88% new). All newly generated sequences were deposited in the GenBank database.
Sequence alignments and phylogenetic analyses
To determine the phylogeny of Laetiporus, we compiled two datasets: the ITS sequences matrix and a concatenated dataset (ITS + nrLSU + nrSSU + mtSSU + EF-1α + RPB2). In the combined dataset, Antrodia serialis (Fr.) Donk and Fomitopsis pinicola (Sw.) P. Karst. were used as outgroups; the sequences of ITS, nrLSU, nrSSU, mtSSU, EF-1α and RPB2 were aligned initially by using MAFFT 6 [45] using the “G-INS-I” strategy and then manually optimized in BioEdit [46]. Ambiguously aligned regions were excluded from subsequent analyses. Finally, the six gene fragments were concatenated with SEAVIEW 4 [47] for further phylogenetic analyses. One thousand partition homogeneity test (PHT) replicates of the ITS, nrLSU, nrSSU, mtSSU, EF-1α and RPB2 sequences were tested using PAUP* version 4.0b10 [48] to determine whether the partitions were homogeneous. The PHT results indicated that all the DNA sequences had a congruent phylogenetic signal (P value =0.19). The ITS dataset included more samples compared to the combined dataset. It contained 100 sequences, of which 98 were Laetiporus sequences; Wolfiporia dilatohypha Ryvarden & Gilb. was used as an outgroup. The sequences were aligned using the same method as that used for the combined dataset. Sequence alignments were deposited at TreeBase (submission ID 20418, 20,419; www.treebase.org).
ML analysis was conducted using RAxML-HPC2 [49] on Abe through the Cipres Science Gateway [50]. To estimate the branch support with an alternative method, we performed BI and MP analyses. For the ML and BI analyses, the optimal substitution models for ITS and the combined dataset were determined using the Akaike information criterion (AIC) as implemented in MrModeltest v2.3 [51, 52]. The selected substitution models for both the combined dataset and ITS dataset were general time reversible + proportion invariant + gamma (GTR + I + G).
In the ML analysis, the concatenated dataset was partitioned into six parts by sequence region, and 1000 ML searches were run under the GTR + GAMMA model with all model parameters estimated using the RAxML-HPC2 program. The best fit maximum likelihood tree from all searches was kept. In addition, 1000 rapid bootstrap replicates were run with the GTR + CAT model to assess the reliability of the nodes.
BI was performed using MrBayes 3.1.2 [53] with 2 independent runs, each beginning from random trees with 4 simultaneous independent chains, performing 5,000,000 replicates each for the concatenated dataset and the ITS dataset, sampling one tree every 1000 generations. Chain convergence was determined using Tracer v1.5 (http://tree.bio.ed.ac.uk/software/tracer/) to confirm sufficiently large ESS values (>200). The first 25% of the sampled trees were discarded as burn-in, and the remaining trees were used to reconstruct a majority rule consensus and calculate BPP of the clades.
MP analysis was performed in PAUP* version 4.0b10 [48]. All characters were equally weighted, and gaps were treated as missing data. Trees were inferred using the heuristic search option with TBR branch swapping and 1000 random sequence additions. Max-trees were set to 5000, branches of zero length were collapsed, and all parsimonious trees were saved. Clade robustness was assessed using a bootstrap (BT) analysis with 1000 replicates [54].
Branches of the consensus tree that received bootstrap support for MP, ML and BPP greater than or equal to 75% (MP/ML) and 0.95 (BPP) were considered to be significantly supported.
Molecular dating analysis
Given that fossil records of fungi are limited, it is difficult to choose a reliable calibration point to estimate the divergence time for any fungal groups. Therefore, extensive sampling of outgroup species for which fossils were available was performed in order to estimate the divergence time of Laetiporus. Two primary calibration points were included in our analyses: (1) the divergence between Ascomycota and Basidiomycota, 582 Mya, by placing P. devonicus in the subphylum Pezizomycotina [55]; and (2) the divergence between Hymenochaetaceae and Fomitopsidaceae based on a 125 million-year-old fossil of Q. cranhamii [56]. The parameter settings for the two calibrations were the same as those used in Feng et al. [3]. As the identifications of the two fossils were fairly ambiguous, the estimated divergence time was constrained by the following two values: the estimated divergence time between Ascomycota and Basidiomycota is at least 400 Mya (the divergence time of P. devonicus), and the initial diversification of Laetiporus should be close to the divergence times of their host plants as suggested by the co-evolution of the fungi and the plants [57]. The calibration point for which the estimated results met these two criteria was eventually chosen for our subsequent analyses.
Three nuclear ribosomal RNA genes (ITS, nrLSU and nrSSU) and two protein coding genes (EF-1α and RPB2) were concatenated for molecular dating using the phylogenetic framework described in James et al. [58]. ITS1, ITS2, and the introns in EF-1α and RPB2 were excluded for a conservation analysis. All of the outgroup sequences were retrieved from GenBank and are listed in Additional file 1: Table S2. MrModeltest v2.3 [51, 52] was used to select the best models of evolution using the hierarchical likelihood ratio test. The selected evolutionary models for the two combined datasets were GTR + I + G. The origin time of Laetiporus was estimated in BEAST v1.8.0 [59] with the molecular clock and substitution models unlinked but with the trees linked for each gene partition. The BEAST input files were constructed using BEAUti (within BEAST). The lognormal relaxed molecular clock and the Yule speciation prior set were used to estimate the divergence time and the corresponding credibility intervals. The posterior distributions of parameters were obtained using MCMC analysis for 50 million generations with a burn-in percentage of 10%. The convergence of the chains was confirmed using Tracer v1.6. Samples from the posterior distributions were summarized on a maximum clade credibility tree with the maximum sum of posterior probabilities listed on its internal nodes using TreeAnnotator v1.8.0 [59] with the posterior probability limits set to 0.5 to summarize the mean node heights. FigTree v1.4.2 [60] was used to visualize the resulting tree and to obtain the means and 95% HPD [59]. A 95% HPD marks the shortest interval that contains 95% of the values sampled.
We also estimated the divergence time of the main nodes in Laetiporus using the ITS dataset, which contained one or two representatives of all of the Laetiporus species that were included in our analyses. The estimated crown age of Laetiporus based on the combined ITS + nrLSU + nrSSU and EF-1α + RPB2 datasets was used as the calibration point to date the ITS phylogeny by setting the prior to a normal distribution. The other procedures were the same as those applied in the estimation using the combined dataset.
Biogeographic analysis
The reconstruction of ancestral areas in a phylogeny is important for understanding the biogeographic diversification history of a lineage, as this reconstruction makes it possible to infer the original location and dispersal routes of the organisms. To infer ancestral areas, the DEC [61] model was used in RASP 3.2 [62], allowing a maximum of two areas per node. The ancestral area analyses were conducted using the posterior distributions of the dated ITS phylogeny that were estimated from BEAST. The geographic distributions for the Laetiporus were delimited into seven areas: (A) East Asia, (B) Europe, (C) North America, (D) Central America, (E) South America, (F) South Africa, and (G) Hawaii. ArcGIS v10.1 [63] was used to visualize the geographic distribution and possible dispersal routes of Laetiporus.
Acknowledgments
The authors extend special thanks to Drs. Shuang-Hui He (BJFC, China) and Xiao-Lan He (SAAS, China) for collecting specimens, as well as to Drs. Mario Rajchenberg (CIEFAP, Argentina) and Michal Tomšovský (BRNU, Czech Republic) for forwarding specimens for our study. We thank Drs. Beatriz Ortiz-Santana (CFMR, USA) and Wanda Daley (PDD, New Zealand), Mr. Pertii Salo (H, Finland) and the curator of the Queensland Herbarium (BRI, Australia) for the loaning of specimens.
Funding
This research was financed by the National Natural Science Foundation of China (Project Nos. 31422001, 31170018) and the Fundamental Research Funds for the Central Universities (No. 2016ZCQ04).
Availability of data and materials
Sequence alignments are deposited at TreeBase (submission ID 20418, 20,419; www.treebase.org). All other data generated or analyzed during this study are included in this published article. Requests for materials should be made to the corresponding authors.
Authors’ contributions
BK C and J S designed the experiment; JS conducted the molecular experiments; and J S and BK C analyzed the data and drafted the manuscript. All of the authors approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- BI
Bayesian inference
- BJFC
Herbaria of the Institute of Microbiology, Beijing Forestry University
- BPP
Bayesian posterior probabilities
- DEC
Dispersal-Extinction-Cladogenesis
- EF-1α
Translation elongation factor 1-α
- GTR + I + G
General time reversible + proportion invariant + gamma
- HMAS
Institute of Microbiology, Chinese Academy of Sciences
- HPD
Higher posterior densities
- IFP
Institute of Applied Ecology, Chinese Academy of Sciences
- ITS
Internal transcribed spacer
- ML
Maximum likelihood
- MP
Maximum parsimony
- mtSSU
Mitochondrial small subunit rDNA
- Mya
Million years ago
- nrLSU
Nuclear large subunit rDNA
- nrSSU
Nuclear small subunit rDNA
- RPB2
DNA-directed RNA polymerase II subunit 2
Additional file
Estimated divergence times of the main nodes correspond with the dating analysis of ITS + nrLSU + nrSSU and EF-1α + RPB2 datasets. Table S2. Information about the samples used in this study. Table S3. Estimated divergence times of the main groups correspond with the dating analysis of ITS datasets. (DOCX 56 kb)
Contributor Information
Jie Song, Email: super_jasone@yahoo.com.
Bao-Kai Cui, Phone: +86-10-62336309, Email: baokaicui2013@gmail.com.
References
- 1.Manos PS, Stanford AM. The historical biogeography of Fagaceae: tracking the tertiary history of temperate and subtropical forests of the northern hemisphere. Int J Plant Sci. 2001;162:S77–S93. doi: 10.1086/323280. [DOI] [Google Scholar]
- 2.Percy DM, Garver AM, Wagner WL, James HF. Progressive island colonization and ancient origin of Hawaiian Metrosideros (Myrtaceae) Proc R Soc B. 2008;275:1479–1490. doi: 10.1098/rspb.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng B, Xu JP, Wu G, Zeng NK, Li YC, Tolgor B, et al. DNA sequences analyses reveal abundant diversity, endemism and evidence for Asian origin of the porcini. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckert AJ, Hall BD. Phylogeny, historical biogeography, and patterns of diversification for Pinus (Pinaceae): phylogenetic tests of fossil-based hypotheses. Mol Phylogenet Evol. 2006;40:166–182. doi: 10.1016/j.ympev.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Milne RI. Northern hemisphere plant disjunctions: a window on tertiary land bridges and climate change? Ann Bot. 2006;98:465–472. doi: 10.1093/aob/mcl148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Q, Tulloss RE, Tang LP, Tolgor B, Zhang P, Chen ZH, et al. 2014. Multi-locus phylogeny of lethal amanitas: implications for species diversity and historical biogeography. BMC Evol Biol. 2014;14:143. [DOI] [PMC free article] [PubMed]
- 7.Chen JJ, Cui BK, Zhou LW, Korhonen K, Dai YC. Phylogeny, divergence time estimation, and biogeography of the genus Heterobasidion (Basidiomycota, Russulales) Fungal Divers. 2015;71:185–200. doi: 10.1007/s13225-014-0317-2. [DOI] [Google Scholar]
- 8.Song J, Chen JJ, Wang M, Chen YY, Cui BK. Phylogeny and biogeography of the remarkable genus Bondarzewia (Basidiomycota, Russulales) Sci Rep. 2016;6:34568. doi: 10.1038/srep34568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murrill WA. The Polyporaceae of North America 9 Inonotus, Sesia and monotypic genera. Bull Bot Club. 1904;31:593–610. doi: 10.2307/2478612. [DOI] [Google Scholar]
- 10.Burdsall HH, Banik MT. The genus Laetiporus in North America. Harv Pap Bot. 2001;6:43–55. [Google Scholar]
- 11.Lindner DL, Banik MT. Molecular phylogeny of Laetiporus and other brown-rot polypore genera in North America. Mycologia. 2008;100:417–430. doi: 10.3852/07-124R2. [DOI] [PubMed] [Google Scholar]
- 12.Vasaitis R, Menkis A, Lim YW, Seok S, Tomšovský M, Jankovský L, et al. Genetic variation and relationships in Laetiporus sulphureus s. Lat., as determined by ITS rDNA sequences and in vitro growth rate. Mycol Res. 2009;113:326–336. doi: 10.1016/j.mycres.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Ota Y, Hattori T, Banik MT, Hagedorn G, Sotome K, Tokuda S, et al. The genus Laetiporus (Basidiomycota, Polyporales) in East Asia. Mycol Res. 2009;113:1283–1300. doi: 10.1016/j.mycres.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Banik MT, Lindner DL, Ortiz-Santana B, Lodge DJ. A new species of Laetiporus (Basidiomycota, Polyporales) from the Caribbean basin. Kurtziana. 2012;37:15–21. [Google Scholar]
- 15.Song J, Chen YY, Cui BK, Liu HG, Wang YZ. Morphological and molecular evidence for two new species of Laetiporus (Basidiomycota, Polyporales) from southwestern China. Mycologia. 2014;106:1039–1050. doi: 10.3852/13-402. [DOI] [PubMed] [Google Scholar]
- 16.Dai YC, Cui BK, Yuan HS, Li BD. Pathogenic wood-decaying fungi in China. Forest Pathol 2007;37: 105–120.
- 17.Sinclair W A, Lyon HH, Johnson WT. Diseases of trees and shrubs. Cornell University Press; 1987.p. 251–306.
- 18.Kovács D, Vetter J. Chemical composition of the mushroom Laetiporus sulphureus (Bull.) Murill. Acta Aliment Hung. 2015;44:104–110. doi: 10.1556/AAlim.44.2015.1.10. [DOI] [Google Scholar]
- 19.Saba E, Son Y, Jeon BR, Kim S, Lee I, Yun B, et al. Acetyl eburicoic acid from Laetiporus sulphureus var. miniatus suppresses inflammation in murine macrophage RAW 264.7 cells. Mycobiology. 2016;43:131–136. doi: 10.5941/MYCO.2015.43.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez M, Torrez AS, Campi MG, Bravo JA, Vila JL. Ergosterol from the mushroom Laetiporus sp.: isolation and structural characterization. Revista Boliviana de Química. 2015;32:90–94. [Google Scholar]
- 21.Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, et al. The Paleozonic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 22.Ran JH, Wei XX, Wang XQ. Molecular phylogeny and biogeography of Picea (Pinaceae): implications for phylogeographical studies using cytoplasmic haplotypes. Mol Phylogenet Evol. 2006;41:405–419. doi: 10.1016/j.ympev.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 23.Aguirre-Planter É, Jaramillo-Correa JP, Gómez-Acevedo S, Khasa DP, Bousquet J, Eguiarte LE. Phylogeny, diversification rates and species boundaries of Mesoamerican firs (Abies, Pinaceae) in a genus-wide context. Mol Phylogenet Evol. 2012;62:263–274. doi: 10.1016/j.ympev.2011.09.021. [DOI] [PubMed] [Google Scholar]
- 24.Cronk QCB, Needham I, Rudall PJ. Evolution of catkins: inflorescence morphology of selected Salicaceae in an evolutionary and developmental context. Front Plant Sci. 2015;6:1030. doi: 10.3389/fpls.2015.01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du SH, Wang ZS, Ingvarsson P, Wang DS, Wang JH, Wu ZQ, et al. Multilocus analysis of nucleotide variation and speciation in three closely related Populus (Salicaceae) species. Mol Ecol. 2015;24:4994–5005. doi: 10.1111/mec.13368. [DOI] [PubMed] [Google Scholar]
- 26.Han ML, Chen YY, Shen LL, Song J, Vlasák J, Dai YC, et al. Taxonomy and phylogeny of the brown-rot fungi: Fomitopsis and its related genera. Fungal Divers. 2016;80:343–73.
- 27.Pires RM, Motato-Vásquez V, Gugliotta ADM. A new species of Laetiporus (Basidiomycota) and occurrence of L. gilbertsonii Burds. In Brazil. Nova Hedwigia. 2016;102:477–490. doi: 10.1127/nova_hedwigia/2016/0320. [DOI] [Google Scholar]
- 28.Harvey CA, Komar O, Chazdon RL, Ferguson BG, Finegan B, Griffith DM, et al. Integrating agricultural landscapes with biodiversity conservation in the Mesoamerican hotspot. Conserv Biol. 2008;22:8–15. doi: 10.1111/j.1523-1739.2007.00863.x. [DOI] [PubMed] [Google Scholar]
- 29.White TJ, Bruns TD, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols, a guide to methods and applications. California Academic Press: San Diego; 1990. pp. 315–322. [Google Scholar]
- 30.Tiffney BH, Manchester SR. The use of geological and paleontological evidence in evaluating plant phylogeographic hypotheses in the northern hemisphere tertiary. Int J Plant Sci. 2001;162:S3–S17. doi: 10.1086/323880. [DOI] [Google Scholar]
- 31.Zachos J, Pagani M, Sloan L, Thomas E, Billups K. Trends, rhythms, and aberrations in global climate 65 ma to present. Science. 2001;292:686–693. doi: 10.1126/science.1059412. [DOI] [PubMed] [Google Scholar]
- 32.Burnham RJ, Graham A. The history of neotropical vegetation: new developments and status. Ann Mo Bot Gard. 1999;86:546–589. doi: 10.2307/2666185. [DOI] [Google Scholar]
- 33.Cortés-Ortiz L, Bermingham E, Rico C, Rodriguez-Luna E, Sampaio I, Ruiz-Garcia M. Molecular systematics and biogeography of the neotropical monkey genus, Alouatta. Mol Phylogenet Evol. 2003;26:64–81. doi: 10.1016/S1055-7903(02)00308-1. [DOI] [PubMed] [Google Scholar]
- 34.Dick CW, Abdul-Salim K, Bermingham E. Molecular systematic analysis reveals cyptic tertiary diversification of a widespread tropical rain forest tree. Am Nat. 2003;162:691–703. doi: 10.1086/379795. [DOI] [PubMed] [Google Scholar]
- 35.Morley RJ. Interplate dispersal paths for megathermal angiosperms. Perspect Plant Ecol. 2003;6:5–20. doi: 10.1078/1433-8319-00039. [DOI] [Google Scholar]
- 36.Zamora-Tavares MDP, Martínez M, Magallón S, Guzmán-Dávalos L, Vargas-Ponce O. Physalis and physaloids: a recent and complex evolutionary history. Mol Phylogenet Eovl. 2016;100:41–50. doi: 10.1016/j.ympev.2016.03.032. [DOI] [PubMed] [Google Scholar]
- 37.Crame JA, Owen AW. Palaeobiogeography and biodiversity change: the Ordovician and Mesozoic-Cenozoic radiations. Geological society special publication. 2002. [Google Scholar]
- 38.Cerling TE, Harris JM, MacFadden BJ, Leakey MG, Quade J, Eisenmann V, et al. Global vegetation change through the Miocene/Pliocene boundary. Nature. 1997;389:153–158. doi: 10.1038/38229. [DOI] [Google Scholar]
- 39.Hopper SD, Gioia P. The southwest Australian floristic region: evolution and conservation of a global hot spot of biodiversity. Annu Rev Ecol Evol S. 2004;35:623–650. doi: 10.1146/annurev.ecolsys.35.112202.130201. [DOI] [Google Scholar]
- 40.Swee-Hock S. The population of Malaysia. Institute of Southeast Asian Studies. 2007. [Google Scholar]
- 41.Vilgalys R, Hester M. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J Bacteriol. 1990;172:4238–4246. doi: 10.1128/jb.172.8.4238-4246.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hibbett DS. Phylogenetic evidence for horizontal transmission of group I introns in the nuclear ribosomal DNA of mushroom-forming fungi. Mol Biol Evol. 1996;13:903–917. doi: 10.1093/oxfordjournals.molbev.a025658. [DOI] [PubMed] [Google Scholar]
- 43.Rehner SA, Buckley E. A Beauveria phylogeny inferred from nuclear ITS and EF-1α sequences: evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia. 2005;97:84–98. doi: 10.3852/mycologia.97.1.84. [DOI] [PubMed] [Google Scholar]
- 44.Liu YJ, Wheelen S, Hall BD. Phylogenetic relationships among ascomycetes: evidence from an RNA polymerase II subunit. Mol Biol Evol. 1999;16:1799–1808. doi: 10.1093/oxfordjournals.molbev.a026092. [DOI] [PubMed] [Google Scholar]
- 45.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 46.Hall TA. Bioedit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 47.Gouy M, Guidon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 48.Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods) Sunderland: Sinauer Associates; 2002. [Google Scholar]
- 49.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 50.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. New Orleans: Proceedings of the Gateway Computing Environments Workshop (GCE); 2010. pp. 1–8. [Google Scholar]
- 51.Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 52.Nylander JAA. MrModeltest v2. Program distributed by the author: Evolutionary Biology Centre, Uppsala University; 2004. [Google Scholar]
- 53.Ronquist F, Huelsenbeck JP. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 54.Felsenstein J. Confidence limits on phylogenetics: an approach using bootstrap. Evolution. 1985;39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 55.Berbee ML, Taylor JW. Dating the molecular clock in fungi – how close are we? Fungal Biol Rev. 2010;24:1–16. doi: 10.1016/j.fbr.2010.03.001. [DOI] [Google Scholar]
- 56.Smith SY, Currah RS, Stockey RA. Cretaceous and Eocene poroid hymenophores from Vancouver Island, British Columbia. Mycologia. 2004;96:180–186. doi: 10.1080/15572536.2005.11833010. [DOI] [PubMed] [Google Scholar]
- 57.Hibbett DS, Matheny PB. The relative ages of ectomycorrhizal mushrooms and their plant hosts estimated using Bayesian relaxed molecular clock analyses. BMC Biol. 2009;7:13. doi: 10.1186/1741-7007-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ, et al. Reconstructing the early evolution of fungi using a six-gene phylogeny. Nature. 2006;443:818–822. doi: 10.1038/nature05110. [DOI] [PubMed] [Google Scholar]
- 59.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214–221. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evolution M. Phylogenetics and epidemiology. 2007. [Google Scholar]
- 61.Ree RH, Smith SA. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol. 2008;57:4–14. doi: 10.1080/10635150701883881. [DOI] [PubMed] [Google Scholar]
- 62.Yu Y, Harris AJ, Blair C, He XJ. RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol Phylogenet Evol. 2015;87:46–49. doi: 10.1016/j.ympev.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 63.ArcGIS Platform. http://esri.com/arcgis. Accessed 18 Sep 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequence alignments are deposited at TreeBase (submission ID 20418, 20,419; www.treebase.org). All other data generated or analyzed during this study are included in this published article. Requests for materials should be made to the corresponding authors.